Introduction
Ischemic stroke is a leading cause of disability and
mortality worldwide, which is triggered by vascular occlusion via
either in situ thrombosis or embolization of a clot from a
proximal arterial or cardiac source (1–3). The
area of the brain, which is reliant exclusively on the blood supply
from the occluded vessel is termed the ‘ischemic core’ (4,5). The
area surrounding the ischemic core is referred to as the
‘penumbra’, which is perfused, in part, by collateral blood flow
(4,5). Usually, vascular occlusion induces
the deprivation of glucose and O2 and leads to
deprivation in the ischemic brain areas, particularly at the
ischemic core (4–6). This finally results in the damage of
neurons in the ischemic brain areas by initiating a complex cascade
of cellular events, including glutamate-induced excitotoxicity,
free radical-mediated injury and inflammation (4–8).
Therefore, following ischemic stroke, patients usually exhibit
motor, sensory or cognitive function, which leads to serious
financial burden for the patient and their families (4–8). At
present, the most successful therapeutic strategy for ischemic
stroke is reperfusion (6,7). However, restoration of perfusion to
ischemic brain areas can exacerbate tissue damage by mechanisms,
including the generation of reactive oxygen species (ROS) by
mitochondria and the increased recruitment of inflammatory cells,
(6,7). Thus, inhibiting or reducing the
damage caused by reperfusion is an important issue in the treatment
of ischemic stroke.
Programmed necrosis, termed, necroptosis was
originally reported by Degterev et al in 2005 (9). It is a form of cell death, which is
distinctly different from necrosis and apoptosis. It is
characterized by cell swelling, mitochondria dysfunction, cell
membrane permeabilization and the release of cytoplasmic content to
the extracellular space (10).
However, DNA fragmentation does not occur (10). Degterev et al demonstrated
that middle cerebral artery occlusion (MCAO) induced significant
brain infarction and behavioral defects of mice (9). The necroptosis specific inhibitor,
necrostatin-1 (Nec-1), significantly decreased the volume of brain
infarction and improved the behavior of mice when administered 2 or
6 h following MCAO (9). Similar
results were also detected in cultured hippocampal neurons
following oxygen-glucose deprivation (OGD), an ischemia-reperfusion
model (11). Inhibiting
necroptosis significantly decreases the OGD-induced loss of
cultured hippocampal neurons (11), suggesting that necroptosis is
important in ischemia-reperfusion damage. Additional studies have
revealed that receptor-interacting protein (RIP)1, RIP3 and ROS are
important signaling molecules in necropolis (11,12).
However, the molecular networks underlying necroptosis during
ischemia-reperfusion damage remain to be elucidated.
Histone deacetylase 6 (HDAC6) is a member of the
class IIb histone deacetylases and is present predominantly in the
cytosol (13,14). HDAC6 is expressed widely in the
brain (15). Chen et al
demonstrated that the expression of HDAC6 is markedly upregulated
in the mice cortex at 3 h and 12 h of reperfusion following MCAO
(16). This study also
demonstrated that the expression of HDAC6 increases in cultured
mouse hippocampal neurons following OGD (16). These findings suggested that HDAC6
is important in ischemia-reperfusion damage. Notably, previous
studies have revealed that HDAC6 enhances neuronal oxidative stress
by deacetylating peroxiredoxin-1 and peroxiredoxin-2 (17). HDAC6 also decreases mitochondrial
transport by modulating the acetylation level of α-tubulin
(13,18). Mitochondria-associated oxidative
stress is the major mechanism of damage in neuron necroptosis
during ischemia-reperfusion (6,7).
Therefore, the present study hypothesized that HDAC6 is closely
involved in the necroptosis of neurons during ischemia-reperfusion.
The present study investigated the effects of OGD on the expression
of HDAC6 in cultured rat cortical neurons, and its association with
the necroptosis of neurons following OGD.
Materials and methods
Materials
The primary antibody against HDAC6 (cat. no.
PAB8753) was purchased from Abnova (Taibei, Taiwan), antibodies
against acetylated (Ac)-tubulin (cat. no. T6793), microtubule
associated protein (MAP)2 (cat. no. M9942), necroptosis inhibitor,
Nec-1 (cat. no. N-9037) were purchased from Sigma-Aldrich (St.
Louis, MO, USA) and the HDAC6 specific inhibitor, tubacin was
purchased from ChemieTek (Indianapolis, IN, USA). Propidium iodide
(PI) was purchased from Jackson ImmunoResearch Laboratories, Inc.
(West Grove, PA, USA). The following secondary antibodies were
purchased from Jackson ImmunoResearch, Laboratories, Inc.:
Polyclonal Alexa Fluor-488 AffiniPure donkey anti-rabbit IgG
(1:500; 711-545-152) and polyclonal Alexa Fluor-594 AffiniPure
donkey IgG (1:500; 711-585-152). The Reactive Oxygen Species Assay
kit (cat. no. C13293) was purchased from Invitrogen Life
Technologies (Shanghai, China).
Animals
All experimental procedures were approved by the
Institutional Review Board of the Third Xiangya Hospital of Central
South University (Changsha, China). A total of six pregnant
Sprague-Dawley rats (18 days of pregnancy; ~3 months old) were
purchased from Central South University. All the rats were raised
under controlled environmental conditions on a 12 h light/dark
cycle at 22–24°C with ad libitum access to food and water
and were housed alone.
Primary neuron culture
Primary cultures of rat cortical neurons were
prepared from embryonic day 18 (E18) rats (removed from the
pregnant rats), using a previously described procedure (19). Briefly, the cortex of the E18 rat
was dissected and washed five times in Hank’s balanced salt
solution (HBSS; Invitrogen Life Technologies, Carlsbad, CA, USA).
The cortex tissue was then digested using 0.125% trypsin
(Invitrogen Life Technologies) for 13 min at 37°C. Following three
washes in HBSS, the digested cortex was dissociated from the
meninges and subcortical structures and plated on the coverslips
coated with poly-D-lysine in Dulbecco’s modified Eagle’s medium
(Invitrogen Life Technologies) with 10% fetal bocine serum
(Invitrogen Life Technologies) and 1% penicillin-streptomycin
(Invitrogen Life Technologies). After 4 h, the cultured medium
(37°C) was replaced with neurobasal medium with 1% B27 (Invitrogen
Life Technologies). Half of the maintenance medium (neurobasal
medium + 1% B27) of THE cultured neurons was replaced every 2
days.
Oxygen-glucose deprivation (OGD) and drug
treatments
OGD was prepared according to previously reported
methods (11,20). Briefly, on the fourth day of
culture, the cultured cortical neurons were divided into control,
OGD, OGD+Nec-1 and OGD+tubacin groups. The neurons in the OGD group
were placed in glucose-free deoxygenated buffer medium (OGD medium;
Dulbecco’s modified Eagle’s medium without glucose; Gibco Life
Technologies, Shanghai, China) with solvent inside an anaerobic OGD
chamber with 5% CO2 and residual levels of O2
(Thermo Forma 1029; Thermo Fisher Scientific, Waltham, MA) at 37°C
for 2 h. The neurons in the OGD + Nec-1 group were placed in 2 ml
OGD medium with 2 μl 1% Nec-1 (Sigma-Aldrich) (11), inside an OGD chamber with 5%
CO2 and residual levels of O2 at 37°C for 2
h. The neurons in the OGD + tubacin group were placed in OGD medium
with 2.5 μM tubacin (21),
inside an OGD chamber with 5% CO2 and residual levels of
O2 at 37°C for 2 h. The neurons in the control group
were placed in a similar buffer, containing 25 mM glucose
(Sigma-Aldrich) and 2 µl dimethyl sulfoxide (DMSO), and maintained
for 2 h in a humidified incubator with 5% CO2/95% air at
37°C. Following incubation, the neurons of each group were placed
in their conditioned medium and returned to the normoxic incubator
(Thermo Forma 1029; Thermo Fisher Scientific) for 3 h recovery.
Nuclear morphology
Neuronal death was assessed by analyzing the nuclear
morphology 3 h after OGD. At this time-point, the neurons in each
group were stained using the nuclear dye, PI (2 μg/ml), for
8 min at 37°C. The neurons were then washed, fixed in 4%
paraformaldehyde (Sigma-Aldrich), washed again with 0.01 M
phosphate-buffered saline (PBS) and covered with mounting medium
(Vector Laboratories, Inc., Burlingame, CA, USA) and
4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Inc.).
The PI staining and chromatin condensation of the neurons in each
group were immediately analyzed under fluorescence microscopy
(Nikon 80i; Nikon, Tokyo, Japan). According to a method described
by Vieira (11), ‘positive PI
staining’ was considered a necrotic marker, since membrane leak is
one of the predominant features of necrotic cell death (11). Chromatin condensation and pyknosis,
revealed by the DAPI staining were referred to as apoptotic markers
(11). Necrotic-like neuronal
death was marked by positive PI staining (11). The percentages of necrotic cell
death (PI-positive cells / total number of cells) and of
apoptotic-like cell death (apoptotic-like nuclei / total number of
cells) were calculated.
Immunofluorescence
Following washing with 0.01 M PBS for 10 min three
times, the neurons were incubated in blocking solution
(Sigma-Aldrich), containing 5% bovine serum albumin and 0.3% Triton
X-100 in 0.01 M PBS, for 1 h at room temperature. The neurons were
then incubated with primary antibodies (rabbit anti-HDAC6; 1:500;
cat. no. PAB8753; Abnova), mouse anti-ac-tubulin (1:1,000; cat. no.
T6793; Sigma-Aldrich), mouse anti-MAP2 (1:1000; cat. no. M9942;
Sigma-Aldrich) overnight at 4°C. Following incubation, the neurons
were washed with 0.01 M PBS three times and then incubated with
secondary antibodies labeled with fluorescent dyes (1:500, Jackson
ImmunoResearch Laboratories, Inc.) for 2 h at room temperature.
Following three washes in PBS, the neurons were covered with
mounting medium and DAPI. As negative controls, normal neurons were
processed using the same procedures without the primary antibodies.
The immunofluorescence intensities of the HDAC6 and Ac-tubulin
staining in each group were detected using the Nikon Eclipse 80i
microscope and Image J software, version 1.48.
ROS detection
The level of intracellular ROS was detected using a
Reactive Oxygen Species Assay kit, according to a previously
reported method (22), in which
DCFH-DA (Invitrogen Life Technologies), a fluorescent probe, is
oxidized by ROS in viable cells to 2′,7′-dichlorofluorescein.
Briefly, the cultured neurons were incubated with 100 μM
DCFH-DA dissolved in DMSO for 30 min at 37°C. Following times
washes with PBS, the cultured neurons were covered and detected
using fluorescence microscopy. The ROS fluorescence intensity was
detected using ImageJ software.
Statistical analysis
Data are presented as the mean ± standard deviation
and were analyzed using one-way analysis of varianced followed by a
Student-Newman-Keuls test. P<0.05 was considered to indicate a
statistically significant difference.
Results
OGD upregulates the expression of HDAC6
and induces necroptosis of cultured rat cortical neurons
OGD is a commonly-used in vitro model of
ischemia-reperfusion. The present study investigated necroptosis
and the expression of HDAC6 in cultured rat cortical neurons
following OGD. The results revealed that the percentage of necrotic
cell death, determined by the number of PI-positive cells without
pyknosis / total number of cells, in the OGD group was 60.5±5.8%,
which was significantly higher than that of the control (12.5±2.5%)
and OGD+Nec-1 (40.8±4.3%) groups (P<0.05; Fig. 1). The percentage of necrotic cell
death in the OGD+Nec-1 group (40.8±43%) was significantly higher
than that of the control group (P<0.05; Fig. 1). The percentage of apoptotic-like
cell death, calculated as the number of apoptotic-like nuclei /
total number of cells, in the OGD group (54.7±5.8%) was markedly
higher, compared with that of the control group (18.6±4.5%;
Fig. 1). However, no significant
difference was observed in the percentage of apoptotic-like cell
death between the ODG and OGD+Nec-1 (48.6±6.2%) groups (P>0.05;
Fig. 1). These results suggested
that ODG induced marked necroptosis in The cultured rat cortical
neurons, which was inhibited by Nec-1.
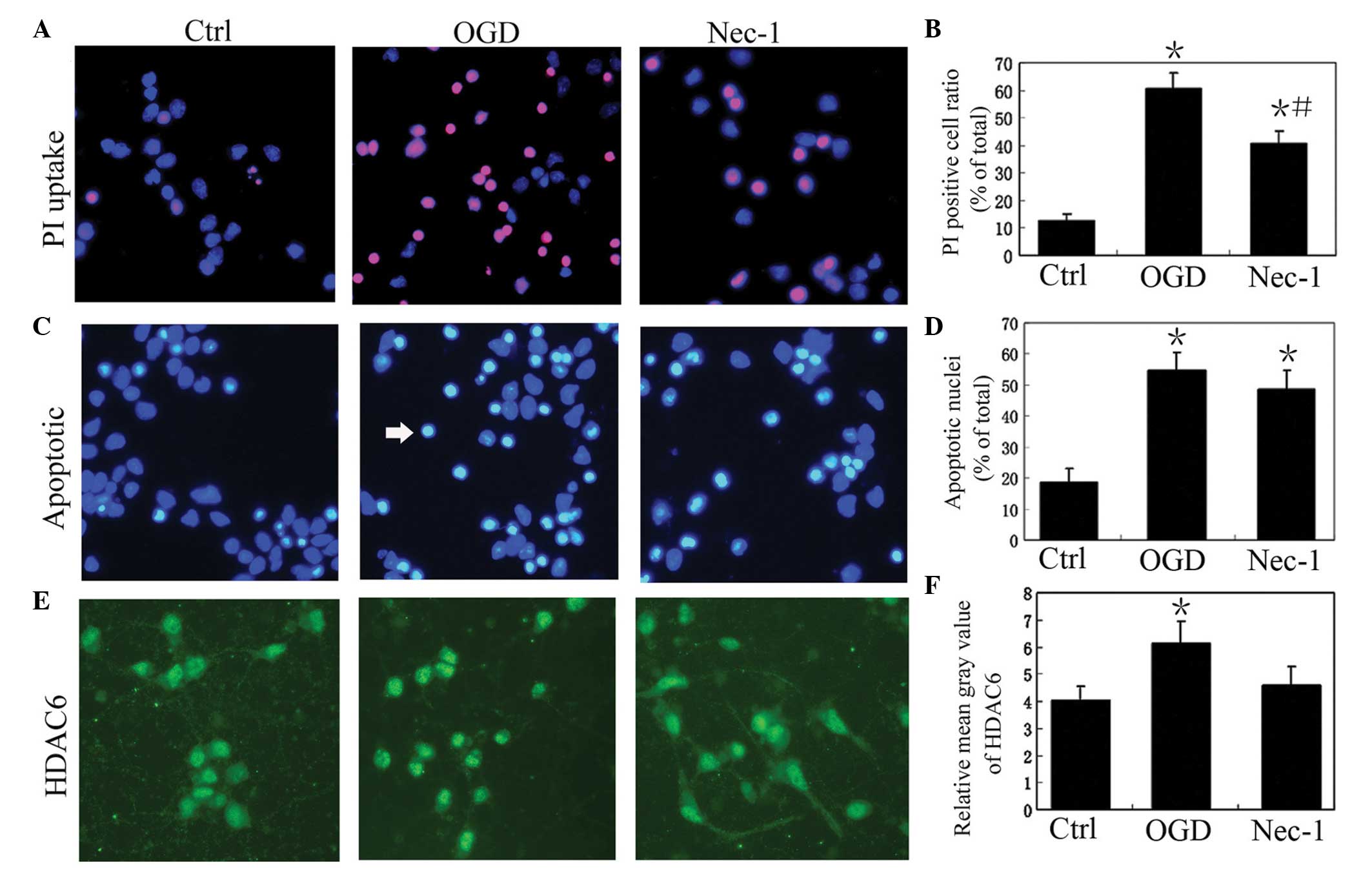 | Figure 1OGD upegulates the expression of HDAC6
and induces the necroptosis of cultured rat cortical neurons. (A)
Representative images of PI staining of cultured rat cortical
neurons in the normal control (Ctrl), OGD and OGD + Nec-1 groups.
Magnification, ×400. (B) PI-positive cell ratio (number of
PI-positive cells / total number of cells) in the Ctrl, OGD and
Nec-1 groups (n=6). Data are presented as the mean ± standard
deviation. *P<0.05, vs. Ctrl; #P<0.05,
vs. OGD. (C) Representative images of apoptotic-like cells in the
cultured rat cortical neurons in Ctrl, OGD and Nec-1 groups. The
white arrow indicates an apoptotic nuclei. DAPI staining;
magnification, ×400. (D) Apoptotic-like cell ratio in the Ctrl, OGD
and Nec-1 groups (n=6). Data are presented as the mean ± standard
deviation. *P<0.05, vs. Ctrl. (E) Representative
images of HDAC6 staining of cultured rat cortical neurons in the
Ctrl, OGD and Nec-1 groups. HDAC6 immunofluorescence;
magnification, ×400. (F) Relative mean gray value of HDAC6 staining
in the Ctrl, OGD and Nec-1 groups (n=6). Data are presented as the
mean ± standard deviation. *P<0.05, vs. Ctrl. OGD,
oxygen-glucose deprivation; HDAC6, histone deacetylase 6; Nec-1,
necrostatin-1; PI, propidium iodide. |
In accordance with the higher level of neuron
necroptosis in the OGD group, the immunofluorescence intensity of
the HDAC6 staining was also increased in the neurons of the OGD
group, compared to that of the control group (P<0.05; Fig. 1). In the OGD+Nec-1 group, the
immunofluorescence intensity of the HDAC6 staining was lower,
compared with that of the OGD group (P<0.05; Fig. 1).
Inhibiting HDAC6 activity reduces
OGD-induced necroptosis in cultured rat cortical neurons
In order to investigate the role of HDAC6 in the
OGD-induced necroptosis of cultured rat cortical neurons, the
present study compared the OGD-induced necroptosis of cultured rat
cortical neurons with and without the HDAC6 specific inhibitor,
Tubacin. The percentage of necrotic cell death in the OGD+Tubacin
group was 45.8±5.3%, which was significantly lower than that of OGD
group (60.5±5.8%; P<0.05), but was not significantly different
to that of the OGD+Nec-1 group (40.8±4.3%; P>0.05; Fig. 2). The percentage of apoptotic-like
cell death in the OGD+Tubacin group was 50.6±5.7%, which was
similar to that of the OGD group (547±5.8%; P>0.05) and
OGD+Nec-1 group (48.6±6.2%; P>0.05; Fig. 2). These results suggested that
inhibiting HDAC6 activity reduced the OGD-induced necroptosis of
cultured rat cortical neurons.
 | Figure 2Inhibiting the activity of HDAC6
reduces the OGD-induced necroptosis of cultured rat cortical
neurons. (A) Representative images of PI staining of cultured rat
cortical neurons in the normal control (Ctrl), OGD, OGD + tubacin
and OGD + Nec-1 treatment groups. Magnification, ×400. (B)
Representative images of apoptotic-like cells in the Ctrl, OGD,
tubacin and Nec-1 groups. The white arrows indicates apoptotic
nuclei. Magnification, x400. (C) PI-positive cell ratio (number of
PI-positive cells / total number of cells) in the Ctrl, OGD,
tubacin and Nec-1 groups (n=6). Data are presented as the mean
±standard deviation. *P<0.05, vs. Ctrl;
#P<0.05, vs. OGD. (D) Apoptotic-like cell ratio in
the Ctrl, OGD, tubacin and Nec-1 groups (n=6). Data are presented
as the mean ±standard deviation. *P<0.05, vs. Ctrl.
OGD, oxygen-glucose deprivation; HDAC6, histone deacetylase 6;
Nec-1, necrostatin-1; PI, propidium iodide. |
Inhibiting HDAC6 activity decreases the
level of ROS and increases the level of acetylated tubulin in
cultured rat cortical neurons following OGD treatment
Mitochondria-associated ROS are important in the
necroptosis of neurons during ischemia-reperfusion. It is now
established that HDAC6 is central in neuronal oxidative stress and
in mitochondrial transport. Acetylated tubulin is closely involved
in mitochondrial transport, therefore, the present study examined
the levels of ROS and acetylated tubulin in each group. Compared
with the control group, the level of acetylated tubulin in the OGD
group was reduced (P<0.05; Fig.
3). Notably, the positive processes of acetylated tubulin in
the PI-positive neurons disappeared (Fig. 3). By contrast, the level of
acetylated tubulin in the OGD+tubacin group increased
significantly, compared with that of the OGD group (P<0.05;
Fig. 3). Compared with the
control, the level of ROS in the OGD group was significantly
increased (P<0.05; Fig. 4).
Following the inhibition of HDAC6 activity, the level of ROS in the
OGD+Tubacin group was significantly lower than that of the OGD
group (P<0.05; Fig. 4).
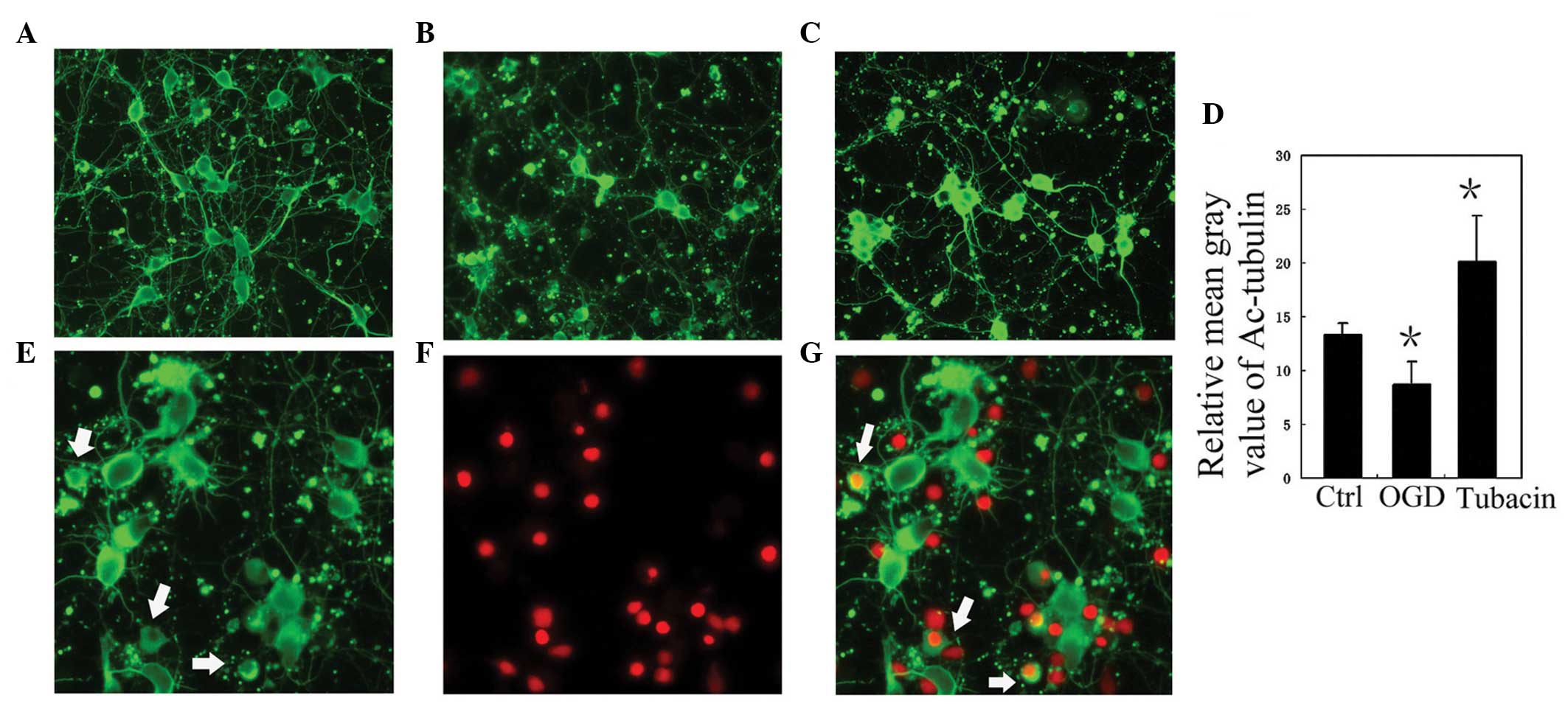 | Figure 3Inhibiting the activity of HDAC6
increases the level of acetylated tubulin in cultured rat cortical
neurons following OGD treatment. (A–C) Representative images of
Ac-tubulin staining of cultured rat cortical neurons in the (A)
normal control (Ctrl), (B) OGD and (C) OGD + tubacin treatment
groups. Magnification, x400. (D) Analysis of the relative mean gray
value of Ac-tubulin staining in the Ctrl, OGD, and tubacin groups.
Data are presented as the mean ±standard deviation. *P<0.05, vs.
Ctrl. (E-G) Co-staining of Ac-tubulin (green) and PI (red), at the
same visual field, in the OGD group. The Ac-tubulin-positive
processes of the PI-positive neurons disappeared (white arrows).
Magnification, ×600. OGD, oxygen-glucose deprivation; HDAC6,
histone deacetylase 6; PI, propidium iodide; Ac, acetylated. |
 | Figure 4Inhibiting the activity of HDAC6
decreases the level of ROS in cultured rat cortical neurons
following OGD treatment. (A–C) Representative images of ROS
staining of cultured rat cortical neurons in the (A) control
(Ctrl), (B) OGD treatment and (C) OGD + tubacin treatment groups.
Magnification, ×400). (D) Co-staining of ROS (red) and
4′,6-diamidino-2-phenylindole (blue), at the same visual field, in
the OGD group. The white arrow indicates an apoptotic-like cell
with a high level of ROS. Magnification, ×600. (E) Analysis of the
relative mean gray value of ROS staining in the Ctrl, OGD and
Tubacin groups. Data are presented as the mean ±standard deviation.
*P<0.05, vs. Ctrl; #P<0.05, vs. OGD.
HDAC6, histone deacetylase 6; ROS, reactive oxygen species. |
Discussion
The aim of the present study was to investigate
whether HDAC6 is involved in the necroptosis of neurons during
ischemia-reperfusion. The results demonstrated that OGD induced the
necroptosis of cultured rat cortical neurons, accompanied by an
increase in the expression of HDAC6. Inhibiting the activity of
HDAC6 reduced the necroptosis of the cultured rat cortical neurons
following OGD treatment. In addition, inhibiting the activity of
HDAC6 also decreased the level of ROS and increased the level of
acetylated tubulin in the cultured rat cortical neurons following
OGD treatment. These results suggested that HDAC6 was involved in
the necroptosis of neurons during ischemia-reperfusion by
modulating the levels of ROS and acetylated tubulin.
Multiple cellular processes are rapidly activated
during ischemic-reperfusion. At the ischemic core, artery occlusion
leads to rapid energy depletion and results in the necrosis of
neurons. However, in the ischemic penumbra, metabolism and
intracellular signaling cascades are maintained partly by a
collateral blood supply during the ischemia (23). Therefore, neurons in the ischemic
penumbra are usually damaged by necroptosis or apoptosis. Notably,
Xu et al demonstrated that the specific necroptosis
inhibitor, Nec-1, significantly reduced the volume of infarction
between 59.3±2.6 and 47.1±1.5% in mice with MCAO/reperfusion
(23). In addition, the
neuroprotective effect of Nec-1 remained when it was used 6 h after
MCAO/reperfusion (9). These
results revealed that anti- necroptosis therapy is of benefit
following ischemic stroke. In the present study, OGD also induced
the necroptosis of cultured rat cortical neurons and upregulated
the expression of HDAC6 in the cultured rat cortical neurons
(Fig. 1). These observations were
consistent with previous reports. Vieira et al demonstrated
that OGD for 2 h leads to a marked loss of cultured neurons by
necroptosis, and Chen et al observed that the expression of
HDAC6 in neurons was markedly upregulated following OGD treatment
or MCAO (16). In order to
determine the role of HDAC6 in the OGD-induced necroptosis of
cultured rat cortical neurons, the present study exposed the
neurons to tubacin, HDAC6 specific inhibitor, and detected the
damage of neurons. The results demonstrated that, similar to the
effects of Nec-1, tubacin significantly decreased the necrotic
neuron death induced by OGD, but had no significant affect on
apoptotic-like neuron death. It is well known that necrosis cannot
be modulated, however, necroptosis can. Thus, tubacin reduced
OGD-induced necrotic neuron death, possibly by inhibiting
necroptosis of the neurons. This suggested that HDAC6 may modulate
the OGD-induced necroptosis of cultured rat cortical neurons.
Further investigations revealed that inhibiting the activity of
HDAC6 reversed the increased levels of ROS and the decreased levels
of acetylated tubulin in the OGD-treated neurons. Previous studies
demonstrated that mitochondria-associated oxidative stress is a
major mechanism of damage in neuron necroptosis (23), and the acetylation level of
α-tubulin modulates mitochondrial transport (24). Thus, HDAC6 may function in
OGD-induced necroptosis of cultured rat cortical neurons by
modulating the levels of ROS and acetylated tubulin. However, the
signaling pathway of HDAC6 in the modulation of necroptosis remains
to be elucidated. Parmigiani et al (17) observed that HDAC6 enhances neuronal
oxidative stress by deacetylating peroxiredoxin-1 and
peroxiredoxin-2. In addition, Rip1 and Rip3 are key molecules in
necroptosis (25) and may be
direct or indirect targets of HDAC6. Collectively, the results of
the present study demonstrated that HDAC6 is an important molecule
in the necroptosis of neurons during ischemia-reperfusion, and
additionally suggest HDAC6 as a possible therapeutic target for the
protection of neurons during ischemia-reperfusion.
Acknowledgments
This study was supported by grants from the
Scientific Research Funds of the Health Department of Hunan
Province (no. 120303), the Hunan Provincial Natural Science
Foundation of China (nos. 13JJ3058 and 14JJ4032) and the Scientific
Research Program of Hunan Provincial Higher Education Institutes
(no. 13C541).
References
|
1
|
Hsieh FI and Chiou HY: Stroke: Morbidity,
risk factors and care in taiwan. J Stroke. 16:59–64. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Joo H, George MG, Fang J and Wang G: A
literature review of indirect costs associated with stroke. J
Stroke Cerebrovasc Dis. 23:1753–1763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pabon M, Tamboli C, Tamboli S, et al:
Estrogen replacement therapy for stroke. Cell Med. 6:111–122. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bright R and Mochly-Rosen D: The role of
protein kinase C in cerebral ischemic and reperfusion injury.
Stroke. 36:2781–2790. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sanderson TH, Reynolds CA, Kumar R, et al:
Molecular mechanisms of ischemia-reperfusion injury in brain:
Pivotal role of the mitochondrial membrane potential in reactive
oxygen species generation. Mol Neurobiol. 47:9–23. 2013. View Article : Google Scholar :
|
|
6
|
Fisher M: New approaches to
neuroprotective drug development. Stroke. 42:s24–s27. 2011.
View Article : Google Scholar
|
|
7
|
Kalogeris T, Bao Y and Korthuis RJ:
Mitochondrial reactive oxygen species: A double edged sword in
ischemia/reperfusion vs preconditioning. Redox Biol. 2:702–714.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Broussalis E, Trinka E, Killer M, et al:
Current therapies in ischemic stroke. Part B. Future candidates in
stroke therapy and experimental studies. Drug Discov Today.
17:671–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Degterev A, Huang Z, Boyce M, et al:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
10
|
Giampietri C, Starace D, Petrungaro S, et
al: Necroptosis: molecular signalling and translational
implications. Int J Cell Biol. 2014:4902752014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vieira M, Fernandes J, Carreto L, et al:
Ischemic insults induce necroptotic cell death in hippocampal
neurons through the up-regulation of endogenous RIP3. Neurobiol
Dis. 68:26–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galluzzi L, Kepp O, Krautwald S, et al:
Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol.
35:24–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Govindarajan N, Rao P, Burkhardt S, et al:
Reducing HDAC6 ameliorates cognitive deficits in a mouse model for
Alzheimer’s disease. EMBO Mol Med. 5:52–63. 2013. View Article : Google Scholar :
|
|
14
|
Gräff J and Tsai LH: The potential of HDAC
inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol.
53:311–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Simões-Pires C, Zwick V, Nurisso A, et al:
HDAC6 as a target for neurodegenerative diseases: what makes it
different from the other HDACs? Mol Neurodegener. 8:72013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen YT, Zang XF, Pan J, et al: Expression
patterns of histone deacetylases in experimental stroke and
potential targets for neuroprotection. Clin Exp Pharmacol Physiol.
39:751–758. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parmigiani RB, Xu WS, Venta-Perez G, et
al: HDAC6 is a specific deacetylase of peroxiredoxins and is
involved in redox regulation. Proc Natl Acad Sci USA.
105:9633–9638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Valenzuela-Fernández A, Cabrero JR,
Serrador JM and Sánchez-Madrid F: HDAC6: A key regulator of
cytoskeleton, cell migration and cell-cell interactions. Trends
Cell Biol. 18:291–297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bi F, Huang C, Tong J, et al: Reactive
astrocytes secrete lcn2 to assist neurons in degeneration. Proc
Natl Acad Sci USA. 110:4069–4074. 2013. View Article : Google Scholar
|
|
20
|
Chen WW, Yu H, Fan HB, Zhan CC, Zhang M,
Zhang C, Cheng Y, Kong J, Liu CF, Geng D and Xu X: RIP1 mediates
the protection of geldanamycin on neuronal injury induced by
oxygen-glucose deprivation combined with zVAD in primary cortical
neurons. J Neurochem. 120:70–7. 2012. View Article : Google Scholar
|
|
21
|
Salmi M, Bruneau N, Cillario J, Lozovaya
N, Massacrier A, Buhler E, Cloarec R, Tsintsadze T, Watrin F, et
al: Tubacin prevents neuronal migration defects and epileptic
activity caused by rat Srpx2 silencing in utero. Brain. 136(Pt 8):
2457–2473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Ye X, Li X, Sun X, Liang Q, Tao L,
Kang X and Chen J: Salidroside protects against MPP(+)-induced
apoptosis in PC12 cells by inhibiting the NO pathway. Brain Res.
1382:9–18. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu X, Chua KW, Chua CC, et al: Synergistic
protective effects of humanin and necrostatin-1 on hypoxia and
ischemia/reperfusion injury. Brain Res. 1355:189–194. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen S, Owens GC, Makarenkova H and
Edelman DB: HDAC6 regulates mitochondrial transport in hippocampal
neurons. PLoS One. 5:e108482010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei B, Yue P and Qingwei L: The molecular
mechanism of necroptosis. Yi Chuan. 36:519–524. 2014.In Chinese.
PubMed/NCBI
|














