Introduction
Hepatocellular carcinoma (HCC) is a type of tumour
with increasing incidence and a high mortality rate worldwide,
ranking as the second most prevalent cancer in China (1–4).
However, since it remains clinically asymptomatic until the late
stages, therapeutic options are limited. Even at the early stages,
treatments, including partial liver resection, chemotherapy,
radiotherapy and molecular targeted therapy have demonstrated only
a modest clinical benefit (5).
Since HCC has been demonstrated to be immunogenic (6), immunotherapy, which aims at inducing
the immune system to eradicate malignant, transformed cells, is
considered to be a potential approach for the treatment HCC
(7). However, in clinical
practice, chemotherapy (8,9) or immunotherapy (10) alone cannot achieve satisfactory
therapeutic efficacy. Thus, the use of chemoimmunotherapy has been
proposed. The putative theory is that traditional chemotherapy can
lower the tumour burden and promote antigen presentation, which
elicits an immunoresponse. A therapeutic chemodrug strategy using
GOLF (gemcitabine, oxaliplatin, leucovorin and 5-fluorouracil),
followed by subcutaneous granulocyte macrophage colony-stimulating
factor and interleukin-2 to treat metastatic colorectal carcinoma
patients resulted in a good therapeutic response and disease
control rate and effectively delayed disease progression (11). In vitro, it was found that
GOLF therapy elicited an antigen-specific cytotoxic T lymphocyte
(CTL) immune response mediated by dendritic cells (DCs) (12). This immune reaction may be due to
the presentation of a ‘risk signal’ induced by chemodrugs (12). It is well established that
chemotherapy causes tumour cell apoptosis (13); however, apoptotic cells stimulate
variable outcomes (i.e., immunotolerance or immunoreactions) under
different conditions. A previous study demonstrated that apoptotic
cells induced by radioactive rays or retrogradation suppress
immunoreactions as the immune system interprets this apoptotic
phenomenon as normal (14). By
contrast, apoptotic cells induced by infection, heat shock or
certain chemodrugs could activate antigen-presenting cells and then
elicit an immune reaction; this apoptosis was considered to be a
‘danger’ and thus abnormal (15).
Therefore, tumour cells were induced to undergo apoptosis and to
express heat shock proteins (HSPs), well-known protein chaperone
molecules, by gene transfection to elicit a ‘danger signal’ and
enhance tumour immunogenicity. Notably, when combined with ‘danger
signal’ HSPs, apoptotic tumour cells elicited an immune response
and subsequently destroyed residual tumour cells (16).
HSPs, including HSP70, gp96, HSP27 and HSP60, are
intracellular molecular chaperones of nascent proteins and function
during protein synthesis, folding, assembly, transport and
stabilization (8). HSPs are
synthesised under different types of stress conditions, including
cell growth and differentiation, infection, inflammation,
malignancy, heat shock and oxygen radicals (17). An immune response is elicited by
HSPs isolated from cancer cells, not from normal cells, due to the
tumour antigen peptide associating with HSPs. In this response,
HSPs act as a ‘danger signal’ to attract and activate DCs (18). Those HSPs eliciting tumour
immunogenicity are marked not by the constitutive but by the
inducible HSPs (19), which may be
exposed under the immune surveillance elements. Thus, the exposure
of HSPs represents an important event in the anti-tumour response
(17).
A comparison of HCC and normal samples demonstrated
HSP deregulation in tumour cells (20). A comparison of HCC and normal
samples demonstrated HSP deregulation in tumour cells (20). HSP70 and gp96 are stress sensitive
members of the HSP family and have been the focus of numerous
studies (21,22). Using flow cytometry, confocol
microscopy, ELISA and reverse transcription quantitative polymerase
chain reaction, the present study examined the potential of
anticancer drugs to induce apoptosis of HepG2 cells and determine
the protein expression levels of HSPs, including HSP70 and gp96, on
the membrane or their release into the extracellular environment,
leading to HSP exposure.
Materials and methods
Cell culture and reagents
The human HCC cell line HepG2 used in the present
study was purchased from the American Type Culture Collection
(Manassas, VA, USA) and routinely maintained in complete Dulbecco’s
modified Eagle’s culture medium (25 mM D-glucose, 4 mM L-glutamine
and 1 mM sodium pyruvate; Gibco-BRL, Carlsbad, CA, USA)
supplemented with 10% foetal bovine serum (Gibco-BRL) at 37°C in 5%
CO2.
Growth inhibition assay
An adenosine triphosphate-tumour chemosensitivity
assay (ATP-TCA; Biothema AB, Handen, Sweden) was used to determine
the growth inhibitory effects of anticancer drugs on HepG2 cells.
All anticancer drugs, including paclitaxel [100% test drug
concentration (TDC) 13.6 μg/ml; Bristol-Myers Squibb Co.,
New York, NY, USA], etoposide (100% TDC 48 μg/ml; Jiangsu
Hengrui Medicine Co., Nanjing, China), pharmorubicin (100% TDC 0.5
μg/ml; Pfizer, Inc., New York, NY, USA), carboplatin (100%
TDC 15.8 μg/ml; Bristol-Myers Squibb Co.), irinotecan
hydrochloride (100% TDC 14 μg/ml; Pfizer, Inc.) and
mitomycin (100% TDC 0.23 μg/ml; Zhejiang Hisun
Pharmaceutical Co., Taizhou, China) were prepared according to the
manufacturer’s instructions of the ATP-TCA kit. In total,
3×103 cancer cells growing in a 96-well plate were
incubated with different anticancer drugs at different
concentrations [6.25, 12.5, 25, 50, 100 and 200% test drug
concentration (TDC)] in complete growth medium at 37°C and 5%
CO2. The cells were collected for the ATP-TCA assay 72 h
later according to the manufacturer’s instructions. Specifically,
100 μl ATP extraction solution was added to the cells, mixed
and incubated for 20–30 min at room temperature. Following that,
500 μl of the mixture was added to the detection plate and
mixed with 50 μl fluorescence-luciferase. It was then
analysed using a Luminescence analyser (Synergy™ MX; BioTek,
Winooski, VT, USA). Data were analysed by Microsoft Excel 2010
software and hyper- and hyposensitive drugs of HepG2 cells were
profiled.
Anticancer drug treatment on HepG2
cells
The hypersensitive drug etoposide and the
hyposensitive drug carboplatin were selected for the experiments.
HepG2 cells were cultured in a 6-well plate at 37°C and 5%
CO2. Etoposide or carboplatin with 100% TDC was added
and co-cultured for the indicated time intervals: 1, 2, 4, 6, 12,
18, 24, 48 or 72 h. The etoposide-treated group was not assessed
for 48 or 72 h as it caused marked apoptosis after 24 h of
treatment. At each time interval, the cells were harvested for flow
cytometry or immunostaining analysis.
Flow cytometry
Following treatment with anticancer drugs, cancer
cells were collected for intracellular and surface immunolabelling
of HSP70/gp96. Briefly, for intracellular labelling, cells were
fixed and permeabilised using 70% ethanol at 4°C overnight and then
incubated with each of the following antibodies:
Phycoerythrin-conjugated mouse anti-HSP70 polyclonal antibody
(1:100; cat. no. sc-1060; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA); rabbit anti-human GRP94 (gp96; 1:100; cat. no.
AHP848; AbD Serotec, Raleigh, NC, USA) followed by the secondary
antibody [goat anti rabbit-fluorescein isothiocyanate (FITC);
1:200; cat. no. 172–1506; Kirkegaard & Perry Laboratories Inc.,
Gaithersburg, MD, USA] for 20 min at room temperature and protected
from light. For surface HSP70 or gp96 staining, tumour cells were
dissociated into a single cell suspension and incubated under the
same conditions as described above without fixation and
permeabilization. All labelled cells were washed and measured
immediately using a FACS Calibur flow cytometer (Beckman-Coulter,
Inc., Miami FL, USA). More than 1×104 cells were
analysed in each analysis and the mean fluorescence intensity (MFI)
was used for the assessment of HSP70 and gp96 expression. Controls
were routinely performed in all cases.
Immunofluorescence
Tumour cells were incubated with hypersensitive or
hyposensitive drugs in a 6-well plate with a coverslip for
different time intervals: Etopiside for 24 h and carboplatin for 72
h. Coverslips with cells were collected for indirect
immunofluorescence staining. Cells were fixed and permeabilised
using 70% ethanol at 4°C overnight and blocked by normal goat serum
confining liquid for 30 min at room temperature. Subsequently, the
cells were incubated with the following specific antibodies for 1 h
at room temperature: Mouse anti-HSP70 (1:100; cat. no. BM0368;
Wuhan Boster Biological Technology, Ltd., Wuhan, China) and rabbit
anti-GRP94 (gp96) monoclonal antibody (1:100; cat.no. AHP848; ABD
Serotec, Inc.). Secondary antibodies used for the visualization of
HSP70 or gp96 were FITC-conjugated goat anti-mouse IgG (Wuhan
Boster Biological Technology, Ltd.) and Cy3-conjugated sheep
anti-rabbit IgG (Sigma-Aldrich, St. Louis, MO, USA). Hoechst was
used for nuclear staining. During incubation with secondary
antibodies and Hoechst, the cells were protected from light.
Control staining was performed in all cases. Following staining,
the cells were observed and images were captured under a
fluorescent confocal microscope (LS< 510 META; Carl Zeiss, Jena,
Germany).
Apoptosis
An annexin V detection kit used to determine
apoptosis was provided by BioVision, Inc. (Milpitas, CA, USA; cat.
no. K102–25). Tumour cells treated with drugs were dissociated into
single cell suspensions in 200 μl binding buffer.
Subsequently, 10 μl FITC-conjugated annexin V and 5
μl propidium iodide were added, and then the cells were
incubated for 30 min at room temperature while protected from
light. Next, 300 μl binding buffer was added and the cells
were analysed using a FACS Calibur flow cytometer (Beckman-Coulter,
Inc.).
ELISA
HSP70 (Surveyor™ IC; R&D Systems, Minneapolis,
MN, USA; cat. no. SUV1663) and gp96 (MyBioSource, San Diego, CA,
USA; cat. no. MBS705033) ELISA kits were used and the quantity of
released HSP70/gp96 from HepG2 cells treated with chemodrugs was
analysed according to the manufacturer’s instructions. Briefly,
culture supernatants in which HepG2 cells were cultured with
etoposide for 24 h or carboplatin for 24 or 72 h were collected and
100 μl of them were added to the microtitre that was
pre-coated with anti-HSP70 or gp96 antibody. The cells were then
incubated for 2 h at room temperature and washed three times with
PBS. Subsequently, 100 μl HSP70 or gp96 detection antibody
was added and incubated for 2 h at room temperature. The HSP70 or
gp96 antibody were washed off and streptavidin horseradish
peroxidase (HRP; HSP70) or HRP avidin (gp96) was added and
incubated at room temperature for an additional 20 min. Following
that, 100 μl substrates were added and incubated for 20 min.
Finally, 50 μl stop solution was added and the optical
density was determined at 450 nm using a Luminescence analyzer. The
HSP70 or gp96 standard was used to make a standard curve by
proportional dilution and the formula was produced for the
concentration and optical density. Subsequently, the HSP70 or gp96
concentration in each sample was calculated.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
HepG2 cells treated with etoposide for 24 h and
carboplatin for 24 or 72 h were harvested. Total RNAs were
extracted and reverse transcribed into cDNA using Superscript II
reverse transcriptase (Invitrogen Life Technologies, Carlsbad, CA,
USA). RT-qPCR was performed using the Lightcycler-Faststart DNA
master SYBR green I PCR kit (Roche Diagnostics, Madison, WI, USA)
in a Roche Lightcycler 1.2 Real Time PCR System (Roche Diagnostics)
according to the manufacturer’s instructions: Initiation with a
10-min denaturation at 95°C, followed by 40 cycles of amplification
with 15 sec of denaturation at 95°C, 5 sec of annealing at 50–60°C,
15 sec of extension at 72°C, and then the plate was read for
fluorescence data collection at 76°C. The primer sequences were as
follows: HSP70, forward 5′ TGGTGGTTCTACTCG TATCCC-3′ and reverse
5′-TGACATCCAAGAGCAGCA AAT-3′; Gp96, forward
5′-GCTTCGGTCAGGGTATCTTTT-3′ and reverse
5′-CACCTTTGCATCAGGGTCAAT-3′; GAPDH, forward
5′-TGTTGCCATCAATGACCCCTT-3′ and reverse 5′-CTCCACGACGTACTCAGCG-3′.
The comparative threshold cycle (CT) method was used for the
calculation of amplification fold. The expression level of each
gene was normalised by dividing by the expression level of the
GAPDH gene transcript.
Statistical analysis
All data are expressed as the mean ± standard
deviation. Statistical analyses were performed using paired
Student’s t-test with Microsoft Excel 2010 (Microsoft, Albuquerque,
NM, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Chemosensitivity of HepG2 cells to
anticancer drugs and apoptosis caused by hyper- or hyposensitive
anticancer drugs
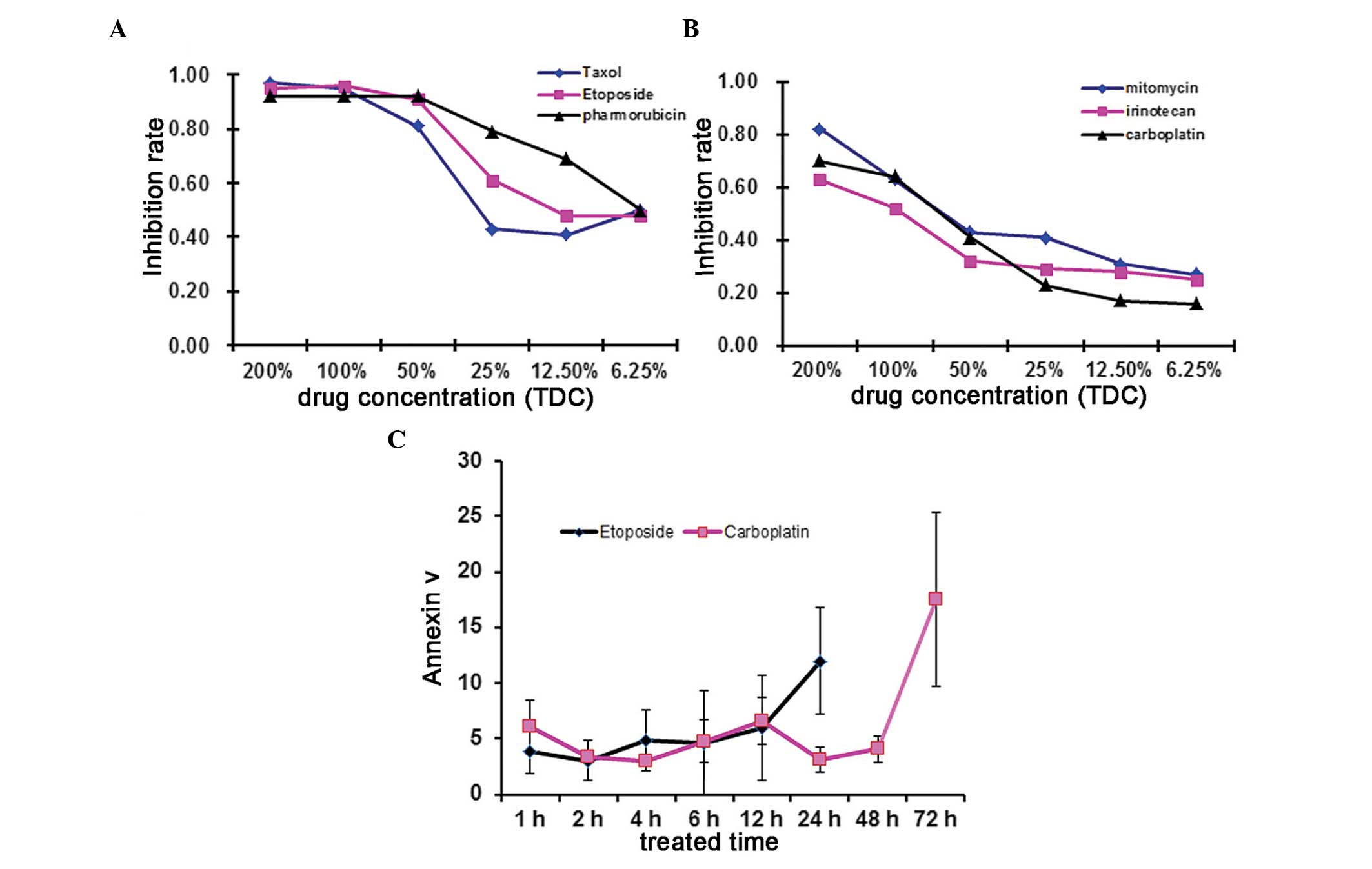
To determine the chemosensitivity of HepG2 cells to
anticancer drugs, the present study selected six commonly used
drugs to treat HepG2 cells and then used those cells to conduct
ATP-TCA assays. For each single anticancer drug, the
chemo-sensitivity was categorised as sensitive (100% TDC>90% and
50% TDC<70%) or resistant (100% TDC<70% and 50% TDC>50%).
Dose-response curves for HepG2 cells following a continuous 72 h
exposure to anticancer drugs at various concentrations using the
ATP-TCA assay are depicted. The results demonstrated that etoposide
(VP-16), taxol (TAX) and pharmorubicin had high inhibitory rates
and were thus classified as sensitive drugs. By contrast,
mitomycin, irinotecan and carboplatin had low inhibitory rates
(Fig. 1A and B) and were
classified as resistant drugs. According to the results, the
hypersensitive drug etoposide (VP-16), a cell cycle-specific
anti-tumour drug that targets the S phase or G2 phase and the
hyposensitive drug carboplatin, a cell cycle-non-specific drug,
which inhibits DNA duplication and transcription, were selected for
subsequent experiments. Subsequently, HepG2 cells were treated with
hypersensitive (etoposide) or hyposensitive (carboplatin) drugs for
the indicated time intervals and cell apoptosis levels were
determined using an annexin V apoptosis detection kit. Apoptosis of
drug-treated HepG2 cells slowly increased with time. Etoposide
caused marked apoptosis in a relatively short time period, with the
maximal annexin V positive rate achieved after 24 h.
Carboplatin-treated cells took longer (72 h) to arrive at the
maximal annexin V positive rate (Fig.
1C).
Alterations in the mRNA level and
secretion of HSP70/gp96 in etoposide- or carboplatin-treated HepG2
cells
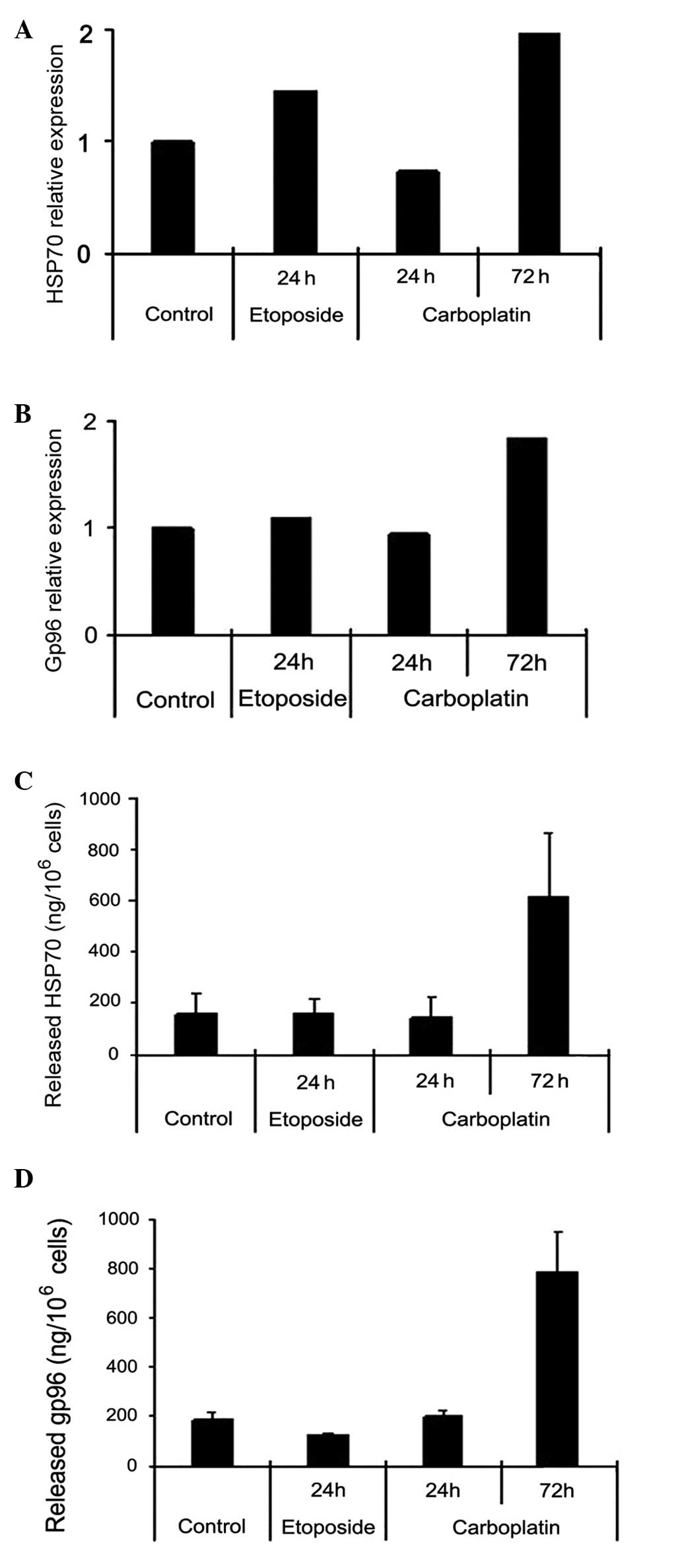
HepG2 cells were treated with etoposide for 24 h or
carboplatin for 72 h. The cells were lysed and the mRNA levels of
HSP70/gp96 were determined by RT-qPCR (Fig. 2A and B). In addition, cell culture
supernatants were collected and the quantity of secretory HSP70 or
gp96 was quantified by ELISA (Fig. 2C
and D). The data indicated that although etoposide and
carboplatin increased the mRNA level of HSP70 and gp96 in HepG2
cells, only carboplatin-treated HepG2 cells produced increased
secretory HSP70 and gp96.
Gradual alterations in cytoplasmic and
surface HSP70/gp96 expression in etoposide- or carboplatin-treated
HepG2 cells
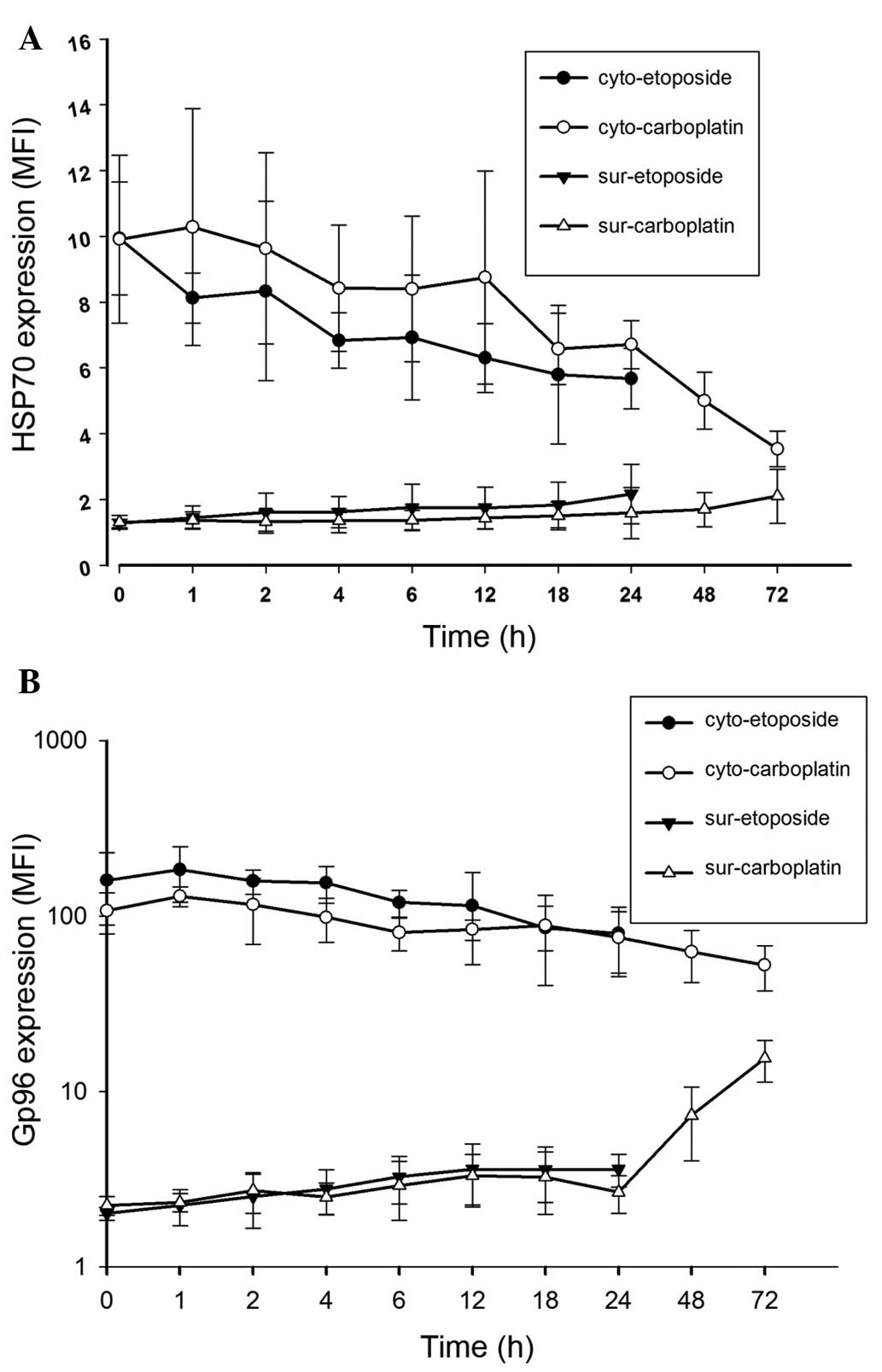
HepG2 cells were treated with etoposide or
carboplatin for the indicated time intervals: 0, 1, 2, 4, 6, 12,
18, 24, 48 and 72 h. The cytoplasmic and surface HSP70 (Fig. 3A) and gp96 (Fig. 3B) expression levels were determined
by flow cytometry and the parameter MFI was used to quantify the
alterations. As shown in Fig. 3,
prior to treatment with chemodrugs, HepG2 cells exhibited strong
intracellular (cytoplasmic) HSP70/gp96 expression but weak surface
expression. However, following treatment with chemodrugs,
cytoplasmic HSP70/gp96 expression decreased and cancer cells
exhibited upregulated surface HSP70/gp96 expression. Marked
alterations were observed following 24 h treatment with etoposide
and 72 h treatment with carboplatin. Following treatment with
carboplatin for 72 h, the surface expression of gp96 in HepG2 cells
was significantly increased.
Cytoplasmic and surface expression
alterations in HSP70/gp96 in drug-treated HepG2 cells
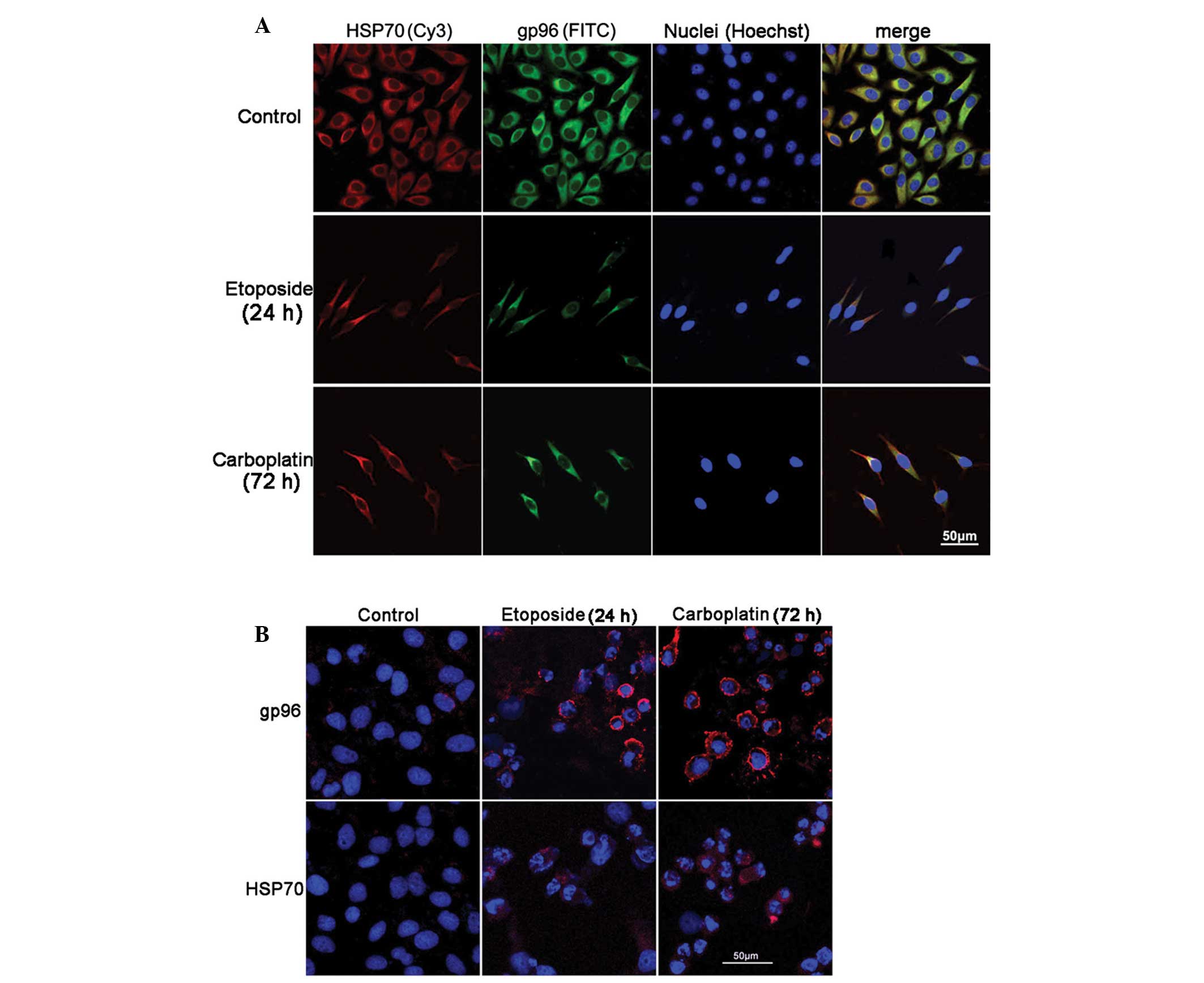
HepG2 cells were treated with etoposide for 24 h or
carboplatin for 72 h and immunofluorescence staining was used to
demonstrate HSP70/gp96 expression. Consistent with flow cytometric
analysis, the confocal images demonstrated a decrease in
cytoplasmic HSP70/gp96 expression (Fig. 4A) and a clear increase in the
surface expression of HSP70/gp96 (Fig.
4B). However, the increase in the surface expression of HSP70
was not significant.
Discussion
Almost all cells contain HSPs, which are present in
a variety of intracellular locations, including the cytosol,
endoplasmic reticulum, nuclei and mitochondria. HSPs function in
normal cells as molecular chaperones to assist protein folding,
unfolding, degradation and assembly (8). When cells become malignant and
proteins mutate or change, HSPs accumulate to adapt to the
condition and protect cells from damage. For example, if tumour
cells suffer from stress, such as following photodynamic therapy
(17), HSPs not only markedly
increase but also alter their distribution. They translocate from
the cytosol to the cell surface and even release into the
extracellular environment (23).
In this circumstance, antigens chaperoned by HSPs are exposed to
immune surveillance elements. Hence, HSP surface expression levels
have been reported to correlate with tumour immunogenicity
(19) and HSPs are considered to
be ‘risk signals’, alerting to the existence of a threat and
priming a self-protection system (24,25).
Thus, upregulating cell surface HSPs in cancer cells or stimulating
HSP release and exposure may be a crucial method to enhance
anti-tumour activity.
Chemotherapy is considered to be an important
treatment for HCC, but its acute and cumulative toxicity limit its
application. However, immunotherapy, with its different functional
mechanism, can be combined with chemotherapy. Chemotherapy is an
immunological priming factor. It reduces tumour burden, augments
tumour immunogenicity and provides the basis for an effective
immunoresponse. A rational combination of chemo- and immunotherapy
would lead to solid tumour regression and generate immunological
memory (26). Furthermore,
immunotherapy based on HSPs has demonstrated such effectiveness
that it is not necessary to identify each tumour-specific antigen
(27). In contrast to previous
studies in which heat shock, ultraviolet radiation or pathogenic
microorganisms were applied as a stressor, the present study
employed chemodrugs as a stressor to examine their effects on HSPs
in the hepatoma cell line HepG2. The present study primarily
detected alterations in HSP70 and gp96 that are well investigated
and can aid in eliciting an anticancer immunity response (28,29).
As the data show, these chemodrugs were able to cause HepG2 cell
apoptosis. The hypersensitive anticancer drug etoposide induced
rapid apoptosis of cancer cells and after a 24 h treatment, the
majority of cells died, thus cancer cell apoptosis and HSP
alterations could only be recorded for 24 h. Conversely, the
hyposensitive anticancer drug carboplatin induced apoptosis
relatively slowly and 72 h were required to reach the peak of
apoptosis. Along with demonstrating apoptosis, the cancer cells
presented the ‘danger signals’ HSP70/gp96. Following treatment with
etoposide for 24 h or carboplatin for 72 h, cancer cells were
collected to determine the expression of HSP70/gp96. RT-qPCR data
demonstrated that the mRNA levels of HSP70/gp96 increased. In
addition, when treated with carboplatin for 72 h, HSP70 expression
and, even more markedly, gp96 expression exhibited a transfer
pattern from the cytoplasm to the cell surface and released into
the extracellular environment. This external exposure of HSP70/gp96
may result in an enhancement of tumour immunogenicity. These
results have greater significance than the simple upregulation of
HSP genes established in previous studies (20). Therefore, it would be beneficial to
re-evaluate the effects of hyposensitive drugs and adjust
therpeutic stratagies.
Previous studies have verified that HSP-peptide
complex could present tumour antigens to activate a specific CTL
response (30–32). These studies claimed that HSPs
could be employed for cancer treatment. However, in cancer
patients, increased HSPs did not elicit an effective anti-tumour
response and the patients often appeared to be immunotolerant
(33). In addition to a large
tumour burden, the possibility that the tumour antigens are not
fully exposed to the immune surveillance system is an important
factor to consider. Therefore, in addition to degrading the tumour
burden, challenging tumour cells with a stressor and eliciting
antigen exposure and presentation are of significance. The present
study successfully employed chemodrugs as a stressor. They induced
cancer cell apoptosis and augmented surface HSP70/gp96 expression,
resulting in HSP70/gp96 release to the microenvironment. Thus,
combined with the HSP70/gp96 ‘danger signal’, apoptotic tumour
cells treated with chemodrugs would elicit an immune response and
subsequently eradicate residual tumour cells. Notably, the present
study found that although the hypersensitive drug etoposide could
promote rapid cancer cell apoptosis, it induced less HSP70/gp96
exposure. The continuous treatment of cancer cells by the
hyposensitive drug carboplatin was more effective, and it induced
stronger surface expression and release of gp96, which suggests a
potential use of hyposensitive drugs for HSP-based DC vaccine
preparation in vitro and chemoimmunotherapy for HCC
patients. The present study provided an experimental basis for
patient-specific chemoimmunotherapy for HCC. The results
demonstrated that chemodrugs boosted surface HSP expression and
release, which may facilitate tumour immunogenicity. Further
studies are required to investigate the potential of this
therapy.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81202076) and the
Natural Science Foundation of Guangdong to Mei Yang (grant no.
S2013010012048) to MY.
References
|
1
|
Tang ZY, Yu YQ, Zhou XD, Ma ZC and Wu ZQ:
Progress and prospects in hepatocellular carcinoma surgery. Ann
Chir. 52:558–563. 1998.PubMed/NCBI
|
|
2
|
Zhou XD, Tang ZY, Yu YQ, et al: Recurrence
after resection of alpha-fetoprotein-positive hepatocellular
carcinoma. J Cancer Res Clin Oncol. 120:369–373. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bismuth H, Chiche L, Adam R, Castaing D,
Diamond T and Dennison A: Liver resection versus transplantation
for hepatocellular carcinoma in cirrhotic patients. Ann Surg.
218:145–151. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jaeck D, Bachellier P, Oussoultzoglou E,
Weber JC and Wolf P: Surgical resection of hepatocellular
carcinoma. Post-operative outcome and long-term results in Europe:
an overview. Liver Transpl. 10(2 Suppl 1): S58–S63. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
El-Serag HB, Marrero JA, Rudolph L and
Reddy KR: Diagnosis and treatment of hepatocellular carcinoma.
Gastroenterology. 134:1752–1763. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kabilova TO, Kovtonyuk LV, Zonov EV, et
al: Immunotherapy of hepatocellular carcinoma with small
double-stranded RNA. BMC Cancer. 14:338–349. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Breous E and Thimme R: Potential of
immunotherapy for hepatocellular carcinoma. J Hepatol. 54:830–834.
2011. View Article : Google Scholar
|
|
8
|
Srivastava P: Roles of heat-shock proteins
in innate and adaptive immunity. Nat Rev Immunol. 2:185–194. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ravoet C, Bleiberg H and Gerard B:
Non-surgical treatment of hepatocarcinoma. J Surg Oncol Suppl.
3:104–111. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zinkernagel RM: Immunity against solid
tumors? Int J Cancer. 93:1–5. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Correale P, Cusi MG, Tsang KY, et al:
Chemo-immunotherapy of metastatic colorectal carcinoma with
gemcitabine plus FOLFOX 4 followed by subcutaneous granulocyte
macrophage colony-stimulating factor and interleukin-2 induces
strong immunologic and antitumor activity in metastatic colon
cancer patients. J Clin Oncol. 23:8950–8958. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Correale P, Cusi MG, Del Vecchio MT, et
al: Dendritic cell-mediated cross-presentation of antigens derived
from colon carcinoma cells exposed to a highly cytotoxic multidrug
regimen with gemcitabine, oxaliplatin, 5-fluorouracil and
leucovorin, elicits a powerful human antigen-specific CTL response
with antitumor activity in vitro. J Immunol. 175:820–828. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Skoberne M, Beignon AS, Larsson M, et al:
Apoptotic cells at the crossroads of tolerance and immunity. Curr
Top Microbiol Immunol. 289:259–292. 2005.PubMed/NCBI
|
|
14
|
Kleinclauss F, Perruche S, Masson E, et
al: Intravenous apoptotic spleen cell infusion induces a
TGF-beta-dependent regulatory T-cell expansion. Cell Death Differ.
13:41–52. 2006. View Article : Google Scholar
|
|
15
|
Zheng L, He M, Long M, Blomgran R and
Stendahl O: Pathogen-induced apoptotic neutrophils express heat
shock proteins and elicit activation of human macrophages. J
Immunol. 173:6319–6326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Feng H, Zeng Y, Whitesell L and Katsanis
E: Stressed apoptotic tumor cells express heat shock proteins and
elicit tumor-specific immunity. Blood. 97:3505–3512. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Korbelik M, Sun J and Cecic I:
Photodynamic therapy-induced cell surface expression and release of
heat shock proteins: relevance for tumor response. Cancer Res.
65:1018–1026. 2005.PubMed/NCBI
|
|
18
|
Liu B, DeFilippo AM and Li Z: Overcoming
immune tolerance to cancer by heat shock protein vaccines. Mol
Cancer Ther. 1:1147–1151. 2002.PubMed/NCBI
|
|
19
|
Clark PR and Ménoret A: The inducible
Hsp70 as a marker of tumor immunogenicity. Cell Stress Chaperones.
6:121–125. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li CL, Meng X, Lang ZW and Zhang LJ: The
detection of heat shock protein gp96 in primary hepatocellular
carcinoma. Zhonghua Gan Zang Bing Za Zhi. 12:569–570. 2004.In
Chinese. PubMed/NCBI
|
|
21
|
Udono H and Srivastava PK: Comparison of
tumor-specific immunogenicities of stress-induced proteins gp96,
hsp90, and hsp70. J Immunol. 152:5398–5403. 1994.PubMed/NCBI
|
|
22
|
Lancaster GI and Febbraio MA:
Exosome-dependent trafficking of HSP70: A novel secretory pathway
for cellular stress proteins. J Biol Chem. 280:23349–23355. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brusa D, Migliore E, Garetto S, Simone M
and Matera L: Immunogenicity of 56 degrees C and UVC-treated
prostate cancer is associated with release of HSP70 and HMGB1 from
necrotic cells. Prostate. 69:1343–1352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Beg AA: Endogenous ligands of Toll-like
receptors: implications for regulating inflammatory and immune
responses. Trends Immunol. 23:509–512. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matzinger P: The danger model: a renewed
sense of self. Science. 296:301–305. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nowak AK, Robinson BW and Lake RA: Synergy
between chemotherapy and immunotherapy in the treatment of
established murine solid tumors. Cancer Res. 63:4490–4496.
2003.PubMed/NCBI
|
|
27
|
Ménoret A, Chaillot D, Callahan M and
Jacquin C: Hsp70, an immunological actor playing with the
intracellular self under oxidative stress. Int J Hyperthermia.
18:490–505. 2002. View Article : Google Scholar
|
|
28
|
Singh-Jasuja H, Scherer HU, Hilf N, et al:
The heat shock protein gp96 induces maturation of dendritic cells
and down-regulation of its receptor. Eur J Immunol. 30:2211–2215.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Millar DG, Garza KM, Odermatt B, et al:
Hsp70 promotes antigen-presenting cell function and converts T-cell
tolerance to autoimmunity in vivo. Nat Med. 9:1469–1476. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tamura Y, Peng P, Liu K, Daou M and
Srivastava PK: Immunotherapy of tumors with autologous
tumor-derived heat shock protein preparations. Science.
278:117–120. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schild H, Arnold-Schild D, Lammert E and
Rammensee HG: Stress proteins and immunity mediated by cytotoxic T
lymphocytes. Curr Opin Immunol. 11:109–113. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sato K, Torimoto Y, Tamura Y, et al:
Immunotherapy using heat-shock protein preparations of leukemia
cells after syngeneic bone marrow transplantation in mice. Blood.
98:1852–1857. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pawel S and Anne MD: The immunosuppressive
activity of heat shock protein 70. Autoimmune Diseases. 2012:1–6.
2012.
|