Introduction
Heart disease is one of the most prevalent diseases
affecting human health worldwide. Heart diseases, including acute
myocardial infarction (AMI) result in a decreased number of
surviving cardiac cells, scar tissue formation, ventricular
remodeling, loss of myocardial contractility, decreased ventricular
ejection fraction and eventually decreased heart function, which
may induce life-threatening heart failure (1). Exogenous cell transplantation into
the infarcted areas has been employed in order to induce these
cells to perform the integrative biological functions, thereby
potentially improving cardiac function (2). Over the past few decades, studies
have investigated the use of cardiac stem cells in the repair of
damaged heart muscle in order to restore heart function. Bone
marrow mesenchymal stem cells (BMSCs) have been a predominant
focus, which have numerous advantages compared with other stem
cells, as they are a rich and extensive source of cells that are
easily accessible. Furthermore, autologous transplantation may be
performed without ethical issues. Culture and isolation methods for
BMSCs are simple as they proliferate rapidly and have high
amplification capability for culture in vitro, as well as
gene stability following multiple passages. In addition, BMSCs have
a high differentiation capacity and low immunogenicity. Therefore,
BMSCs are ideal stem cells for transplantation in the treatment of
cardiovascular disease.
There are three common methods of myocardial cell
differentiation from BMSCs in vitro, which include drug
induction, co-culture with myocardial cells and genetic
modification (3). However, these
methods are inefficient and therefore maximizing the efficiency of
this process has been the focus of numerous studies.
Genetic modification of BMSCs may promote the
transformation into cardiac cells at the molecular level. This
novel method of differentiation is a result of development in
molecular biology, which aims to activate cardiac differentiation
genes and regulatory networks via the activation of one or more key
genes in order to induce the transformation of BMSCs into cardiac
cells (4). Early cardiac
transcription factors are expressed in early heart development, the
most important of which are Nkx2.5, GATA-4 and TBX5 (5–8).
These transcription factors regulate the expression of numerous
cardiac structural protein genes and promote BMSC differentiation
into cardiomyocytes, which is a key step in normal heart
development. Numerous studies have investigated the effectiveness
of drug-induced differentiation of stem cells into cardiomyocytes;
these drugs include 5-azacytidine (5-aza), dimethylsulfoxide
(DMSO), insulin, dexamethasone and ascorbic acid (9–15).
5-Aza is the most commonly used drug to induce differentiation into
cardiomyocytes; however, it has been reported to result in cell
toxicity and therefore may not be appropriate for clinical
treatment (16). At present,
co-culture of BMSCs with cardiomyocytes is the most commonly used
method of in vitro cardiomyocyte differentiation. It is also
the simplest method, which is almost non-cytotoxic and therefore
may be feasible for clinical use.
In the present study, early cardiac transcription
factors Nkx2.5 or GATA-4 were transfected into BMSCs. In addition,
myocardial cell co-culture methods in vitro were used in
order to determine the effects of myocardial cell external culture
on the differentiation capacity of BMSCs into cardiomyocytes. The
present study therefore aimed to provide the experimental basis for
the realization of efficient cardiac cell differentiation of BMSCs,
as well as to provide an effective source and clinical basis for
stem cell transplantation in myocardial cell injury repair.
Materials and methods
Isolation and culture of BMSCs in New
Zealand white rabbits
One-week-old male and female New Zealand white
rabbits (The Animal Experimental Center of Hebei Medical
University, Shijiazhuang, China; weight, 200–250 g) were sacrificed
via intraperitoneal injection of 10% chloral hydrate, and then
placed into 75% ethanol for 5 min. Under strict sterile conditions,
the humerus, femur and tibia were isolated and washed three times
with 0.01 mol/l phosphate-buffered saline (PBS), containing 100
U/ml penicillin and 100 µg/ml streptomycin (both North China
Pharmaceutical Factory, Shijiazhuang, China). The epiphysis of the
humerus, femur and tibia were then cut open and the bone marrow
cavity was exposed. A 5-ml sterile syringe was used to wash the
marrow cavity repeatedly with Dulbecco’s modified Eagle’s medium
(DMEM)-F12 (Gibco-BRL, Grand Island, NY, USA). The cell suspension
washed out was filtered using a 200-hole strainer and centrifuged
at 174 × g for 5 min; the supernatant was then discarded. Cells
were resuspended in complete cell culture medium and the single
cell suspension was produced using 90 µl cell suspension,
which was added to 10 µl trypan blue solution (0.4%; Beijing
Cable Laibao Technology Co. Ltd., Beijing, China) for mixing and
incubated for 5 min. An inverted microscope (AE2000; Motic
Deutschland GmbH, Wetzlar, Germany) and a counting hemocytometer
(Hebei Medical University, Shijiazhuang, China) were then used for
counting. The study was approved by the ethics committee of Hebei
Medical University, Shijiazhuang, China.
Culture and subculture of BMSCs in New
Zealand White rabbits
The isolated cells were seeded in 25-cm2
flasks (1×105 cells/cm2) and then stored at
37°C and 5% CO2 in a humidified incubator thermostat
(Shanghai Lishen Scientific Instrument Co., Ltd., Shanghai, China).
The flasks were kept still and incubated for 48 h in order to
promote cell adhesion. The media was then replaced every 2–3 days
in order to remove non-adherent cells, until primary BMSCs formed.
When the adherent cells were ~80% confluent, the culture medium was
discarded and cells were washed 1–2 times with PBS, followed by the
addition of 1 ml of 0.25% trypsin (Sigma-Aldrich, Shanghai, China)
and 0.02% EDTA (Shanghaiziyi-Reagent, Shanghai, China) for
digestion. The extent of digestion was observed under an inverted
microscope. Total cell culture medium was added to terminate
trypsin digestion when cytoplasmic retraction and increased cell
gaps were observed; the digestion time was <2 min each time.
Pipettes were used to remove the culture liquid from the culture
flask in order to repeatedly and gently collect the cells at the
bottom, air bubbles were avoided. Digested cells were made into a
single cell suspension, and centrifuged at 174 x g for 5 min. The
precipitated cells were then resuspended in total cell culture
medium. Cells were passaged and inoculated into two flasks and
incubated at 37°C and 5% CO2 using a humidified
incubator thermostat for culture. The liquid was replaced every 2–3
days, when adherent cells reached ~80% confluence every 3–5 days
and digestion was performed immediately. Passaging (1:2) was
performed, passages were labeled as P1, P2 and P3. An inverted
microscope was used to observe growth and the morphological
characteristics of cells. P3 cells were selected for subsequent
experiments.
Co-cultured cardiomyocyte establishment
in New Zealand white rabbits
Newborn New Zealand white rabbits (male and female;
weight, 10–20 g), were sacrificed via intraperitoneal injection of
10% chloral hydrate. The hearts were isolated under strict aseptic
conditions and washed three times with 0.01 mol/l PBS solution
containing 100 U/ml penicillin and 100 µg/ml streptomycin.
The apex of the hearts were removed and sectioned into small pieces
(1×1 mm), then placed into a sterile beaker containing 10 ml of
0.25% trypsin and 0.02% EDTA. Cells were then incubated in a water
bath at 37°C with agitation for 5 min; a 5-ml sterile syringe was
used to remove the storage solution in order to repeatedly wash the
cells. The cell suspensions were then added to flasks containing
DMEM-F12, which were stored on ice. This process was repeated until
the apex became white floc; the DMEM-F12 liquid in flasks was
filtered through a 200-hole strainer and the supernatant was
discarded following centrifugation at 174 × g for 5 min. The total
cell culture medium was used to resuspend precipitated cells and
produce a cell suspension. The suspension was placed in sterile
flasks and 10 µl BRDU (Wuhan Boster Biotechnology, Ltd.,
Wuhan, China) was added. The flasks were incubated at 37°C and 5%
CO2 in a humidified incubator thermostat. After 1 h, the
culture liquid was placed into another culture flask for continued
culture and the fluid was changed every 48 h.
Identification of surface molecules on
BMSCs from New Zealand white rabbits using flow cytometry
Cell obtainment
Well grown P3 cells were washed 1–2 times with PBS.
Trypsin (0.125%) was then used to make the single cell suspension,
which was centrifuged at 174 × g for 5 min. The cell pellets were
collected for cell counting (1×106 cells/tube), and
washed twice with PBS, prior to centrifugation at 174 × g for 4
min. PBS (100 µl) was then added to the cell precipitate.
The cell suspension was transferred to a flow tube and divided at
random into experimental group (n=3) and isotype control group
(n=3).
Identification of surface molecules of
BMSCs
Cells were divided into three experimental groups
and three control groups. Each fluorescent antibody (5 µl)
was added to cells in each experimental group [PE-CD29 monoclonal
antibody (BD Biosciences, Shanghai, China), PerCP/Cy5.5-CD90
monoclonal antibody (BioLegend, Shanghai, China), fluorescein
isothiocyanate (FITC)-CD45 monoclonal antibody (BioLegend)] and
each isotype control group [PE-immunoglobulin (Ig)M monoclonal
antibody (BD Biosciences), PerCP/Cy5.5-IgG1 monoclonal antibody
(BioLegend), FITC-IgG1 monoclonal antibody (BioLegend)]. Samples
were then incubated at room temperature in the dark for 30 min.
Flow cytometric analysis using a flow cytometer (Beckman Coulter,
Inc., Brea, CA, USA) was then conducted in order to identify BMSC
surface molecules.
Exogenous gene transfection of Nkx2.5 or
GATA-4, and co-culture with myocardial extracellular environment to
induce the differentiation of BMSCs into myocardial cells
Experimental grouping
Cells in each group underwent transfection and
co-culture with myocardial cells as follows:
A1 group, transfection of p-enhanced green
fluorescent protein (EGFP)-N1-Nkx2.5
A2 group, transfection of pEGFP-N1-Nkx2.5 and
myocardial cell co-culture
A3 group, blank culture group of BMSCs
B1 group, transfection of pVP22-GATA-4/myc-His
B2 group, transfection of pVP22-GATA-4/myc-His and
myocardial cell co-culture
B3 group, blank culture group of BMSCs
Competent E. coli DH5α
preparation
Competent E. coli were selected from single
colonies of newly activated E. coli DH5α (Biovector, Co.,
Ltd., Beijing, China) and incubated in 3–5 ml LB liquid medium
(Gibco-BRL) at 37°C and agitated overnight. When in logarithmic
growth phase, the bacterial suspension was diluted 1:100 or 1:50
and inoculated and agitated in the 100 ml LB liquid medium at 37°C
for amplification. When the suspension appeared cloudy, optical
density 600 was measured every 20 or 30 min until it reached
0.3–0.5 and culturing was stopped.
The cells were then placed on ice for 20 min, and
centrifuged at 4°C at 4,000 × g for 10 min. The supernatant was
discarded and 10 ml CaCl2 (0.1mol/l) was added. The
cells were then resuspended and placed on ice for 15–30 min. This
process was repeated once.
For pcDNA3.1-TBX5 transformation, 200 µl
competent cells were placed on ice. pcDNA3.1-TBX5 was combined with
10 µl deionized water and added to the cell suspension and
placed on ice for 30 min. The cells were then heat shocked at 42°C
in water for 90 sec and placed on ice immediately for 3–5 min. LB
liquid medium (1 ml) was added and mixed gently and incubated at
37°C for 1 h.
The suspension was distributed on the LB plates
(Amp~+), and cultured at 37°C. After 30–60 min, the culture dish
was inverted and the cells were cultured at 37°C for 12–16 h.
Single colonies from LB plates were selected and incubated in LB
liquid medium containing Amp (3–5 ml) at 37°C, Glycerol (300
µl) was added and cells were stored at −70°C. pcDNA3.1-TBX5
was then sequenced by Sangon Biotech (Shanghai) Co., Ltd (Shanghai,
China). Plasmids were extracted using the PureLink®
HiPure Plasmid Midiprep kit (Thermo Fisher Scientific, Rockford,
IL, USA).
pEGFP-N1 (negative control), pEGFP-N1-Nkx2.5,
pcDNA3.1, pVP22-GATA-4/myc-His and pcDNA3.1-TBX5 (all Biovector
Co., Ltd.) were added to LB plates and the plates were cultured
upside down at 37°C for 12–16 h. Single colonies were selected and
incubated in 25 ml LB liquid medium for 16–21 h. Plates containing
pEGFP-N1 or pEGFP-N1-Nkx2.5 were incubated in the LB with Amp, and
the plates containing pcDNA3.1, pVP22-GATA-4/myc-His and
pcDNA3.1-TBX5 were incubated in the LB with Kana.
The bacteria were collected in 50-ml tubes and
centrifuged at 10,000 × g for 10 min. The supernatant was discarded
and 4 ml suspension buffer was added. Cell lysate (4 ml) was
incubated at room temperature for 5 min. Buffer (4 ml) was added to
stop cell lysis and the suspension was centrifuged at 12,000 × g
for 10 min.
Samples were added to a purification column
Wizard® Plus Minipreps DNA Purification System, Promega
(Beijing) Biotech Co., Ltd. (Beijing, China) after equilibrium. The
solution was passed through the column and lysis solution was
discarded. The column was washed twice with washing buffer, 10 ml
each time. The column was washed with 5 ml elution buffer and the
solution containing the plasmids was collected. The DNA was
precipitated with 3.4 ml isopropanol and centrifuged at 12,000 × g
at 4°C for 30 min. The DNA was washed with 5 ml of 70% ethanol, and
centrifuged at 12,000 × g for 5 min, the supernatant was then
discarded. The precipitation was then air dried for 10 min, and
suspended in 120 µl sterile water without nucleases. It was
loaded into 1.5 ml EP tubes, the concentration was detected and the
tubes were stored at −20°C.
DNA transfection. When P3 cells were grown to 80%
confluency, they were selected and serum-free, antibiotic-free
DMEM-F12 medium was used for passage. Cells
(4×104/cm2) were seeded into six-well plates
and incubated for 24 h, until the cells reached 90–95% confluence;
well-grown cells were then selected for subsequent transfection.
Plasmid DNA (4 µg) was diluted in 250 µl serum-free,
antibiotic-free DMEM-F12 medium. Lipofectamine™ 2000 reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) was gently
agitated prior to transfection and diluted in 250 µl
serum-free, antibiotic-free DMEM-F12 medium. Each solution was then
incubated at room temperature for 5 min and combined (total volume,
510 µl); following gentle agitation, they were incubated at
room temperature for 20 min to form DNA-Lipofectamine 2000
complexes. This was repeated for cells in each group. Cells were
centrifuged at 12,000 × g for 5 min and the supernatant was
discarded. The transfected cells were then washed twice with
serum-free antibiotic-free medium and then 500 µl
DNA-Lipofectamine 2000 complexes were added into each well.
Following agitation, the cells were incubated at 37°C and 5%
CO2 for 3–5 h, and then washed twice with PBS. Cells
were then cultured in 10% fetal bovine serum (PAA Laboratories
GmbH, Pashing, Austria) antibiotic-free DMEM-F12 medium for 48 h in
order to allow cells to express the exogenous gene.
Exogenous gene expression
Cell culture of exogenous
gene-transfected BMSCs
All groups were incubated at 37°C in 5%
CO2 in a humidified incubator for 4 weeks, during which
time no passage occurred. An inverted contrast microscope (Olympus
IX83, Olympus, Tokyo, Japan) was used to observe cell
differentiation each day. Immunocytochemistry and western blot
analysis were then used to detect specific cardiac troponin T
(cTnT) and connexin 43 (Cx43) expression in myocardial cells in
each group.
Immunocytochemistry
Cell specimens were cultured for 4 weeks in each
group and the expression of cTnT and Cx43 was analyzed by the SP
method (17). The specimens were
incubated in 3% methanol-H2O2 at room
temperature for 10 min. After washing three times for 5 min in 0.01
mol/l PBS, they were treated with 1% Triton X-100 and then
incubated with 10% normal goat serum. Primary anti-cTnT (1:100;
sc-20025; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA),
primary anti-Cx43 (1:100; #3512; Cell Signaling Technology, Inc.,
Danvers, MA, USA) primary antibodies (dilution, 1:100) were added
for incubation. In the negative control group the primary
antibodies were replaced with PBS. Biotinylated goat anti-mouse IgG
(1:100, BA1001, Boster Bio-engineering Co., Ltd, Wuhan, China) and
goat anti-rabbit (1:100, BA1001, Boster Bio-Engineering Co., Ltd,
Wuhan, China) secondary antibodies and horseradish
peroxidase-labeled chain enzyme avidin fluid were then added and
incubated. Then the samples were stained with 5% DAB (D8230–5;
Beijing Solarbio Science and Technology Co., Ltd., Beijing, China)
and hematoxylin, dehydrated in gradient alcohol, cleared with
xylene and mounted with neutral gum. The image data were analyzed
with Image-pro Plus 6 software (Media Cybernetics, Inc., Rockville,
MD, USA). The integral absorbance (IA) value was measured (Beckman
Coulter, Inc.) and averages were taken.
Western blot analysis
Cells were transfected for 48 h and washed twice in
PBS that had been precooled at 4°C. Then, lysis buffer was added to
each group and centrifuged to save the supernatant containing
proteins. The proteins were separated by SDS-PAGE and transferred
to polyvinylidene fluoride (PVDF) membranes (Hefei United-Bio Co.,
Ltd., Anhui, China) that had been pretreated with methanol. The
primary antibodies, anti-Nkx2.5 (1:500; #8792; Cell Signaling
Technology, Inc.), anti-Myc (1:500; #5605; Cell Signaling
Technology, Inc.) and anti-TBX5 (1:500; sc-17866; Santa Cruz
Biotechnology Inc.), were diluted and added to the groups A1, A2
and A3, and the anti-β-actin (1:1,000; #4967; Cell Signaling
Technology, Inc.) antibody was added to groups B1, B2 and B3. The
groups were then incubated at 4°C overnight. The membranes were
washed three times for 5 min with TBST. The membranes were then
incubated with secondary antibodies (horseradish peroxidase labeled
goat anti-rabbit (1:1,000; #7074; Cell Signaling Technology, Inc.)
and goat anti-mouse (1:1,000; #7076; Cell Signaling Technology,
Inc.) at 37°C for 2 h. The PVDF membrane was placed into the X-ray
film cassette and tablet (Thermo Fisher Scientific, Inc.) in the
darkroom. The film was screened using a scanner (Epson (China) Co.,
Ltd. Beijing, China) and analyzed with BIO-1D software (GoldSim,
Issaquah, WA, USA) and corrected to β-actin, which served as
internal reference. The relative expression of protein was
presented by the ratios of absorbance of target protein to the
absorbance of β-actin.
Statistical analysis
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
was used for statistical analysis and all values are expressed as
the mean ± standard deviation. One way analysis of variance was
used for comparison among groups followed by post hoc tests; when
the homogeneity of variance was met, a least significant
differences test was used and if it was not met, the non-parametric
Tamhane’s T2 method was used. α=0.05 was taken as the standard,
P<0.05 was considered to indicate a statistically significant
difference between values.
Effects of exogenous gene transfection of
Nkx2.5 or GATA-4, and myocardial cells outer environment co-culture
of New Zealand rabbit BMSCs on the repair of infarcted
myocardium
Experimental grouping
Transfected cells co-cultured with myocardial cells
and blank cells were injected into rabbits with induced myocardial
infarction as follows:
A1 group, transfection of pEGFP-N1-Nkx2.5 and
myocardial cell co-culture (n=10)
A2 group, blank group of BMSCs (n=10)
B1 group, transfection with pVP22-GATA-4/myc-His and
myocardial cell co-culture (n=10)
B2 group, blank group of BMSCs (n=10)
Establishment of rabbit models with
acute myocardial infarction
Left anterior descending coronary artery ligation
was used to establish the acute myocardial infarction model, as
previously described (18).
Briefly, New Zealand white rabbits were anesthetized using an
injection of 10% chloral hydrate (3.5 ml/kg) into the ear vein.
Head and limbs were fixed onto a surgical board, and the neck and
chest skin were disinfected using povidone-iodine. The trachea was
then isolated in order to insert a tube for ventilation and the
ventilator parameters were adjusted (expiration:inspiration, 1.5:1;
respiratory rate, 30–35 beats/min); following connection between
the trachea cannula and ventilator, positive pressure ventilation
was performed. A sternal line vertical incision was used to cut
open the skin of New Zealand white rabbits and a thoracotomy was
performed in order to expose the heart. The pericardium was cut
open and a sterile cotton swab was used to gently lift the heart,
while the rabbits were rotated slightly to the right. Approximately
1–2 mm below the junction of the pulmonary cone and left atrial
appendage, 4–0 non-damage ligation lines (Johnson (China)
Investment Co., Ltd. Shanghai, China) were used to line the left
anterior descending coronary artery and left ventricular anterior
wall myocardial infarction was established. A pale left ventricular
anterior was used as an indicator of successful myocardial
infarction induction. The heart was then put back into the chest
cavity and the heartbeat was restored for 1 min.
Mesenchymal stem cell transplantation
of New Zealand white rabbits
Following establishment of the myocardial infarction
model, rabbits were left until their breathing became regular and
then a sterile cotton swab was used to gently lift the heart, while
the rabbits were rotated slightly to the right. A 1-ml syringe was
used to inject the prepared cell suspension into two points (50
µl/point) of the left ventricular myocardium at the border
zone of infarcted myocardium and normal myocardium, avoiding cell
spillage. The heart was then put back, the muscle and skin were
sutured, and the chest was closed immediately. In the event of
cardiac arrest, the heart was massaged in order to restore
heartbeat and non-assisted breathing. Following surgery, ventilator
and oxygen support was provided; following the restoration of
spontaneous breathing, the ventilator was removed and 30 min later,
the oxygen was removed. An intraperitoneal injection of 25,000 U/kg
penicillin was then administered in order to avoid infection.
Tissue sampling, H&E staining and
immunohistochemical staining
Perfusion sampling method
Chloral hydrate (10%) was used to anesthetize the
New Zealand white rabbits through the auricular vein. The thoracic
cavity was opened avoiding the transplantation site. Irrigation
fluid was perfused into the left ventricle, a small incision was
made in the right atrium and the blood and perfusate was allowed to
flow out. Following perfusion of physiological saline, the blood
was washed out and added to 200–400 ml of 4% paraformaldehyde. The
hearts were removed immediately following perfusion and the scar
area and slightly purple cell injection zone were identified; the
tissues were fixed in 4% paraformaldehyde for 24 h. The tissues
were then washed in 0.01 M PBS for 12 h, dehydrated with gradient
ethanol (each step, 2 h incubation) and xylene, and embedded in
paraffin. The paraffin block was sectioned using a Leica RM2125
paraffin machine (Leica Biosystems, Shanghai, China) for continuous
paraffin sections with a thickness of 5 µm.
H&E staining
The sections were dewaxed in xylene I, xylene II,
100% alcohol I and 100% alcohol II for 10 min, and then in 95%
alcohol, 90% alcohol, 80% alcohol, 70%alcohol for 5 min, followed
by a wash with distilled water. The sections were stained with
hematoxylin for 1 min and washed with distilled water.
Differentiation was conducted using 1% hydrochloric acid, followed
by a wash in tap water. Slides were then counterstained with eosin
for 2 min. Slides were then dehydrated in gradient alcohol, 70%
alcohol, 80% alcohol, 90% alcohol, 95% alcohol for 2 min, follwed
by 100% alcohol I and and 100% alcohol II for 10 min. They were
then immersed in xylene for 1 min and covered with neutral gum.
Immunohistochemistry staining
Prior to dewaxing, the tissues were placed in 4%
xylene and then in 20% sucrose solution at 4°C overnight. Tissues
were then fixed in paraffin. The sections were incubated at 68°C
for 20 min and dewaxed in xylene I, xylene II, and gradient
alcohol. The sections were incubated in 3%
H202 at 37°C for 10 min, then washed with PBS
three times. The sections were boiled in citric acid buffer
solution (0.01 M, pH 6.0) at 95°C for 15 to 20 min, cooled for 20
min, washed with cold water and then washed with PBS three times.
Normal goat serum was added for 20 min. Anti-cTnT and anti-Cx43
primary antibodies were added and incubated at 4°C overnight,
followed by three washes with PBS. Biotinylated goat anti-mouse IgG
and goat anti-rabbit secondary antibodies were then added, followed
by washing with PBS. Horseradish peroxidase-labeled avidin and
fluid was added and incubated at 37°C for 30 min, followed by three
washes with PBS. DAB was added for 5 to 10 min and slides were
observed under an inverted microscope. Slides were then washed with
distilled water three times and PBS was added to terminate the
staining reaction. Slides were counterstained with hematoxylin for
2 min, differentiated with 0.1% hydrochloric acid and washed with
water. They were then dehydrated in gradient alcohol, immersed in
xylene for 1 min and covered with neutral gum.
Collagen staining
Tissues were fixed in 10% formalin solution for 1 h
and dehydrated and embedded in paraffin. Dewaxed paraffin slices
were hydrated and stained with Weigert iron hematoxylin for 5–10
min. Excess stain was removed by washing in water. Hydrochloric
ethanol (1%) was added for differentiation, any excess was removed
by washing in water. Samples were then stained with Van Gieson for
2 min. Samples were desiccated with 95% ethanol and put in a
temperature box and dried. Samples were then dehydrated with
ethanol, cleared in xylene and mounted with neutral gum. They were
then analyzed with the AE2000 inverted microscope.
Results
Flow cytometric analysis of surface
molecules detected in BMSCs
The successfully isolated cells were observed under
an inverted microscope, they were found to be round or nearly round
and were mixed with a large number of red blood cells and
non-adherent mononuclear cells. Following 24 h, a small number of
adherent cells were observed, which were round, polygonal or
multilateral spindle shaped. Following 10 min, the non-adherent
cells were removed and the number of adherent cells had markedly
increased, the majority of which had a polygonal and spindle
morphology, with a slow growth rate; the cells observed at this
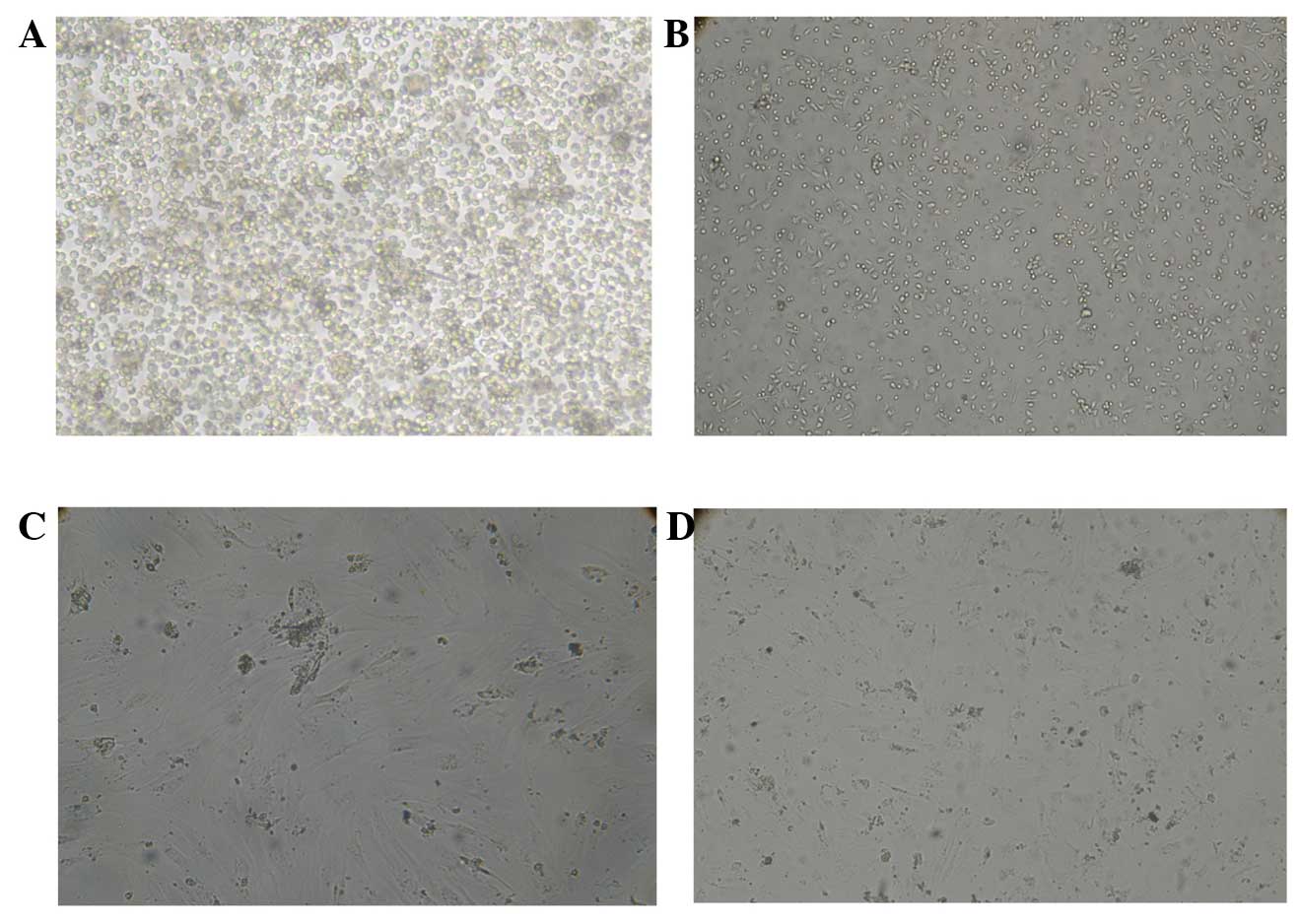
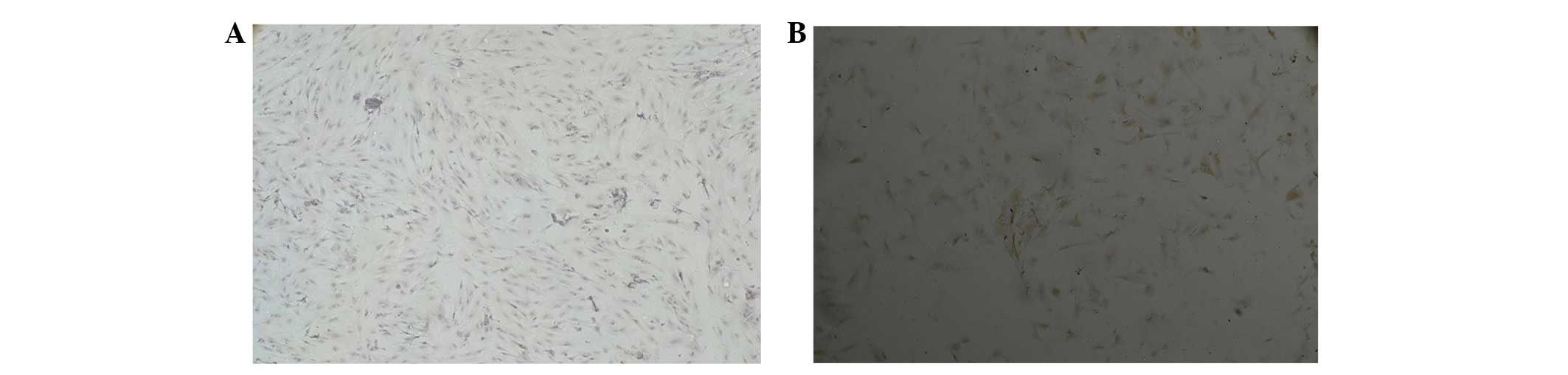
time point were the primary BMSCs (Fig. 1A). Following 2–3 days, adherent
cells had rapidly proliferated and adjacent cells had gradually
converged as agglomerates. In addition, some of the cell clusters
were arranged radially (Fig. 1B).
Following 6–7 days, the confluence of cells reached 80–90% and
cells were arranged in a swirling or radial pattern. At this point,
the primary passage was conducted. Following passage, BMSCs had
more rapid adherent abilities than the primary cells. Within 24 h,
all cells were adherent and demonstrated rapid, as well as evenly
distributed, growth and proliferation. Following 3–5 days, 80–90%
of cells were confluent adherent cells were and subsequent passage
was then performed and cells were labeled as P1, P2 or P3. Over the
three passages and medium replacements, non-adherent cells were
gradually removed, round or oval cells decreased among adherent
cells and adherent cell morphology gradually converged. When the
passage reached the third generation, mesenchymal stem cells were
predominantly in the same fusiform shape (Fig. 1C and D).
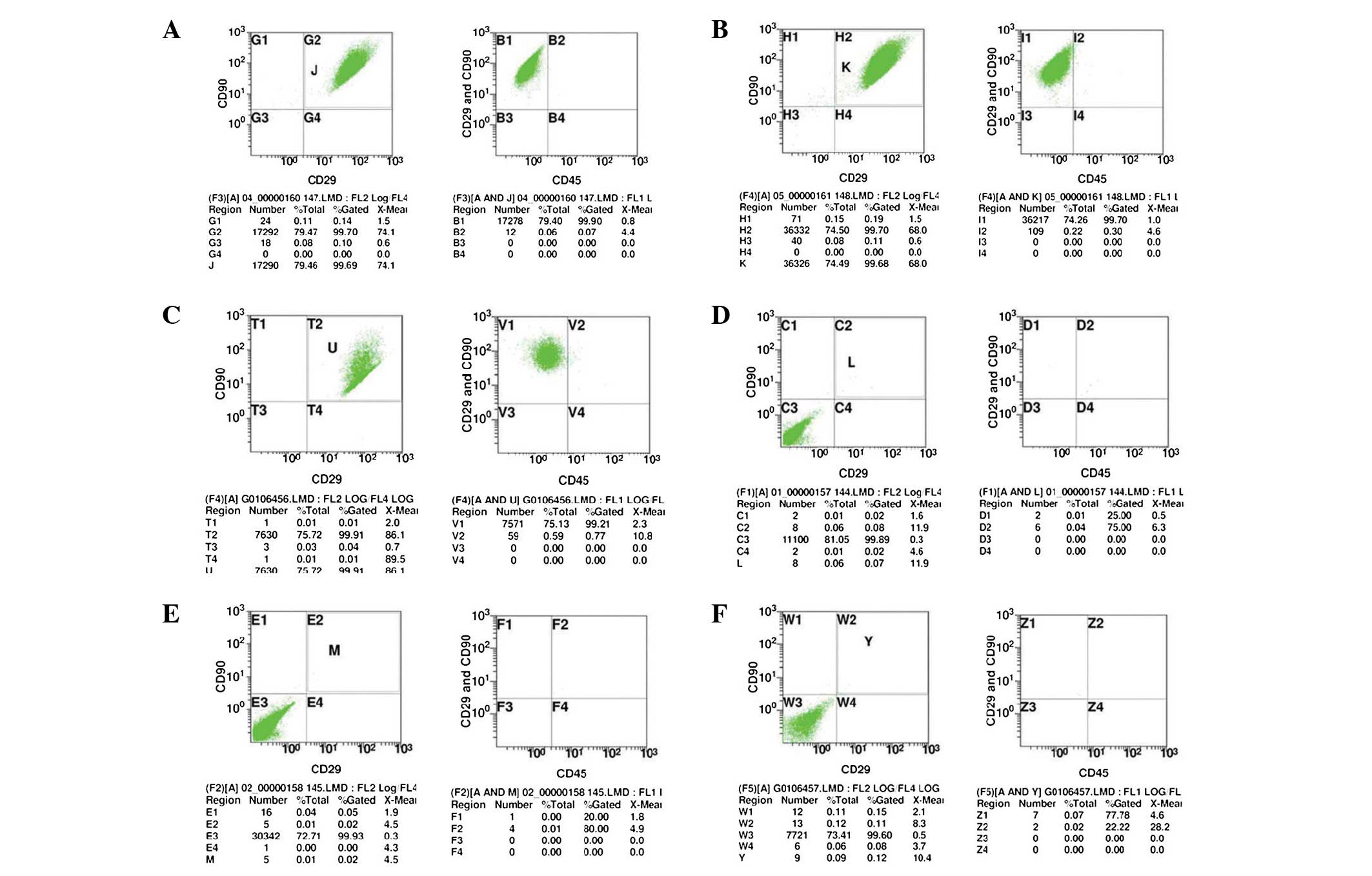
Flow cytometric analysis was used to detect the P3
cells with good growth. The results showed that the percentages of
CD90+/CD29+ cells in the experimental groups
(A1–3) were 99.70, 99.70 and 99.91%, respectively; and the
percentages of CD45− in
CD90+/CD29+ cells were 99.90, 99.70 and
99.21%, respectively (Fig. 2A–C).
The results suggested that in the experimental group (A1-3), the
percentages of CD90+/CD29+/CD45−
cells were 99.61, 99.40 and 99.11%, respectively. The percentages
of CD90+/CD29+ cells in the isotype control
group (B1–3) were 0.08, 0.02 and 0.11%, respectively. Furthermore,
the percentage of CD45+ in
CD90+/CD29+ cells were 25.00, 20.00 and
77.78% (Fig. 2E–F). It suggested
that in the experimental group, the percentages of
CD90+/CD29+/CD45+ cells were 0.02,
0.00 and 0.11%. These results therefore indicated that the obtained
mesenchymal stem cells were uniformly expressed and had high cell
purities, which complied with the basic characteristics of
BMSCs.
Western blot analysis detection of
exogenous gene expression
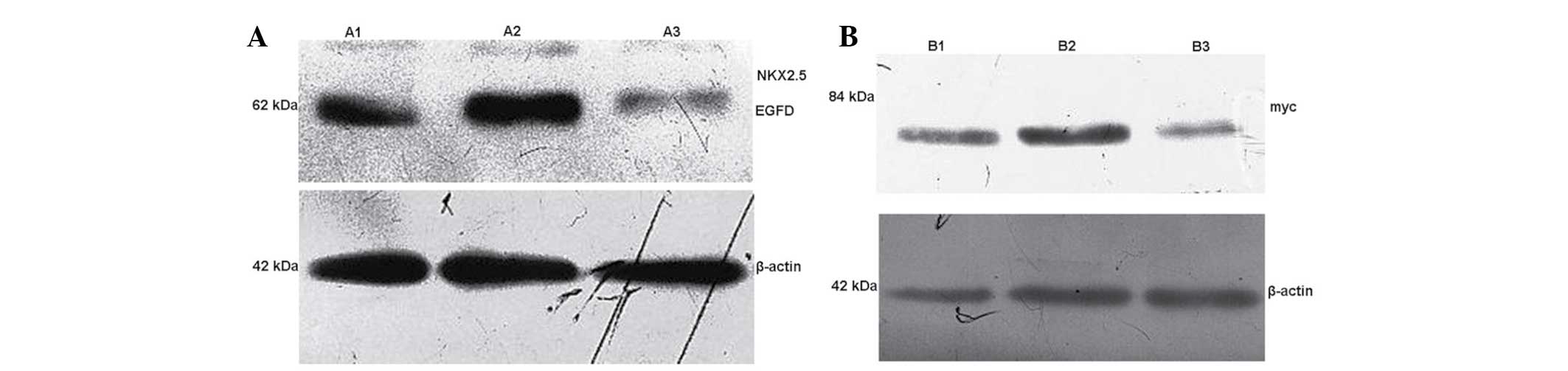
At 48 h post-transfection, the cellular total
protein in A1, A2 and A3 groups were collected. Western blot
analysis was used to detect the Nkx2.5-EGFP fusion protein
expression in A1, A2 and A3 groups. The results showed that the
exogenous expression of Nkx2.5 was observed following transfection
in A1 and A2 groups, whereas NKx2.5 was not expressed in the
control A3 group (Table I;
Fig. 3A). The cell total protein
was collected in groups B1, B2 and B3, and western blot analysis
was used to detect Myc protein expression in each group. The
results showed that exogenous GATA-4 expression was present
following transfection in groups B1 and B2, whereas GATA-4 was not
expressed in the control B3 group (Fig. 3B; Table II).
 | Table IWestern blot analysis was used to
detect the expression of Nkx2.5-EGFP in cells of groups A1, A2 and
A3 group. |
Table I
Western blot analysis was used to
detect the expression of Nkx2.5-EGFP in cells of groups A1, A2 and
A3 group.
| Relative protein
expression | A1 group | A2 group | A3 group |
|---|
|
Nkx2.5-EGFP/β-actin | 0.180±0.029b | 0.184±0.031a | 0.001±0.000a |
| Table IIWestern blot analysis was used to
detect the expression of myc in cells of groups B1, B2 and B3
group. |
Table II
Western blot analysis was used to
detect the expression of myc in cells of groups B1, B2 and B3
group.
| Relative protein
expression | B1 group | B2 group | B3 group |
|---|
| Myc/β-actin | 0.036±0.040b | 0.038±0.039a | 0.000±0.000a |
Detection of cell differentiation of
BMSCs following transgene induction
Cell morphology changes in New Zealand
white rabbit BMSCs after transgene induction
Four weeks following transfection, inverted
microscopy revealed that cells in the A1 and B1 groups were
fusiform in shape; in addition, the growth of the cells showing
overlay and high density features increased in a time-dependent
manner (Fig. 4). When cell density
reached a certain level, no further cell proliferation was observed
and the morphological changes were similar in each group. The
majority of aggregated myocardial cells in groups A2 and B2 were
spindle shaped and certain cells with good growth showed a rhythmic
beat. There was a high density of cells in groups A3 and B3 that
were fusiform shaped, the growth of the local cells showed overlay
and high density features.
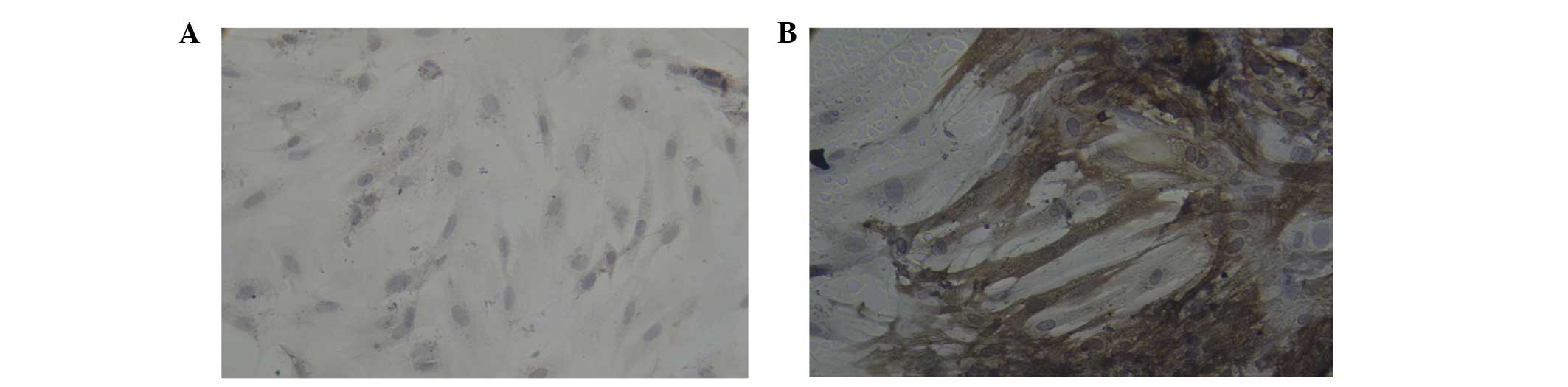
Immunocytochemical analysis of BMSCs
following transgene induction
Four weeks post-transfection and culture,
immuno-cytochemical analysis revealed that certain cells were
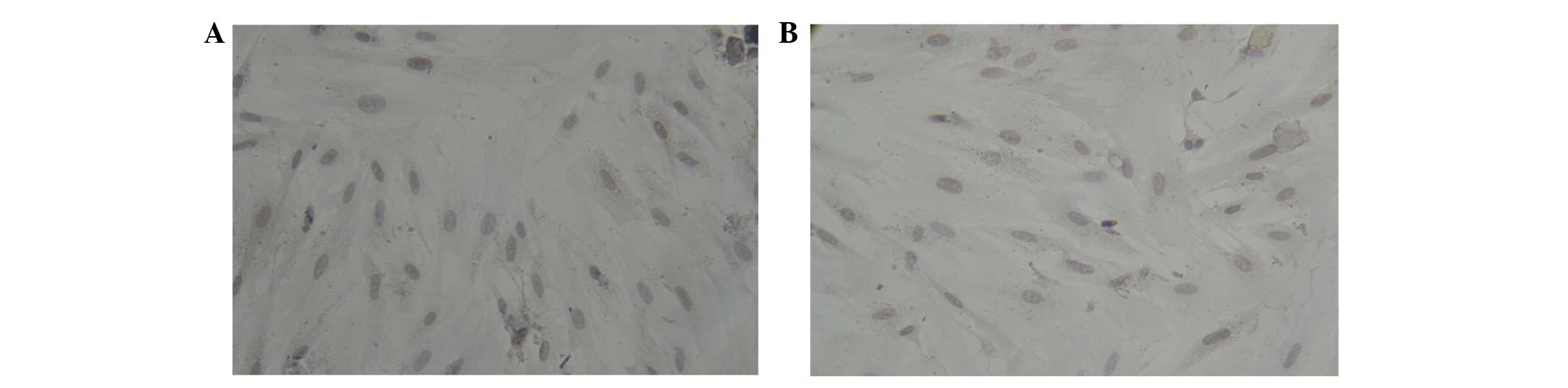
positive for cTnT in the A1 (Fig.
5A), A2 (Fig. 5B), B1
(Fig. 6A) and B2 (Fig. 6B) groups; brown mesh-like and
particle-like structures were observed in the cytoplasm of cTnT
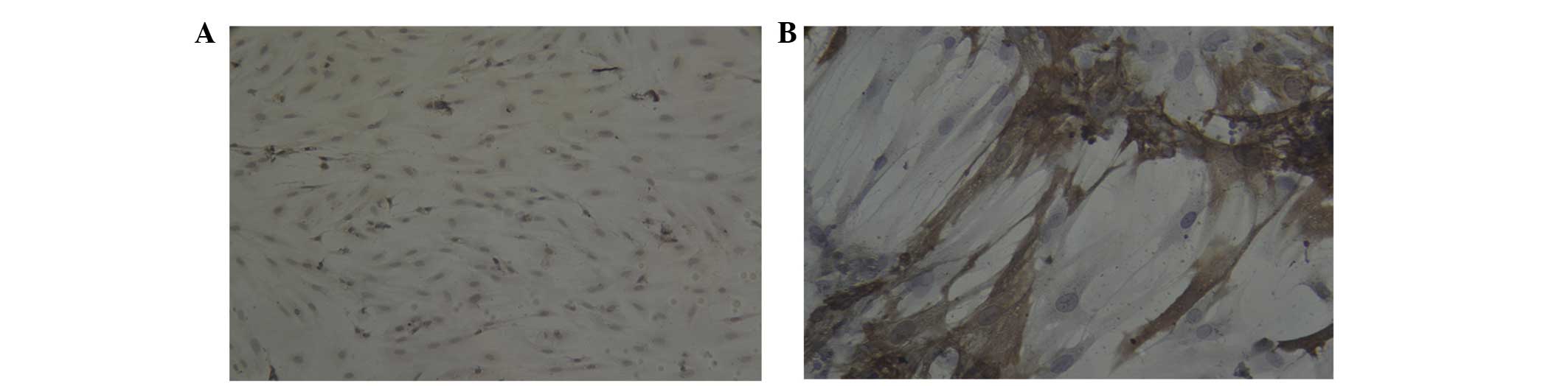
positive cells. In addition, certain cells were positive for Cx43
in groups A1 (Fig. 7A), A2
(Fig. 7B), B1 (Fig. 8A) and B2 (Fig. 8B); brown particles were observed in
the cytoplasm of Cx43-positive cells. However, few cells positively
expressed cTnT and Cx43 in the A3 and B3 group. As shown in
Table III, the integrating
absor-bance value (IA) of each group demonstrated that in groups A1
and A2, cTnT and Cx43 expression levels were significantly
increased following transfection of NKx2.5 compared with that of
the blank transfected A3 group (P<0.05) ; with the highest
expression levels observed in the A2 group (P<0.05) (Fig. 9A and C). In addition, cTnT and Cx43
expression levels were significantly increased in groups B1 and B2
following transfection of GATA-4, compared with that of the
blank-transfected B3 group (P<0.05); furthermore, cTnT and Cx43
expression was significantly increased in the B2 group compared
with that of the B1 group (P<0.05) (Table IV; Fig. 9B and D).
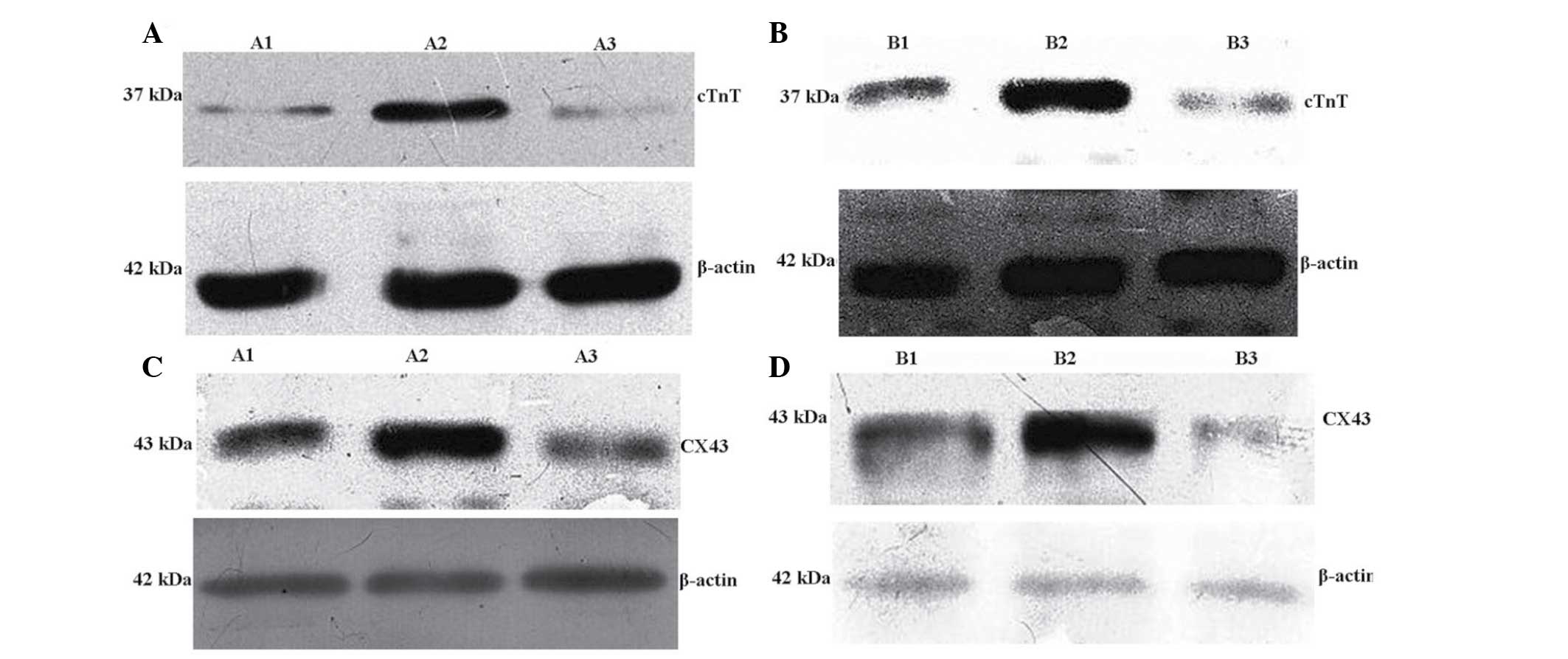 | Figure 9Western blot analysis of the
expression of (A) cTnT in groups A1, A2 and A3, (B) cTnT in groups
B1, B2 and B3, (C) Cx43 in groups A1, A2 and A3 and (D) Cx43 in
groups B1, B2 and B3. cTnT, cardiac troponin T; Cx43, connexin 43;
A1, BMSCs transfected with Nkx2.5; A2, BMSCs transfected with
Nkx2.5 and myocardial cell co-culture; A3, blank culture of BMSCs;
B1, BMSCs transfected with GATA-4; B2, BMSCs transfected with
GATA-4 and myocardial cell co-culture; B3, blank culture of BMSCs;
BMSCs, bone marrow mesenchymal stem cells. |
| Table IIIImmunocytochemical staining and
western blot analysis of cTnT and Cx43 expression in groups A1, A2
and A3. |
Table III
Immunocytochemical staining and
western blot analysis of cTnT and Cx43 expression in groups A1, A2
and A3.
| Antibody | IA
|
|---|
| A1 group | A2 group | A3 group |
|---|
| cTnT | 0.226±0.051b | 0.231±0.050a | 0.155±0.006a |
| Cx43 | 0.271±0.030b | 0.280±0.031a | 0.176±0.024a |
| cTnT/β-actin | 0.490±0.111b | 0.498±0.098a | 0.009±0.001a |
| Table IVImmunocytochemical staining and
western blot analysis of cTnT and Cx43 expression in groups B1, B2
and B3. |
Table IV
Immunocytochemical staining and
western blot analysis of cTnT and Cx43 expression in groups B1, B2
and B3.
| Antibody | IA
|
|---|
| B1 group | B2 group | B3 group |
|---|
| cTnT | 0.222±0.041b | 0.235±0.046a | 0.155±0.006a |
| Cx43 | 0.262±0.022b | 0.275±0.029a | 0.176±0.024a |
| cTnT/β-actin | 0.410±0.124b | 0.437±0.101a | 0.039±0.010a |
Hematoxylin and eosin (H&E) and
immunohistochemical staining of infarcted myocardium repair
Collagen fiber staining
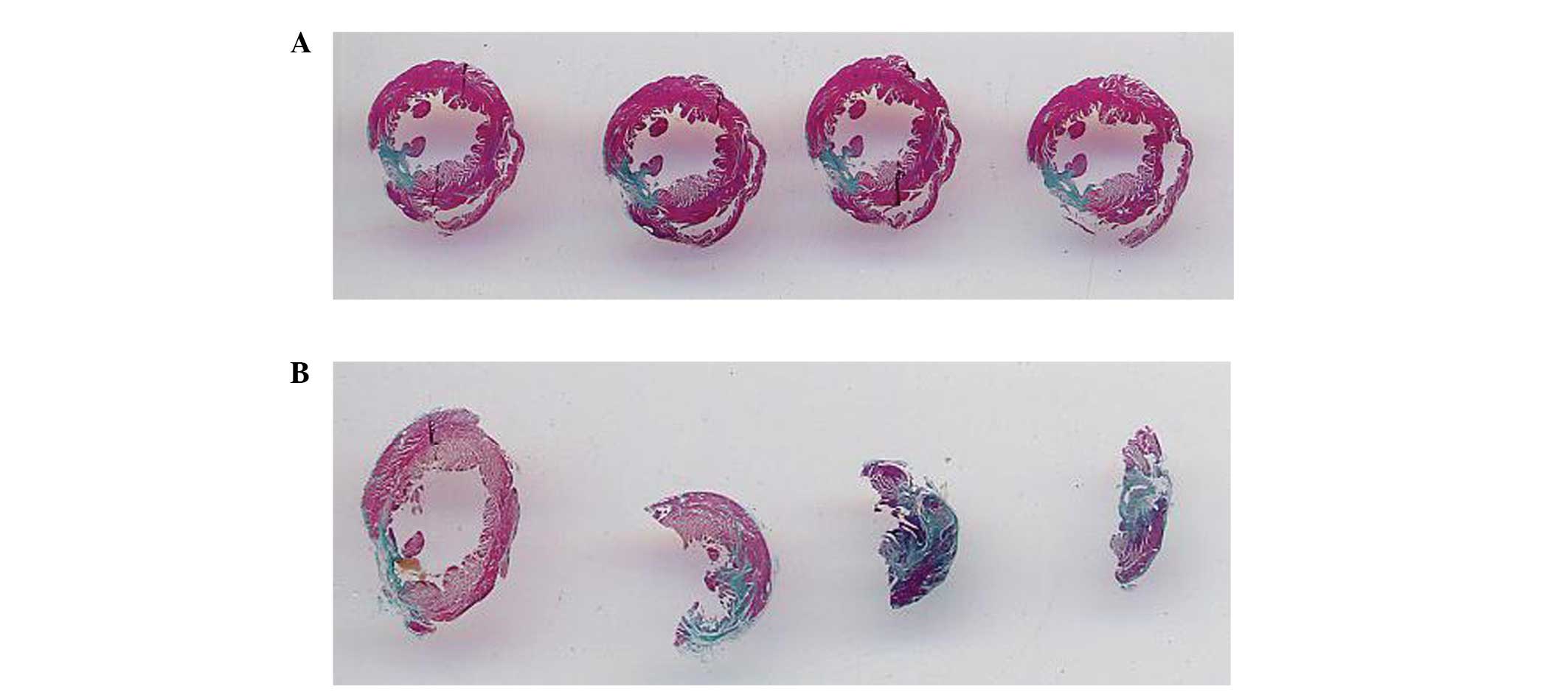
Following the establishment of a myocardial
infarction rabbit model, collagen fiber staining was used in order
to observe the effects of myocardial infarction model establishment
on the heart. As shown in Fig. 10A
and B, normal left ventricular myocardium was stained pink and
myocardial infarct areas were stained gray-blue; these images
demonstrated that the full left ventricular wall was stained
gray-blue. Therefore it was concluded that the establishment of
myocardial infarction models was successful and reached the
requirement of experiments (Fig. 10A
and B).
H&E staining
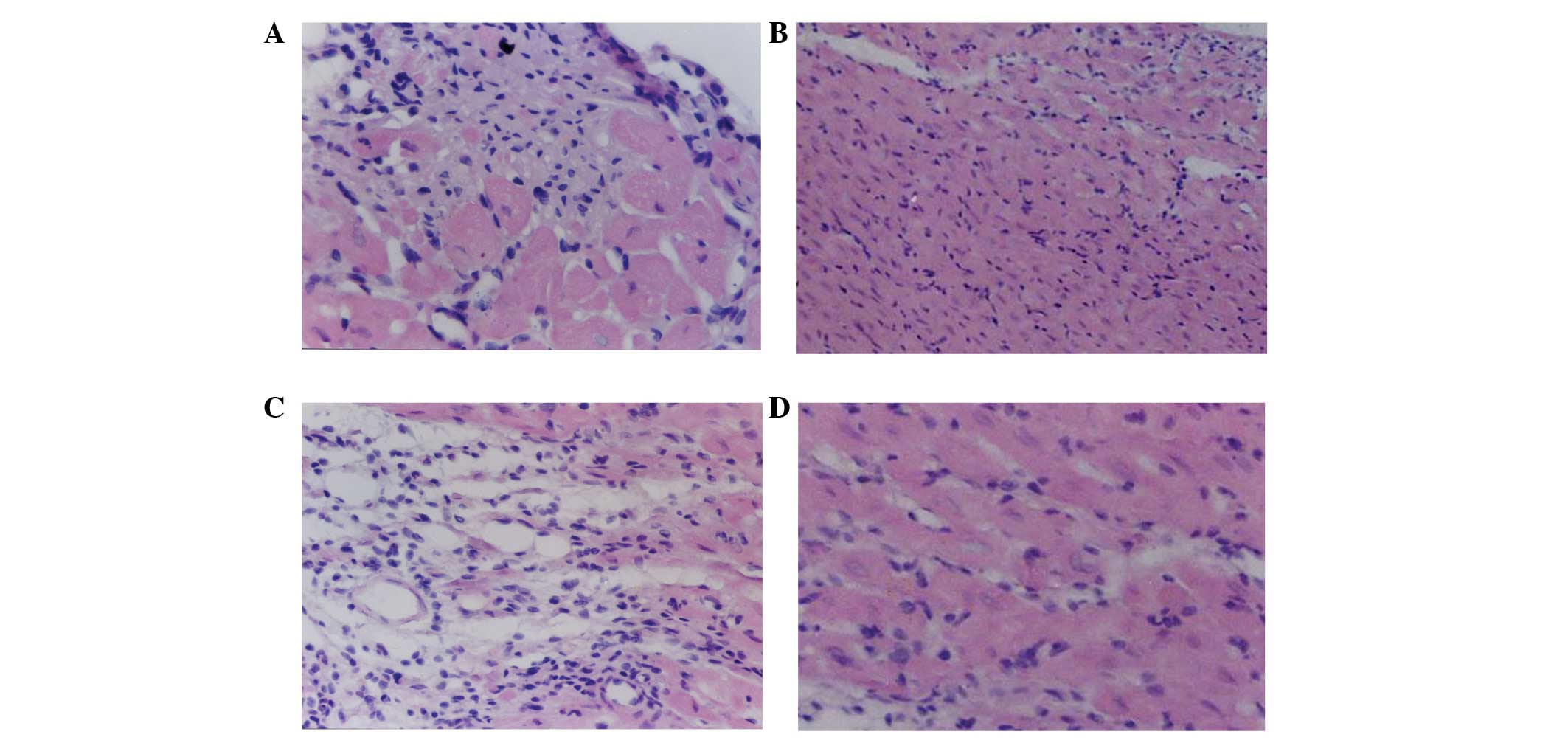
As shown in Fig. 11A
and C, in the A1 and B1 groups, respectively, transplanted
cells were observed to have survived and grown among the myocardial
cells; in addition, these cells also showed radial and infiltrating
growth into the surrounding area, where the transplanted cells at
the junction between the transplanted area and the normal
myocardial cell area gradually shifted to a spindle-shape.
Fibroblasts and angiogenesis were observed and connections between
cells were found at the junction between BMSCs and myocardial
cells. In the A2 and B2 groups, transplanted cardiac cell
differentiation and neovascularization were rarely observed
(Fig. 11B and D); therefore
indicating that the experimental effects of A1 and B1 treatments
were markedly more beneficial compared with those of A2 and B2
(Fig. 11).
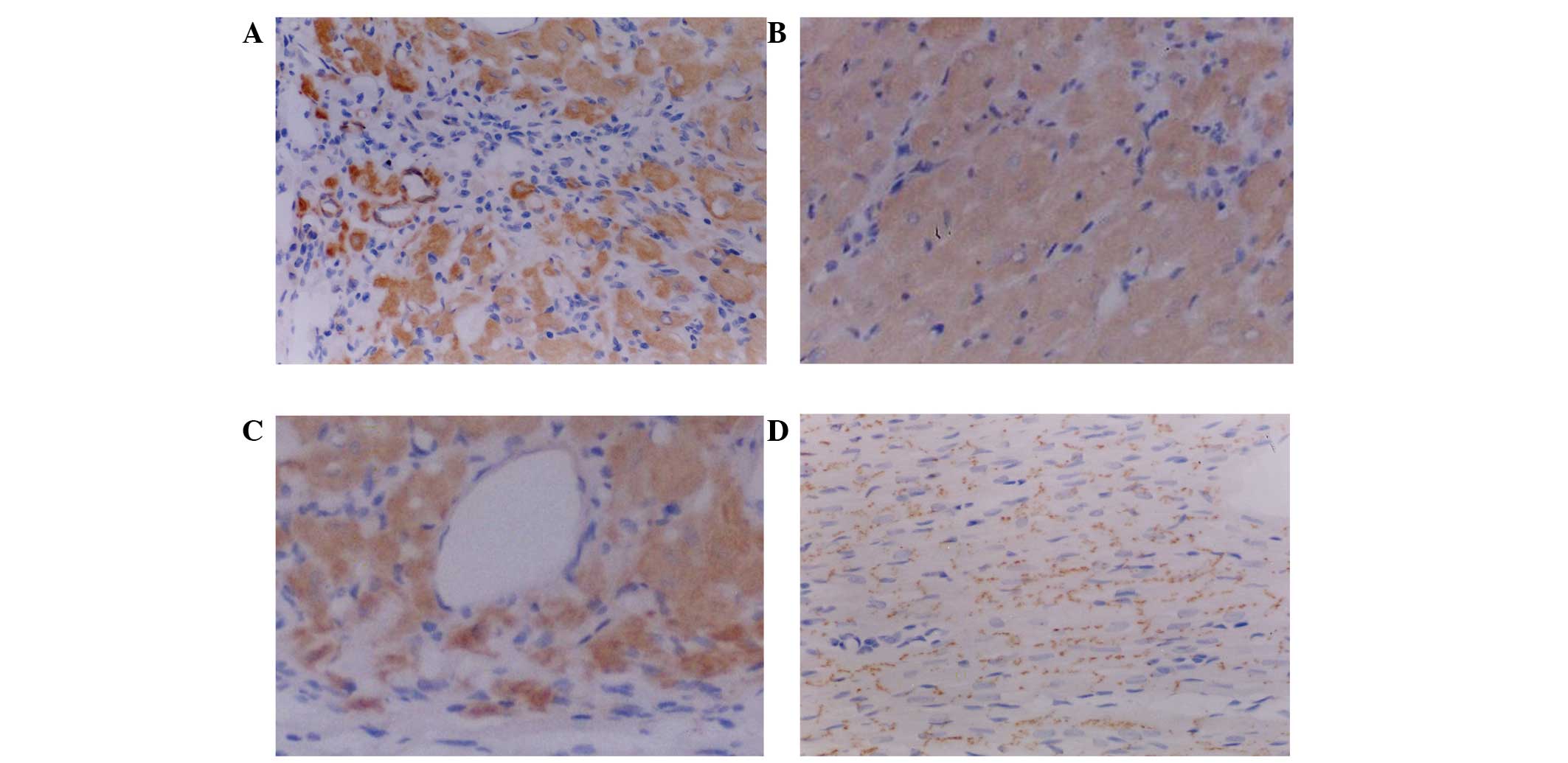
Immunohistochemical staining
Following transfected-BMSC transplantation in the A1
and B1 rabbit groups, immunohisto-chemical analysis revealed that
the transplanted cells survived and grew among the myocardial cells
(Fig. 12A and C). Brown stained
substances were visible in the peripheral areas of the cells, which
suggested that the transplanted cells successfully survive among
myocardial cells or had been induced to differentiate into
cardiomyocyte-like cells. Connections between myocardial cells were
increased and the repair of damaged myocardial tissue was promoted.
Certain brown-stained cells were able to self assemble in order to
form vessel-like structures; in addition, to a certain extent,
blood circulation within the damaged myocardium was increased and
damaged heart muscle tissue repair was promoted. Transplanted cells
showed overall growth of the transfected area as well as radial and
infiltrating growth into the surrounding area. At the junction
between transplanted cells and normal myocardium, the transplanted
cells gradually shifted into spindle-shapes and the boundaries
between the two areas became indistinguishable, where the brown
staining was observed. This therefore indicated that the
transplanted cells were able to partially or completely replace the
function of the damaged portion of myocardial cells and novel cell
connections were noticed between transplanted cells and myocardial
cells. In A2 and B2 groups, the surviving transplanted cells
stained brown and were distributed in the myocardium (Fig. 12B and D). These transplanted cells
did not show signs of improving myocardial function; however, they
showed improved cell connections between the myocardial cells. This
therefore suggested that induced and myocardial cell co-cultured
rabbit BMSCs had improved adaptive abilities in the myocardial
microenvironment following transplantation compared with those of
blank-transfected cells; therefore, indicating that these methods
of BMSCs pre-treatment may induce the function of transplanted
cells more rapidly and improve the survival of transplanted
cells.
Discussion
As a result of the rapid progress in the development
of molecular biology techniques over recent years, gene
transfection technology is currently used in various fields of
biology. Gene transfection technology is used in order to transfer
and deliver genes, which are then maintained and expressed in the
cells. There are several types of transgenic plants and animals
that are widely known to the public (18,20);
however, gene trans-fection technology is also commonly used for
gene therapy studies (21). In
studies on the use of mesenchymal stem cell (MSC) therapy to repair
damaged areas of the myocardium, MSCs transfected with particular
genes were used to further improve the efficacy of bone marrow
mesenchymal stem cell therapy. For example, certain studies have
focused on improving the survival of BMSCs following
transplantation, in which the heme oxygenase-1 (HO-1) was used to
genetically modify BMSCs in order to enhance cell tolerance to
hypoxia (22,23,24).
In addition, angiogenin has been used to genetically modify BMSCs
in order to protect against cardiomyocyte damage and promote the
process of angiogen-esis. The overexpression of HO-1 or angiogenin
in marrow MSCs was shown to increase the survival of transplanted
cells and neovascularization, and improve cardiac function.
Furthermore, insulin like growth factor, vascular endothelial
growth factor, hepatocyte growth factor and Akt have been used to
modify the gene expression of BMSCs (14–16,18,20,
25,26).
Gene transfection methods for eukaryotic cells are
performed according to different mechanisms, which are divided into
physical transfection methods, chemical transfection methods and
viral transfection methods. Chemical transfection methods include
the diethylaminoethyl cellulose (DEAE)-dextran method, the calcium
phosphate method and the artificial liposome method (25–28);
the most widely used method is the artificial liposome method
(29). DEAE-dextran reagent was
one of the earliest transfection reagents used in mammalian cells.
It is a cationic polymer, which can be combined with negatively
charged nucleic acids in order to be absorbed by the cell membrane
(30). DEAE-dextran transfection
is not reliable for stable transfection (30). The calcium phosphate
co-precipitation transfection method is widely used in studies for
transient and stable transfection as it is cheap and reagents are
readily available. The method is as follows: DNA and calcium
chloride are combined and then added to PBS in order to slowly form
a DNA calcium phosphate precipite. The suspension containing the
precipitate is then added to cultured cells and the DNA is uptaken
by endocytosis of the cell membrane (18). In the artificial liposome method,
the liposomes may also transport DNA and RNA in vivo in
order to transfect into animals and humans for gene therapy;
synthetic cationic liposomes bind with negatively charged nucleic
acids to form a complex, which becomes surrounded by the cell
membrane and is taken into the cytoplasm via endocytosis where the
DNA complexes are released into the nucleus (31). Physical transfection methods
include electroporation, microinjection and the gene gun method.
The electroporation method involves high-voltage electroporation
for interference of the cell membrane, so that pores are formed for
easy entrance of nucleic acid into the cytoplasm. Using the
electroporation method it is possible to inject millions of cells
at a time, without using glass microin-jectors, technical training
or expensive equipment. Compared with chemical transfection
methods, electroporation has few biological or chemical side
effects. As a physical method, elec-troporation is also less
dependent on cell type, and is widely used and highly efficient
(32). Microinjection methods are
more expensive, but are effective for the induction of nucleic acid
into human cells or nuclei. This method is used to produce
transgenic animals; however, it is not suitable for studies that
require a large quantity of transfected cells (33). The gene gun method uses high-speed
particles to carry nucleic acids into cells; this method is
applicable to cultured cells and cells in the body (34). The viral transfection method is an
efficient gene delivery system; therefore, it is the most effective
transfer method for the transfection of exogenous genes into a
large number of cells in the human body. Viral infections have high
efficiency, stable inheritance, may be applied to a variety of
cells from different sources via simple methods, and can be
conducted using retroviruses or adenoviruses. However, most viruses
are potentially dangerous; therefore, operators are required to be
experienced and have access high standard virus facilities
(35). At present, the most widely
used method of transfection is the artificial liposomes method as
it has fewer issues compared with other methods. Lipofectamine 2000
is the third generation of multivalent cationic lipo-somes, which
has significantly higher transfection efficiency, specificity and
reproducibility compared with those of other liposomes. In
addition, it is a non-immunogenic, non-infectious, non-carcinogenic
and simple method for transfection. It has passed the authorization
of NIH and recombinant DNA Advisory Committee (RAC) as a gene
therapy vector for PhaseII clinical trials and has been used for
the treatment of certain types of cancer (36). A previous study has shown that
Lipofectamine 2000 successfully achieved the exogenous genes
transfection of BMSCs (37).
Therefore the present study used Lipofectamine 2000 for BMSC
transfection. Western blot analysis revealed that liposomes
successfully mediated plasmid transfection of pEGFP-N1-Nkx2.5 or
pVP22-GATA-4/myc-His into BMSCs.
The specific mechanisms of cardiomyocyte
differentiation of BMSCs in vitro remain to be elucidated.
One of the primary aims of cardiomyocytes differentiation studies
at present, is to identify the key genes and factors required to
initiate the directed differentiation of BMSCs in vitro in
order to cultivate a variety of cells and even organs. The present
study investigated the transfection of Nkx2.5 and GATA-4, two
closely associated transcription factors, which were reported to be
involved in heart development (17,38).
Nkx2.5 was found to participate in cardiac formation, including the
right heart cyclization, differentiation of cardiac chambers,
maturation of cardiac interval function, as well as the maintenance
of the myocardium and the cardiac conduction system (39). GATA-4 has been shown to have an
important role in cardiac proliferation, differentiation and
survival as well as the cardiac hypertrophy process (40). The development process of the heart
is not completed by a single or small number of genes, but a
variety of genes which form regulatory networks, these genes
include Nkx2.5 and GATA-4, which jointly regulate the normal
development of the heart (41). At
present, the gene regulatory mechanisms of heart development
remained to be elucidated and require further investigation. The
present study used the cationic liposome method to transfect Nkx2.5
or GATA-4 into BMSCs in order to determine whether the
over-expression of transcription factors was able to induce BMSCs
to differentiate into cardiomyocytes. Transfected BMSCs were
co-cultured with cardiomyocytes in order to compare the effect of
the two methods; previous studies on these methods are limited. The
results of this experiment showed that the effect of co-culture in
combination with transfection was more effective than transfection
alone.
cTnT is a cardiac-specific troponin T, which is only
expressed in the myocardium. In the present study, the results of
the western blot and immunocytochemical analysis were consistent in
the transfection only and transfection with co-culture groups,
indicating that Nkx2.5 and GATA-4 upregulated the expression of
cardiac-specific protein-cTnT following four weeks of culture.
Myocardial infarction animal models are commonly
established using mechanical ventilation, chest coronary artery
ligation or myocardial freezing injury (42–44).
The myocardial freezing injury model forms identical infarct sizes
as those of transmural myocardial infarction and may therefore be
easily compared and observed; however, controlling the freezing
time in the method is difficult. Freezing for too long may result
in destruction of the cardiac tissue, whereas freezing for not long
enough prevents effective transmural myocardial infarction; control
of freezing time is particularly difficult in small animals,
including mice and rabbits, due to the small volume of the heart.
Therefore, this method is not commonly used (45–49).
In the present study, coronary artery ligation was used to
establish a rabbit myocardial infarction model as the cardiac
structure of rabbits and humans are similar. The clinical and
pathological mechanisms underlying coronary artery ligation in
order to establish myocardial infarction in rabbits were similar to
that of the mechanisms of myocardial infarction. This method is
cheap and the surgical procedure is simple, with high success rates
when performed by skilled surgeons; however, the disadvantage of
this method is different infarct sizes due to the size of different
animals. Human intervention cannot control the size of the infarct
area following myocardial infarction; therefore, the efficacy has
not been well observed. In the present study, the transplanted New
Zealand rabbit BMSCs grew and survived among the myocardial cells.
Cells were found to accumulate in the central transplantation area
as a mass; however, they also demonstrated radial infiltrative
growth in the peripheral transplant area. The cells at the junction
between the transplanted area and normal myocardium gradually
changed to a spindle shape. In addition, novel cell connections
were formed between the transplanted cells and the rabbit
myocardial cells, indicating that this connection may cause the
reactivation of myocardial cells within the infarction area, which
may restore the basic function of myocardial cells in the infarct
area. In addition, in the infarct area, an increase in the number
of fibroblasts and in angiogenesis was observed, which may have
limited the continuous expansion of the infarct area and the impact
of myocardial infarction on heart function. Novel blood vessel
formation may have also improved the blood microcirculation and
cellular microenvironment within the infarcted myocar-dium, which
may increase the survival of myocardial cells. In addition,
angiogenesis may increase the nutritional supply to transplanted
exogenous cells and re-activate cardiac cells, therefore increasing
their effectiveness. The differentiation and angiogenesis in the
transfected BMSCs and co-cultured myocardial cell group was
markedly increased compared with that in the control group. A
degree of cardiomyocyte differentiation and angiogenesis were
observed in the control group; however, these effects were improved
in rabbits treated with BMSCs and co-cultured myocardial cells
following transfection.
The results of the present study showed that
following transplantation into areas with myocardial cells, the
growth and survival of the transplanted cells was observed;
immu-nohistochemical staining revealed brown-stained substances at
the periphery of certain cells, suggesting that transplanted cells
successfully survived among the myocardial cells or certain
transplanted cells had been induced to differentiate into
cardiomyocyte-like cells. Connections were observed between the
myocardial cells, and certain cells with brown-staining were able
to self-assemble to form vessel-like structures, which resulted in
increased blood circulation within the damaged myocardium. In
addition, the repair of damaged myocardial tissues was promoted in
transplanted cell region and transplanted cells grew as a mass, as
well as radial infiltrating growth into the surrounding area. The
transplanted cells at the junction between the transplanted area
and the normal myocardium area gradually shifted towards a
spindle-shaped morphology and the boundaries between the two
regions became indistinguishable. In addition, immunohistochemical
staining indicated that the transplanted cells were able to
partially or completely restore the function of the damaged portion
of the myocardium. Novel cell connections were observed between the
transplanted cells and myocardial cells, which suggested that
transfection and co-culture of BMSCs prior to transplantation
increased the adaptive ability of cells to the myocardial
microenvironment as well as improved cell survival, compared with
that of the control BMSCs. The results of the present study were
only observed by microscopy. Further studies are required in order
to determine the actual experimental results and survival rates of
transplanted cells, as well as their role in the cardiac
microenvironment.
In conclusion, the exogenous gene transfection of
Nkx2.5 or GATA-4 in combination with myocardial extracellular
environment co-culture of BMSCs, resulted in improved myocardial
cell differentiation compared with that of gene transfection only.
In addition, exogenous gene transfection of Nkx2.5 or GATA-4 into
myocardial cell extracellular environment co-cultured BMSCs was
able to enhance the ability to repair, mitigate the death of the
myocardial cells and activate myocardium in myocardial
infarction-induced rabbits.
References
|
1
|
Arrington ED, Smith WJ, Chambers HG,
Bucknell AL and Davino NA: Complications of iliac crest bone graft
harvesting. Clin Orthop Relat Res. 329:300–309. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blum B, Moseley J, Miller L, Richelsoph K
and Haggard W: Measurement of bone morphogenetic proteins and other
growth factors in demineralized bone matrix. Orthopedics. 27(1
Suppl): S161–S165. 2004.PubMed/NCBI
|
|
3
|
Fernandes KJ, Toma JG and Miller FD:
Multipotent skin-derived precursors: adult neural crest-related
precursors with therapeutic potential. Philos Trans R Soc Lond B
Biol Sci. 363:185–198. 2008. View Article : Google Scholar
|
|
4
|
Gimble JM, Guilak F, Nuttal ME,
Sathishkumar S, Vidal M and Bunnel BA: In vitro differentiation
potential of mesenchymal stem cells. Transfus Med Hemother.
35:228–238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gronthos S, Mankani M, Brahim J, Robey PG
and Shi S: Postnatal human dental pulp stem cells (DPSCs) in vitro
and in vivo. Proc Natl Acad Sci USA. 97:13625–13630. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gronthos S, Brahim J, Li W, Fisher LW,
Cherman N, Boyde A, DenBesten P, Robey PG and Shi S: Stem cell
properties of human dental pulp stem cells. J Dent Res. 81:531–535.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeon O, Rhie JW, Kwon IK, Kim JH, Kim BS
and Lee SH: In vivo bone formation following transplantation of
human adipose-derived stromal cells that are not differentiated
osteogenically. Tissue Eng Part A. 14:1285–1294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jeon BG, Kang EJ, Kumar BM, Maeng GH, Ock
SA, Kwack DO, Park BW and Rho GJ: Comparative analysis of telomere
length, telomerase and reverse transcriptase activity in human
dental stem cells. Cell Transplant. 20:1693–1705. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaiser S, Hackanson B, Follo M, Mehlhorn
A, Geiger K, Ihorst G and Kapp U: BM cells giving rise to MSC in
culture have a heterogeneous CD34 and CD45 phenotype. Cytotherapy.
9:439–450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kajahn J, Gorjup E, Tiede S, von Briesen
H, Paus R, Kruse C and Danner S: Skin-derived human adult stem
cells surprisingly share many features with human pancreatic stem
cells. Eur J Cell Biol. 87:39–46. 2008. View Article : Google Scholar
|
|
11
|
Kanematsu D, Shofuda T, Yamamoto A, Ban C,
Ueda T, Yamasaki M and Kanemura Y: Isolation and cellular
properties of mesenchymal cells derived from the deciduas of human
term placenta. Differentiation. 82:77–88. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kang EJ, Byun JH, Choi YJ, Maeng GH, Lee
SL, Kang DH, Lee JS, Rho GJ and Park BW: In vitro and in vivo
osteogenesis of porcine skin-derived mesenchymal stem cell-like
cells with a demineralized bone and fibrin glue scaffold. Tissue
Eng Part A. 16:815–827. 2010. View Article : Google Scholar
|
|
13
|
Kasten P, Luginbühl R, van Griensven M,
Barkhausen T, Krettek C, Bohner M and Bosch U: Comparison of human
bone marrow stromal cells seeded on calcium-deficient
hydroxy-apatite, beta-tricalcium phosphate and demineralized bone
matrix. Biomaterials. 24:2593–2603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lindroos B, Mäenpää K, Ylikomi T, Oja H,
Suuronen R and Miettinen S: Characterisation of human dental stem
cells and buccal mucosa fibroblasts. Biochem Biophys Res Commun.
368:329–335. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu G, Sun J, Li Y, Zhou H, Cui L, Liu W
and Cao Y: Evaluation of partially demineralized osteoporotic
cancellous bone matrix combined with human bone marrow stromal
cells for tissue engineering: an in vitro and in vivo study. Calcif
Tissue Int. 83:176–185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu G, Li Y, Sun J, Zhou H, Zhang W, Cui L
and Cao Y: In vitro and in vivo evaluation of osteogenesis of human
umbilical cord blood-derived mesenchymal stem cells on partially
demineralized bone matrix. Tissue Eng Part A. 16:971–982. 2010.
View Article : Google Scholar
|
|
17
|
Suda J, Suda T and Ogawa M: Analysis of
differentiation of mouse hemopoietic stem cells in culture by
sequential replating of paired progenitors. Blood. 64:393–399.
1984.PubMed/NCBI
|
|
18
|
Ogawa M: Stochastic model revisited. Int J
Hematol. 69:2–5. 1999.
|
|
19
|
Imabayashi H, Mori T, Gojo S, Kiyono T,
Sugiyama T, Irie R, Isogai T, Hata J, Toyama Y and Umezawa A:
Redifferentiation of dedifferentiated chondrocytes and
chondrogenesis of human bone marrow stromal cells via chondrosphere
formation with expression profiling by large-scale cDNA analysis.
Exp Cell Res. 288:35–50. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dezawa M, Ishikawa H, Itokazu Y, Yoshihara
T, Hoshino M, Takeda S, Ide C and Nabeshima Y: Bone marrow stromal
cells generate muscle cells and repair muscle degeneration.
Science. 309:314–317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Umezawa A, Tachibana K, Harigaya K,
Kusakari S, Kato S, Watanabe Y and Takano T: Colony-stimulating
factor 1 expression is down-regulate d during the adipocyte
differentiation of H-1/A marrow stromal cells and induced by
cachectin/tumor necrosis factor. Mol Cell Biol. 11:920–927.
1991.PubMed/NCBI
|
|
22
|
Liu XH, Bai CG, Xu ZY, et al: Therapeutic
potential of angiogenin modified mesenchymal stem cells: Angiogenin
improves mesenchymal stem cells survival under hypoxia and enhances
vasculogenesis in myocardial infarction. Microvasc Res. 76:23–30.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang YL, Tang Y, Zhang YC, et al: Improved
graft mesenchymal stem cell survival in ischemic heart with a
hypoxia-regulated heme oxygenase-1 vector. J Am Coll Cardiol.
46:1339–1350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsubokawa T, Yagi K, Nakanishi C, et al:
Impact of anti-apoptotic and anti-oxidative effects of bone marrow
mesenchymal stem cells with transient overexpression of heme
oxygenase-1 on myocardial ischemia. Am J Physiol Heart Circ
Physiol. 298:H1320–H1329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kohyama J, Abe H, Shimazaki T, Koizumi A,
Nakashima K, Gojo S, Taga T, Okano H, Hata J and Umezawa A: Brain
from bone: efficient ‘meta-differentiation’ of marrow
stroma-derived mature osteoblasts to neurons with Noggin or a
demethylating agent. Differentiation. 68:235–244. 2001. View Article : Google Scholar
|
|
26
|
Mori T, Kiyono T, Imabayashi H, Takeda Y,
Tsuchiya K, Miyoshi S, Makino H, Matsumoto K, Saito H, et al:
Combination of hTERT and bmi-1, E6, or E7 induces prolongation of
the life span of bone marrow stromal cells from an elderly donor
without affecting their neurogenic potential. Mol Cell Biol.
25:5183–5195. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gojo S, Gojo N, Takeda Y, Mori T, Abe H,
Kyo S, Hata J and Umezawa A: In vivo cardiovasculogenesis by direct
injection of isolated adult mesenchymal stem cells. Exp Cell Res.
288:51–59. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hakuno D, Fukuda K, Makino S, Konishi F,
Tomita Y, Manabe T, Suzuki Y, Umezawa A and Ogawa S: Bone
marrow-derived regenerated cardiomyocytes (CMG Cells) express
functional adrenergic and muscarinic receptors. Circulation.
105:380–386. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hoang T: The origin of hematopoietic cell
type diversity. Oncogene. 23:7188–7198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lemischka IR, Raulet DH and Mulligan RC:
Developmental potential and dynamic behavior of hematopoietic stem
cells. Cell. 45:917–927. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Watt FM and Hogan BL: Out of Eden: stem
cells and their niches. Science. 287:1427–1430. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tenen DG, Hromas R, Licht JD and Zhang DE:
Transcription factors, normal myeloid development, and leukemia.
Blood. 90:489–519. 1997.PubMed/NCBI
|
|
33
|
Sharov AA, Piao Y, Matoba R, Dudekula DB,
Qian Y, VanBuren V, Falco G, Martin PR, Stagg CA, Bassey UC, et al:
Transcriptome analysis of mouse stem cells and early embryos. PLoS
Biol. 1:E742003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matsumoto S, Shibuya I, Kusakari S, Segawa
K, Uyama T, Shimada A and Umezawa A: Membranous osteogenesis system
modeled with KUSA-A1 mature osteoblasts. Biochim Biophys Acta.
1725:57–63. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Makino S, Fukuda K, Miyoshi S, Konishi F,
Kodama H, Pan J, Sano M, Takahashi T, Hori S, et al: Cardiomyocytes
can be generated from marrow stromal cells in vitro. J Clin Invest.
103:697–705. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Simpson P, McGrath A and Savion S: Myocyte
hypertrophy in neonatal rat heart cultures and its regulation by
serum and by catecholamines. Circ Res. 51:787–801. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sano M, Umezawa A, Abe H, Akatsuka A,
Nonaka S, Shimizu H, Fukuma M and Hata J: EAT/mc l-1 expression in
the human embryonal carcinoma cells undergoing differentiation or
apoptosis. Exp Cell Res. 266:114–125. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dennis JE and Charbord P: Origin and
differentiation of human and murine stroma. Stem Cells. 20:205–214.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vincentz JW, Barnes RM, Firulli BA, et al:
Cooperative interaction of Nkx2.5 and Mef2c transcription factors
during heart development. Dev Dyn. 237:3809–3819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Leong FT, Freeman LJ and Keavney BD: Fresh
fields and pathways new: recent genetic insights into cardiac
malformation. Heart. 95:442–447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schluterman MK, Krysiak AE, Kathiriya IS,
et al: Screening and biochemical analysis of GATA4 sequence
variations identified in patients with congenital heart disease. Am
J Med Genet A. 143:817–823. 2007. View Article : Google Scholar
|
|
42
|
Muraglia A, Cancedda R and Quarto R:
Clonal mesenchymal progenitors from human bone marrow differentiate
in vitro according to a hierarchical model. J Cell Sci. 113(Pt 7):
1161–1166. 2000.PubMed/NCBI
|
|
43
|
Santi DV, Norment A and Garrett CE:
Covalent bond formation between a DNA-cytosine methyltransferase
and DNA containing 5-azacytosine. Proc Natl Acad Sci USA.
81:6993–6997. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oh H, Bradfute SB, Gallardo TD, et al:
Cardiac progenitor cells from adult myocardium: homing,
differentiation, and fusion after infarction. Proc Natl Acad Sci
USA. 100:12313–12318. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kitakaze M, Asanuma H, Funaya H, Node K,
Takashima S, Sanada S, Asakura M, Ogita H and Kim J: role of
bradykinin. J Am Coll Cardiol. 40:162–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Weber KT: Extracellular matrix remodeling
in heart failure: a role for de novo angiotensin II generation.
Circulation. 96:4065–4082. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sutton MG and Sharpe N: Left ventricular
remodeling after myocardial infarction: pathophysiology and
therapy. Circulation. 101:2981–2988. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Szárszoi O, Malý J, Ostádal P, Netuka I,
Besík J, Kolár F and Ostádal B: Effect of acute and chronic
simvastatin treatment on post-ischemic contractile dysfunction in
isolated rat heart. Physiol Res. 57:793–796. 2008.PubMed/NCBI
|
|
49
|
Tiefenbacher CP, Kapitza J, Dietz V, Lee
CH and Niroomand F: Reduction of myocardial infarct size by
fluvastatin. Am J Physiol Heart Circ Physiol. 285:H59–H64. 2003.
View Article : Google Scholar : PubMed/NCBI
|