Introduction
Ovarian cancer is the most life-threatening type of
gynecological cancer, with a mortality rate of almost 14,000 in the
United States alone in 2010 (1).
The five-year survival rate for all stages of ovarian cancer is 47%
(2). The poor prognosis results
from the lacks of early detection or screening assessments, which
leads to the majority of cases being undiagnosed until they have
reached advanced stages.
Platinum-based cancer chemotherapy has been the
general treatment approach for ovarian cancer for decades (3). However, >80% of patients
eventually relapse with fully chemoresistant disease (4). The antitumor activity of cisplatin is
based upon DNA damage via the formation of cisplatin-DNA adducts
(5). The accumulation of DNA
lesions can lead to steric obstruction of DNA-binding proteins,
which are necessary for vital intracellular functions, and
recognition of the lesions by high mobility group and mismatch
repair proteins eventually lead to p53-initiated apoptosis
(6–8). In addition, activation of the
endoplasmic reticulum stress pathway also causes activation of
apoptotic caspases (9).
Reduced drug uptake, decreased binding of cisplatin
to DNA, DNA repair, decreased mismatch repair and impaired
apoptosis have been considered as potential molecular mechanisms
responsible for the platinum-based drug resistance (10–12).
Lee et al observed that activation of the phosphoinositide
3-kinase/Akt pathway by phosphatase and tensin homolog reduction
contributed to cisplatin resistance in an ovarian cancer cell line
(13). Yang et al indicated
that Akt leads to resistance via modulation of the action of p53 on
the caspase-dependent mitochondrial death pathway (14). Li et al examined epigenetic
changes and reported that DNA methylation is key in chemoresistance
in ovarian cancer (15).
To further investigate altered gene expression
profiles and relevant biological pathways, the present study
performed a global and comparative analysis of the gene expression
data between cisplatin-resistant ovarian cancer cells and
cisplatin-sensitive ovarian cancer cells using bioinformatic tools,
including functional enrichment analysis and protein-protein
interaction (PPI) network analysis. The findings may advance
current understanding of the molecular mechanisms underlying
cisplatin resistance, and thus benefit the development of more
effective approaches in the treatment of ovarian cancer.
Materials and methods
Gene expression data
The gene expression data (accession no. GSE15372)
were downloaded from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), and included
five biological replicates of cisplatin-sensitive A2780 epithelial
ovarian cancer cells and five biological replicates of
cisplatin-resistant Round5 A2780 epithelial ovarian cancer cells
(Table I). The gene expression
profiles were acquired using the Affymetrix Human Genome U133 Plus
2.0 array (Affymetrix Inc., Santa Clara, California, USA).
 | Table ISummary of the five
cisplatin-sensitive and five cisplatin-resistant replicates,
obtained from the Gene Expression Omnibus. |
Table I
Summary of the five
cisplatin-sensitive and five cisplatin-resistant replicates,
obtained from the Gene Expression Omnibus.
| Accession | Description |
|---|
| GSM385721 | Parental A2780
(cisplatin-sensitive), biological replicate 1 |
| GSM385722 | Parental A2780
(cisplatin-sensitive), biological replicate 2 |
| GSM385723 | Parental A2780
(cisplatin-sensitive), biological replicate3 |
| GSM385724 | Parental A2780
(cisplatin-sensitive), biological replicate 4 |
| GSM385725 | Parental A2780
(cisplatin-sensitive), biological replicate 5 |
| GSM385726 | Round5 A2780
(cisplatin-resistant), biological replicate 1 |
| GSM385727 | Round5 A2780
(cisplatin-resistant), biological replicate 2 |
| GSM385728 | Round5 A2780
(cisplatin-resistant), biological replicate 3 |
| GSM385729 | Round5 A2780
(cisplatin-resistant), biological replicate 4 |
| GSM385730 | Round5 A2780
(cisplatin-resistant), biological replicate 5 |
Pre-treatment of raw data and
differential analysis
The raw data in CEL format were read using the affy
package in R (http://www.r-project.org) (16). Normalization was performed using a
Robust Multi-array which consisted of three steps: Background
adjustment, quantile normalization, and summarization (17). Gene expression values were averaged
to calculate the final expression value for multiple probes
corresponding to the same gene symbols. mRNAs, which were not
detected in all samples were removed using the Affymetrix
Microarray Suite 5 calls (MAS5CALLS) algorithm (Affymetrix,
Inc.).
Differential analysis was performed using the limma
package in R (18).
P<0.05 and |log2 (fold change)|>1 were set as the
cut-off values to screen out the differentially expressed genes
(DEGs).
Functional enrichment analysis
To determine the biological pathways altered in
cisplatin-resistant ovarian cancer, Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses
were performed on the DEGs using Database for Annotation,
Visualization and Integration Discovery (DAVID; http://david.abcc.ncifcrf.gov/) (19). P<0.05 was set as the cut-off
value.
Construction of the protein-protein
interaction (PPI) network
The PPI network was constructed for the DEGs using
information provided by the Search Tool for the Retrieval of
Interacting Genes (STRING) (http://string-db.org/) (20), and was subsequently visualized
using Cytoscape (http://cytoscape.org) (21). Interactions with a score >0.4
were retained in the network. Proteins in the network served as the
‘nodes’, and each pairwise protein interaction, referred to as an
‘edge’, was presented as an undirected link. The sub-networks were
then analyzed by Clustering with Overlapping Neighborhood Expansion
(ClusterONE) (http://www.paccanarolab.org/clusterone) (22).
Results
Differentially expressed genes
A total of 69,954 transcripts were obtained from the
raw data using the affy package and annotation files. Following
removal of blank transcripts using the MAS5CALLS algorithm, 47,643
transcripts with expression levels were retained, from which 1,887
differentially expressed transcripts were identified in the
cisplatin-sensitive ovarian cancer cells, including 815 upregulated
transcripts, corresponding to 246 genes, and 1,072 downregulated
transcripts, corresponding to 310 genes.
Functional enrichment analysis
results
The KEGG pathway enrichment analysis revealed that
the metabolism-associated pathways, hsa00900 (terpenoid backbone
biosynthesis), hsa00100 (steroid biosynthesis), hsa00020 (citrate
cycle), hsa03030 (DNA replication) and hsa04110 (cell cycle) were
enriched in the downregulated genes (Fig. 1). These pathways were associated
with cell proliferation, which was inhibited by drugs in the
cisplatin-sensitive cells. A total of 118 significant GO biological
pathway terms were identified in the downregulated genes, which
were divided into 12 clusters, of which two were associated with
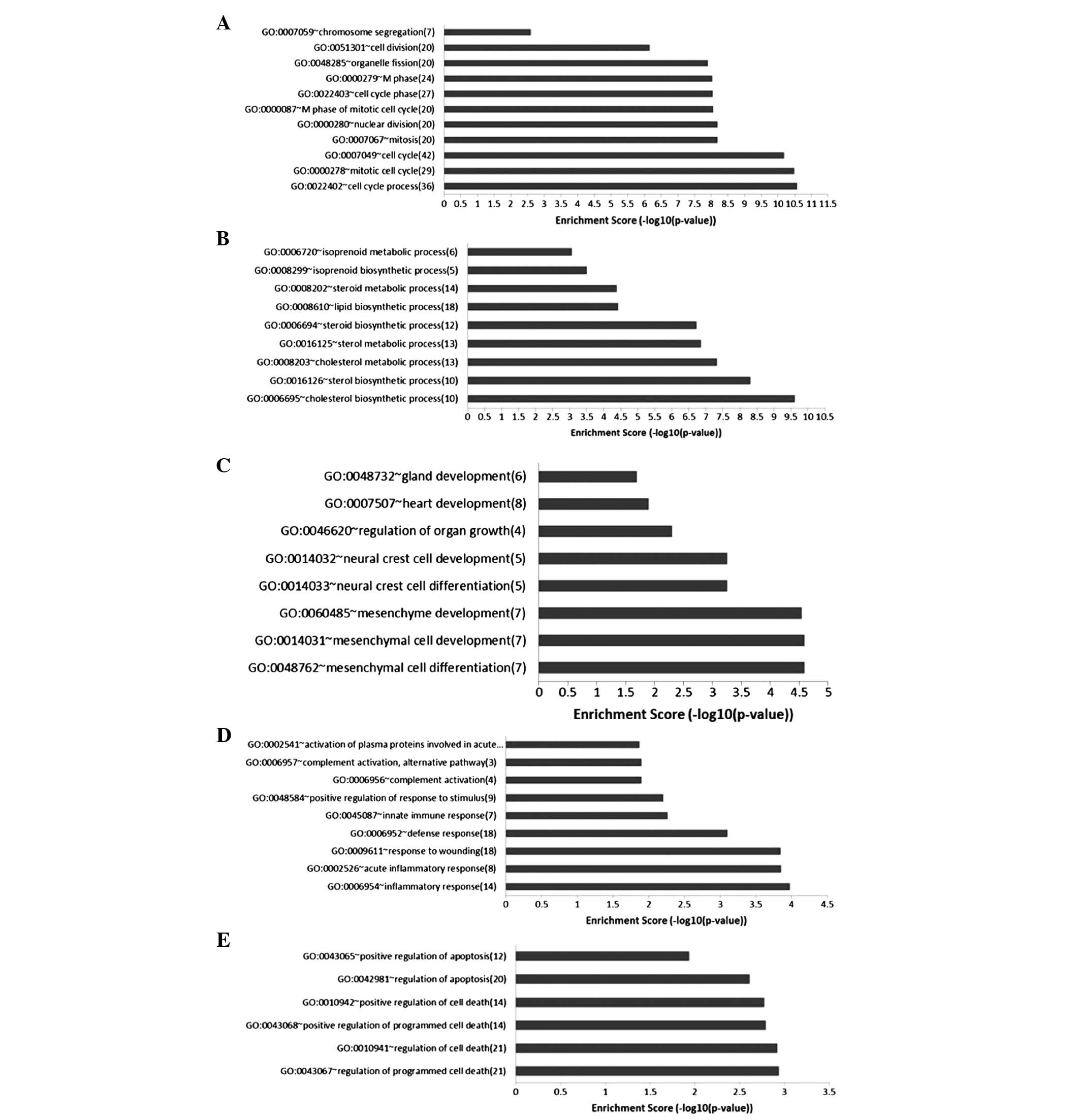
the cell cycle and metabolic process (Fig. 2).
Only one significant KEGG pathway was identified in
the upregulated genes (Fig. 1),
whereas a total of 163 GO biological pathway terms were
significantly enriched in the upregulated genes. These terms were
divided into 20 clusters, of which three were associated with cell
growth and differentiation, responses to stimuli and apoptosis
(Fig. 2).
PPI network of the DEGs
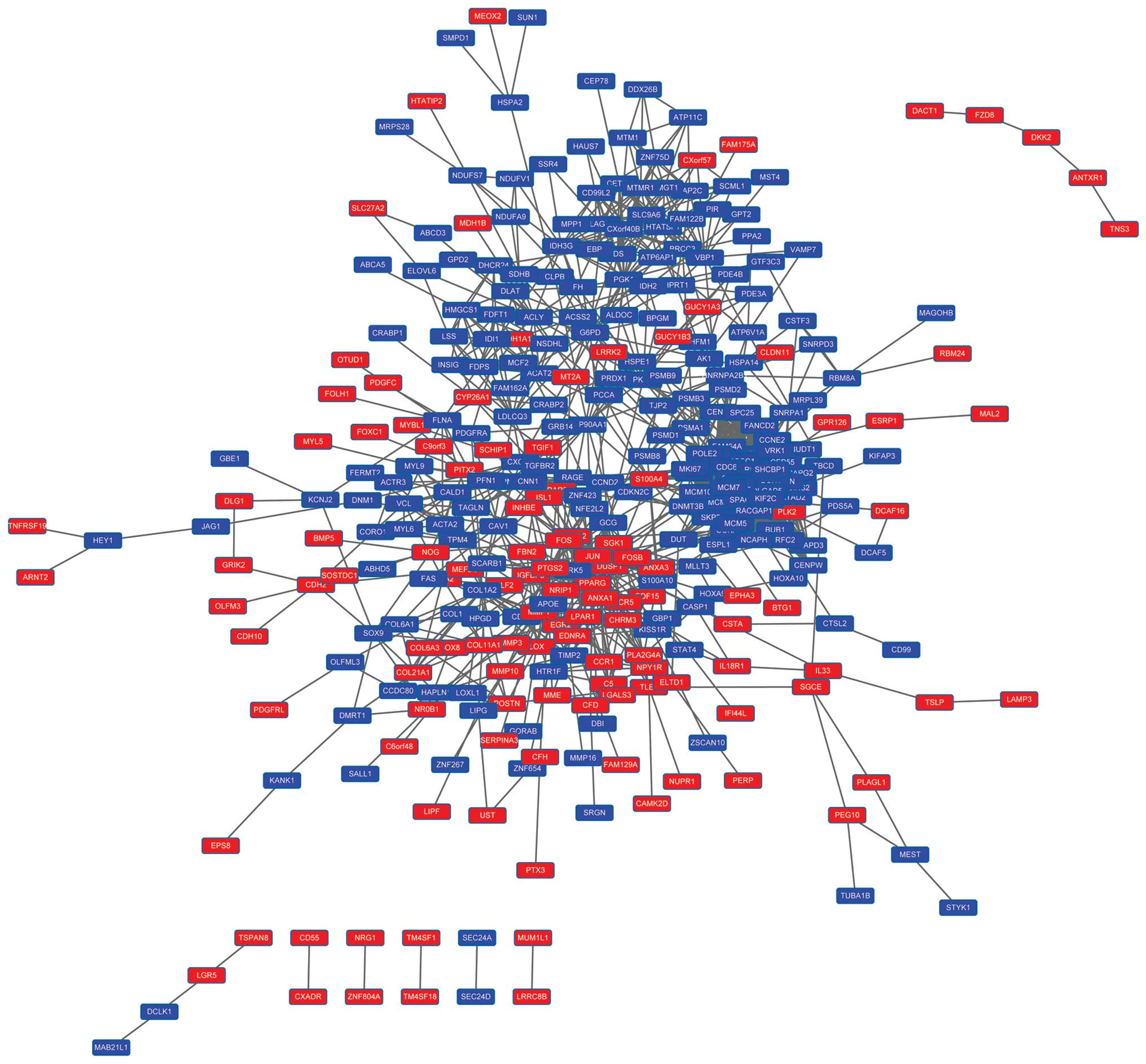
A PPI network consisting of 342 nodes was
constructed for the DEGs (Fig. 3).
A total of nine subnetworks were identified by ClusterONE
(P<0.01). The top four subnetworks are shown in Fig. 4. Functional enrichment analysis
indicated that subnetwork 1 (Fig.
4A) was predominantly associated with the cell cycle,
subnetwork 2 (Fig. 4B) was
associated with phosphoric acid metabolism and subnetwork 4
(Fig. 4D) was linked with the
formation of central body and microtubules. They were all
associated with cell division (Table
II). No GO terms or pathways were enriched in subnetwork 3
(Fig. 4C).
| Table IIFunctional enrichment analysis of the
differentially expressed genes in the subnetworks. |
Table II
Functional enrichment analysis of the
differentially expressed genes in the subnetworks.
| Subnetwork | Term | P-value | Gene |
|---|
| 1 | GO:0007049: cell
cycle | 1.11E-23 | FOXM1, ANLN, CEP55,
AURKB, CCNE2, KIF2C, SPC25, CDC45, NCAPH, MCM7, NCAPG2, BUB1,
CCNA2, CDC6, MKI67, PDS5A, DSN1, GMNN, DLGAP5, SKP2, ESPL1,
RACGAP1, RAD54L, NCAPD3, MCM6, PLK2, SPAG5, FANCD2, DSCC1 |
| GO:0000279: M
phase | 2.85E-19 | CDC6, MKI67, PDS5A,
DSN1, DLGAP5, ANLN, ESPL1, AURKB, CEP55, RAD54L, NCAPD3, SPC25,
KIF2C, NCAPH, NCAPG2, FANCD2, SPAG5, BUB1, CCNA2, DSCC1 |
| GO:0022403: cell
cycle phase | 8.57E-19 | CDC6, MKI67, PDS5A,
DSN1, DLGAP5, SKP2, ANLN, ESPL1, AURKB, CEP55, RAD54L, NCAPD3,
SPC25, KIF2C, NCAPH, NCAPG2, FANCD2, SPAG5, BUB1, CCNA2, DSCC1 |
| GO:0007067:
mitosis | 8.67E-18 | CDC6, PDS5A, DSN1,
DLGAP5, ANLN, ESPL1, AURKB, CEP55, NCAPD3, SPC25, KIF2C, NCAPH,
NCAPG2, SPAG5, BUB1, CCNA2, DSCC1 |
| GO:0000280: nuclear
division | 8.67E-18 | CDC6, PDS5A, DSN1,
DLGAP5, ANLN, ESPL1, AURKB, CEP55, NCAPD3, SPC25, KIF2C, NCAPH,
NCAPG2, SPAG5, BUB1, CCNA2, DSCC1 |
| hsa04110: ell
cycle | 9.91E-11 | CCNE2, CDC6, CDC45,
MCM7, SKP2, BUB1, ESPL1, CCNA2, MCM5, MCM6 |
| hsa03030: DNA
replication | 6.04E-06 | MCM7, POLE2, RFC2,
MCM5, MCM6 |
| hsa00240: yrimidine
metabolism | 2.86E-04 | POLE2, RRM2, RRM1,
TK1, DUT |
| 2 | GO:0006793:
phosphorus metabolic process | 0.03 | MTM1, MTMR1,
ATP6AP1, PGK1, MST4 |
| GO:0006796:
phosphate metabolic process | 0.03 | MTM1, MTMR1,
ATP6AP1, PGK1, MST4 |
| 4 | GO:0051297:
centrosome organization | 0.039 | CETN2, HAUS7 |
| GO:0031023:
microtubule organizing center organization | 0.043 | CETN2, HAUS7 |
Association between enhancer of zeste
homolog 2 (EZH2) and cisplatin-resistance in ovarian cancer
Previous studies have indicated that (EZH2) is
involved in resistance of ovarian cancer cells to platinum-based
drugs, such as cisplatin, carboplatin and paclitaxel (23,24).
The present study found that EZH2 was down-regulated in
cisplatin-sensitive cells (P=0.007; logFC=−0.35) and upregulated in
cisplatin-resistant cells. A total of 34 DEGs (score ≥0.4)
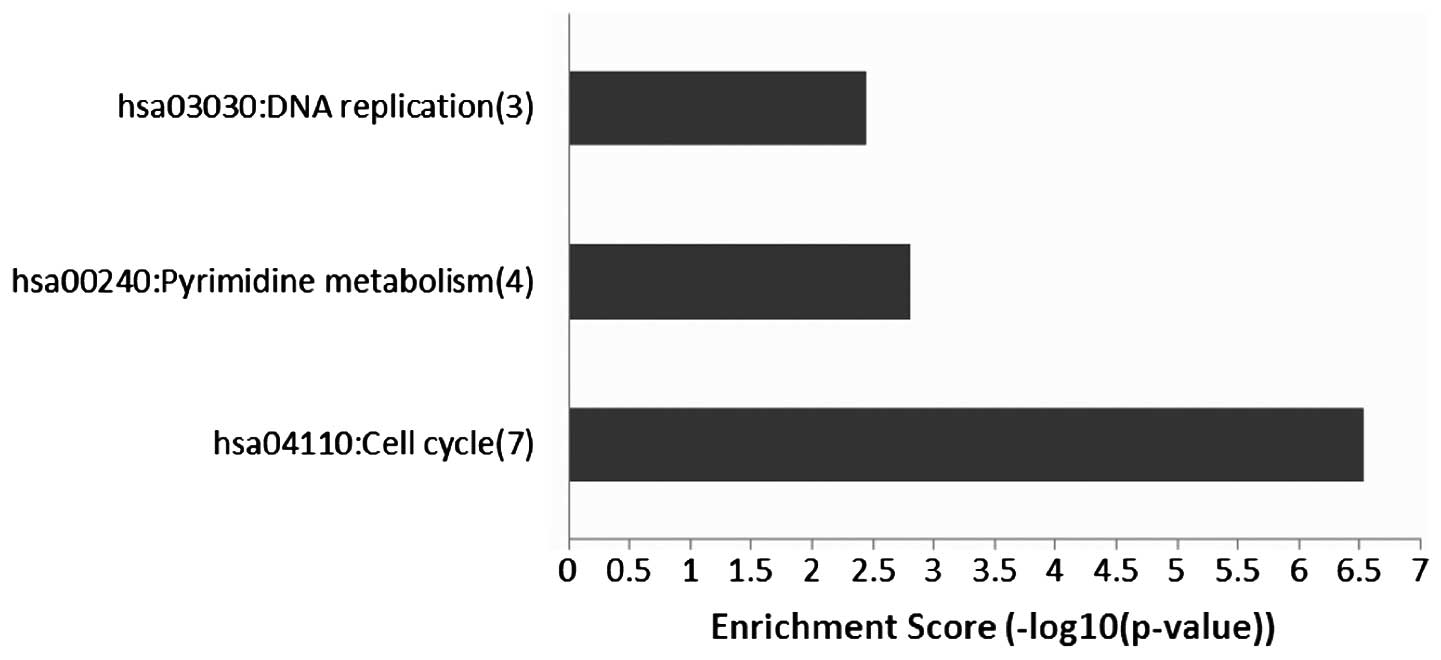
interacting with EZH2 were identified by STRING (Fig. 5). A total of three KEGG pathways
were significantly enriched in the DEGs: DNA replication,
pyrimidine metabolism and cell cycle (Fig. 6). Similar results were obtained in
the GO enrichment analysis, in which 38 GO biological pathway terms
were identified and divided into three clusters. Of these three
clusters, two were associated with the cell cycle and the third was
associated with DNA replication (Table III). These results suggested that
EZH2 affected the cisplatin-resistance of ovarian cancer cells via
modulation of the cell cycle.
| Table IIIGene Ontology functional enrichment
analysis of the genes interacting with enhancer of zeste homolog
2. |
Table III
Gene Ontology functional enrichment
analysis of the genes interacting with enhancer of zeste homolog
2.
| Term | P-value |
|---|
| Cluster 1 |
| GO:0007049: cell
cycle | 5.50E-12 |
| GO:0000279: M
phase | 1.79E-10 |
| GO:0051301: cell
division | 1.24E-09 |
| GO:0000280:
nuclear division | 1.86E-09 |
| GO:0007067:
mitosis | 1.86E-09 |
| GO:0022403: cell
cycle | 2.04E-09 |
| GO:0000087: M
phase of mitotic cell cycle | 2.18E-09 |
| GO:0048285:
organelle fission | 2.64E-09 |
| GO:0022402: cell
cycle process | 3.98E-09 |
| GO:0000278:
mitotic cell cycle | 1.68E-07 |
| Cluster 2 |
| GO:0000075: cell
cycle checkpoint | 6.75E-05 |
| GO:0051726:
regulation of cell cycle | 1.30E-04 |
| GO:0007346:
regulation of mitotic cell cycle | 0.006 |
| GO:0031570: DNA
integrity checkpoint | 0.007 |
| Cluster 3 |
| GO:0051726:
regulation of cell cycle | 1.30E-04 |
| GO:0045934:
negative regulation of nucleobase, nucleoside, nucleotide and
nucleic acid metabolic process | 0.001 |
| GO:0051172:
negative regulation of nitrogen compound metabolic process | 0.001 |
| GO:0010558:
negative regulation of macromolecule biosynthetic process | 0.002 |
| GO:0008156:
negative regulation of DNA replication | 0.002 |
| GO:0031327:
negative regulation of cellular biosynthetic process | 0.002 |
| GO:0009890:
negative regulation of biosynthetic process | 0.002 |
| GO:0051053:
negative regulation of DNA metabolic process | 0.004 |
| GO:0010605:
negative regulation of macromolecule metabolic process | 0.008 |
| GO:0006275:
regulation of DNA replication | 0.01 |
| GO:0051052:
regulation of DNA metabolic process | 0.031 |
Discussion
In the present study, the gene expression profiles
of cisplatin-sensitive ovarian cancer cells were compared with
those of cisplatin-resistant ovarian cancer cells. A total of 556
DEGs were identified in the cisplatin-sensitive ovarian cancer
cells, of which 246 were upregulated and 310 were downregulated.
Functional enrichment analysis revealed that metabolism-associated
pathways, DNA replication and the cell cycle were significantly
enriched in the downregulated genes, while cell growth and
differentiation, responses to stimuli and apoptosis were
significantly enriched in the upregulated genes. These findings
were in accordance with known biochemical mechanisms of cisplatin
cytotoxicity (6,7,25,26).
In addition, a PPI network, including 342 nodes, was constructed
for the DEGs. Subnetworks linked to the cell cycle, phosphoric acid
metabolism and formation of central body and microtubules were
extracted from the entire network. These findings may assist in
further elucidating the molecular mechanisms of cisplatin
cytotoxicity and cisplatin resistance.
EZH2, a member of the polycomb-group family, is a
specific histone 3 lysine 27 methylt ransferase, and is important
in tumorigenesis and cancer progression through epigenetic gene
silencing and chromatin remodeling (27). Hu et al reported that the
overexpression of EZH2 contributes to acquired cisplatin resistance
in ovarian cancer cells (28).
Rizzo et al observed the EZH2 is overexpressed in ovarian
cancer stem cell-like side populations and is associated with drug
resistance (29). A similar role
for EZH2 has been reported in lung cancer (30). The present study further
investigated EZH2 and found that it was downregulated in
cisplatin-sensitive ovarian cancer cells. A total of 34 DEGs
directly interacting with EZH2 were identified. Functional
enrichment analysis suggested that DNA replication, pyrimidine
metabolism and cell cycle were significantly enriched in the 34
DEGs. Cyclin E2 (CCNE2) is involved in the cell cycle G1/S
transition and it has been reported that the overexpression of
CCNE2 is associated with endocrine resistance in human breast
cancer cells (31,32). A study by Tu et al further
indicated that the inhibition of CCNE2 can reduce tamoxifen
resistance in breast cancer cells (33). Cyclin A2 (CCNA2) is also closely
associated with tamoxifen resistance, as its expression is
positively associated with genes overexpressed in endocrine therapy
resistant samples (34).
Minichromosome maintenance complex component 5 (MMC5) and MMC6,
members of the minichromosome maintenance (MCM) family of
chromatin-binding proteins, are essential for the initiation of
eukaryotic genome replication. Gao et al suggested that
genes involved in genome stability may contribute significantly to
the development of camptothecins resistance in melanoma, with MCM5
as one of the candidates (35).
The present study hypothesized that these genes may be involved in
the cisplatin-resistance of ovarian cancer cells in a similar way.
BUB1 mitotic checkpoint serine/threonine kinase (BUB1) not only
regulates chromosome segregation (36), but also mediates cell death in
response to chromosome missegregation (37). Overexpression of BUB1 contributes
tothe cytogenetic and morphologic progression of clear cell kidney
carcinomas (38). The present
study demonstrated that it is upregulated in cisplatin-resistant
ovarian cancer cells, suggesting it may be involved in the
acquisition of drug resistance. These findings indicated that EZH2
may lead to drug resistance via regulation of the cell cycle.
In conclusion, the present study identified a number
of DEGs in cisplatin-sensitive ovarian cancer cells, compared with
cisplatin-resistant ovarian cancer cells. These findings may
advance current understanding of the molecular mechanisms
underlying cisplatin cytotoxicity and cisplatin resistance. EZH2
and its interactors were also identified, which may be used as
targets to modulate drug resistance and thus benefit the treatment
of ovarian cancer.
Acknowledgments
This study was supported by grants from the Seed
Fund of the Second Hospital of Shandong University (grant. no.
S20130100019) and the Foundation of Science and Technology
Commission of Shandong Province (grant. no. 2013ZRE27255).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer. J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2
|
Ries L, Melbert D, Krapcho M, et al: SEER
cancer statistics review, 1975–2005. Bethesda, MD: National Cancer
Institute; pp. 1975–2005. 2008
|
|
3
|
McGuire WP III and Markman M: Primary
ovarian cancer chemotherapy: current standards of care. Br J
Cancer. 89:S3–S8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agarwal R and Kaye SB: Ovarian cancer:
strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer. 3:502–516. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Crul M, Van Waardenburg R, Beijnen J and
Schellens J: DNA-based drug interactions of cis platin. Cancer
Treat Rev. 28:291–303. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cepeda V, Fuertes MA, Castilla J, Alonso
C, Quevedo C and Pérez JM: Biochemical mechanisms of cisplatin
cytotoxicity. Anticancer Agents Med Chem. 7:3–18. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rabik CA and Dolan ME: Molecular
mechanisms of resistance and toxicity associated with platinating
agents. Cancer Treat Rev. 33:9–23. 2007. View Article : Google Scholar :
|
|
8
|
Sedletska Y, Giraud-Panis MJ and Malinge
JM: Cisplatin is a DNA-damaging antitumour compound triggering
multifactorial biochemical responses in cancer cells: importance of
apoptotic pathways. Curr Med Chem Anticancer Agents. 5:251–265.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mandic A, Hansson J, Linder S and Shoshan
MC: Cisplatin induces endoplasmic reticulum stress and
nucleus-independent apoptotic signaling. J Biol Chem.
278:9100–9106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stewart DJ: Mechanisms of resistance to
cisplatin and carboplatin. Crit Rev Oncol Hematol. 63:12–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galluzzi L, Senovilla L, Vitale I, et al:
Molecular mechanisms of cisplatin resistance. Oncogene.
31:1869–1883. 2012. View Article : Google Scholar
|
|
13
|
Lee S, Choi EJ, Jin C and Kim DH:
Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA
amplification contributes to cisplatin resistance in an ovarian
cancer cell line. Gynecol Oncol. 97:26–34. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang X, Fraser M, Moll UM, Basak A and
Tsang BK: Akt-mediated cisplatin resistance in ovarian cancer:
modulation of p53 action on caspase-dependent mitochondrial death
pathway. Cancer Res. 66:3126–3136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li M, Balch C, Montgomery JS, et al:
Integrated analysis of DNA methylation and gene expression reveals
specific signaling pathways associated with platinum resistance in
ovarian cancer. BMC Med Genomics. 2:342009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Team RC: R:A language and environment for
statistical computing. R foundation for Statistical Computing;
2005
|
|
17
|
Irizarry RA, Hobbs B, Collin F, et al:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smyth GK: Limma: Linear models for
microarray data. Bioinformatics and computational biology solutions
using R and Bioconductor Springer. pp. 397–420. 2005
|
|
19
|
Sherman BT, Huang da W, Tan Q, et al:
DAVID Knowledgebase: a gene-centered database integrating
heterogeneous gene annotation resources to facilitate
high-throughput gene functional analysis. BMC Bioinformatics.
8:4262007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Franceschini A, Szklarczyk D, Frankild S,
et al: STRING v9.1: protein-protein interaction networks, with
increased coverage and integration. Nucleic Acids Res.
41:D808–D815. 2013. View Article : Google Scholar :
|
|
21
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar :
|
|
22
|
Nepusz T, Yu H and Paccanaro A: Detecting
overlapping protein complexes in protein-protein interaction
networks. Nat Methods. 9:471–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rizzo S, Hersey JM, Mellor P, et al:
Ovarian cancer stem cell-like side populations are enriched
following chemotherapy and overexpress EZH2. Mol Cancer Ther.
10:325–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu S, Yu L, Li Z, et al: Overexpression of
EZH2 contributes to acquired cisplatin resistance in ovarian cancer
cells in vitro and in vivo. Cancer Biol Ther. 10:788–795. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bragado P, Armesilla A, Silva A and Porras
A: Apoptosis by cisplatin requires p53 mediated p38α MAPK
activation through ROS generation. Apoptosis. 12:1733–1742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qin LF and Ng IO: Induction of apoptosis
by cisplatin and its effect on cell cycle-related proteins and cell
cycle changes in hepatoma cells. Cancer Lett. 175:27–38. 2002.
View Article : Google Scholar
|
|
27
|
Tsang DP and Cheng AS: Epigenetic
regulation of signaling pathways in cancer: Role of the histone
methyltransferase EZH2. J Gastroenterol Hepatol. 26:19–27. 2011.
View Article : Google Scholar
|
|
28
|
Hu S, Yu L, Li Z, et al: Overexpression of
EZH2 contributes to acquired cisplatin resistance in ovarian cancer
cells in vitro and in vivo. Cancer Biol Ther. 10:788–795. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rizzo S, Hersey JM, Mellor P, et al:
Ovarian cancer stem cell-like side populations are enriched
following chemotherapy and overexpress EZH2. Mol Cancer Ther.
10:325–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lv Y, Yuan C, Xiao X, et al: The
expression and significance of the enhancer of zeste homolog 2 in
lung adenocarcinoma. Oncol Rep. 28:147–154. 2012.PubMed/NCBI
|
|
31
|
Caldon CE, Sergio CM, Kang J, et al:
Cyclin E2 overexpression is associated with endocrine resistance
but not insensitivity to CDK2 inhibition in human breast cancer
cells. Mol Cancer Ther. 11:1488–1499. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng L, Zhao Y, Feng H and Liu Y:
Endocrine resistance in breast cancer. Climacteric. 1–7. 2013.
|
|
33
|
Tu SH, Ho CT, Liu MF, et al: Luteolin
sensitises drug-resistant human breast cancer cells to tamoxifen
via the inhibition of cyclin E2 expression. Food Chem.
141:1553–1561. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao T, Han Y, Yu L, Ao S, Li Z and Ji J:
CCNA2 is a prognostic biomarker for ER+Breast cancer and tamoxifen
resistance. Plos One. 9:e917712014. View Article : Google Scholar
|
|
35
|
Gao K, Lockwood WW, Li J, Lam W and Li G:
Genomic analyses identify gene candidates for acquired irinotecan
resistance in melanoma cells. Int J Oncol. 32:1343–1349.
2008.PubMed/NCBI
|
|
36
|
Klebig C, Korinth D and Meraldi P: Bub1
regulates chromosome segregation in a kinetochore-independent
manner. J Cell Biol. 185:841–858. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jeganathan K, Malureanu L, Baker DJ,
Abraham SC and van Deursen JM: Bub1 mediates cell death in response
to chromosome missegregation and acts to suppress spontaneous
tumorigenesis. J Cell Biol. 179:255–267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pinto M, Vieira J, Ribeiro FR, et al:
Overexpression of the mitotic checkpoint genes BUB1 and BUBR1 is
associated with genomic complexity in clear cell kidney carcinomas.
Cell Oncol. 30:389–395. 2008.PubMed/NCBI
|