Introduction
The podocyte is one of the resident cells of the
glomerulus, which is often attacked as the target cell in the
glomerulus by a variety of immune or non-immune inflammatory
factors. Podocytopathy refers to the various degrees of podocyte
injury occurring in pathological conditions. Due to their highly
differentiated state and limited proliferative capability,
podocytes are not able to recover from damage effectively.
Therefore, podocyte injury is a pivotal factor during the
progression of glomerular diseases (1–3).
During the disease process of glomerulonephritis,
numerous interconnected network signaling cascades are activated in
renal cells. The mammalian nuclear factor (NF)-κB signaling
pathway, which is a fundamental intracellular transcription factor
system, is induced in response to various sources of extracellular
stimulation. An indicator of NF-κB activation is the nuclear
translocation of dimeric Rel protein 8, which regulates numerous
NF-κB-dependent genes involved in inflammation, immunity,
apoptosis, cell proliferation and differentiation (4–6).
Accumulating lines of evidence have indicated that the activation
of NF-κB is a critical response to kidney diseases. Upregulation of
the canonical (RelA/p50) NF-κB isoform in macrophages, mesangial
cells, tubular epithelial cells and podocytes has a pathogenic role
in mediating acute and chronic inflammatory nephropathy (7–13). A
number of in vivo studies of podocytes in nephrotic
glomerular diseases have demonstrated that NF-κB and RelA are
markedly upregulated. For example, NF-κB is activated within
podocytes in passive Heymann nephritis, which contributes to
autologous phase proteinuria (14). Furthermore, in patients with Lupus
nephritis (LN), activation of NF-κB (predominantly at p65) and
upregulation of interleukin (IL)-1, IL-4 and tumor necrosis factor
(TNF)-α are co-localized in diseased podocytes. The staining score
of activated NF-κB p65 has been observed to be positively
correlated with the severity of proteinuria (11). Treatment with genistein, which is
able to inhibit pro-inflammatory cytokines through downregulation
of the NF-κB pathway, prevented pathological changes in podocytes,
including extensive disruption and effacement of processes
(13). In vitro, NF-κB is
activated in podocytes in response to their exposure to various
factors, which may cause kidney damage. For example, NF-κB was
upregulated in murine podocytes exposed to either a Shiga toxin or
protein overload, and mediated the increase of endothelin-1
production (15,16). Angiotensin II is capable of
upregulating the expression of Toll-like receptor 4 in murine
podocytes, which may effectively lead to NF-κB activation (17). Transforming growth factor (TGF)-β1
may induce podocyte damage by upregulating transient receptor
potential cation channel, subfamily C, member 6 protein, most
likely through the Smad3-extracellular signal-regulated
kinase-NF-κB pathway (18).
Therefore, it was suggested that the podocyte injury was induced
mainly via NF-κB activation, which may further mediate a large
number of NF-κB target genes and cause morphological and functional
abnormalities of podocytes.
A previous in vitro study by our group
demonstrated that ubiquitin carboxy-terminal hydrolase L1 (UCH-L1)
is a downstream target protein of NF-κB. The activation of NF-κB by
inflammatory factors may increase the expression of UCH-L1 in
murine podocytes and affect morphological changes in podoctyes
(19).
UCH-L1 is a member of the deubiquitinating enzyme
family, which is important in the regulation of the
ubiquitin-proteasome system (20,21).
Although it is mainly localized to the brain and testis, UCH-L1 is
also expressed in the kidney and involved in nephrogenesis
(22–24). Previous studies by our group and
others reported that UCH-L1 is involved in podocyte differentiation
and injury. The level of expression was increased in podocytes in a
variety cases of immune complex-mediated glomerulonephritis
(1,2,25).
In cultured HEK293T cells, using a luciferase assay, it was further
demonstrated that NF-κB upregulates UCH-L1 via binding to the −300
bp and −109 bp sites of the UCH-L1 promoter. This supported the
hypothesis that UCH-L1 was upregulated in podocytes through the
NF-κB signaling pathways (19).
The present study further investigated the
involvement of NF-κB in the regulation of expression of UCH-L1 in
podocytes under inflammatory conditions. Expression levels of NF-κB
and UCH-L1 were detected using immunohistochemistry in kidney
biopsy tissues from specific forms of immune complex-mediated
glomerulonephritis, including LN, immunoglobulin A nephropathy
(IgAN) and membranous glomerulonephritis (MGN). These proteins were
also assessed in vivo in murine podocytes co-cultured with
mesangial cells and treated with rabbit anti-rat thymocyte serum
(ATS), which may interact with mesangial cells to form immune
complexes.
Patients and methods
Immunohistochemistry
Tissues from 56 individuals, including 1 normal
control, 19 patients with LN, 15 patients with IgAN, 15 patients
with MGN and 6 patients with minimal change disease (MCD), were
collected from renal needle biopsies from the Nephrosis Laboratory,
Department of Pathology, (Fudan University, Shanghai, China)
between 2010 and 2013. Permission to use the tissue sections for
research purposes was obtained and approved by the Ethics Committee
from the College of Basic Medicine, Fudan University, and a written
consent form was obtained from all patients. Sections (4 μm)
were deparaffinized in 100% xylene for 10 min then rehydrated
gradually in an alcohol series. They were then incubated in a 0.3%
hydrogen peroxide/methanol buffer for 30 min to quench endogenous
peroxidase activity. Antigen retrieval was performed by immersing
the sections in 0.5 mol/l ethylenediaminetetraacetic acid buffer
(pH 8.0) for 3 min, followed by boiling in a water bath for 7 min.
The sections were rinsed in phosphate-buffered saline (PBS) and
subsequently incubated with rabbit polyclonal anti-UCH-L1 antibody
(1:100; cat. no. AB1761; Millipore, Billerica, MA, USA) overnight
at 4°C in a humidified chamber. Following incubation, the sections
were washed three times with PBS containing 0.05% Tween-20 for 5
min each time. The sections were then incubated with 100 μl
horseradish peroxidase conjugated-anti-rabbit immunoglobulin G,
maintaining the tissue section in a moist chamber for 30 min at
37°C and then the sections were washed three times, as previously.
Immobilized antibodies were detected using a two step EnVision+
system peroxidase kit (Dako, Carpinteria, CA, USA).
3,3′-diaminobenzidine was used as the chromogen and hematoxylin as
the nuclear counterstain (Sangon Biotech Co., Ltd., Shanghai,
China). The sections were rinsed thoroughly with tap water for 1–2
min, then they were rinsed with distilled water, followed by tap
water. The protocol for assessing activated NF-κB p65 (rabbit
monoclonal anti-p-p65 antibody; 1:100; cat. no. 3033; Cell
Signaling Technology, Beverly, MA, USA) and Wilms tumor 1 (WT1;
rabbit polyclonal anti-WT1 antibody; 1:100; cat. no. P-0526;
Changdao Biotech Co., Ltd., Shanghai, China) in successive sections
was similar to that described above. The positive cell number of
all slides was counted by two individuals.
Cell culture
The conditionally thermosensitive SV40-transfected
immortalized murine podocyte cell line MPC5 (a gift from Professor
Xu Hong; Affiliated Children’s Hospital of the Medical College of
Fudan University, Shanghai, China) was cultured as described
previously (19). Briefly, MPC5
cells were cultured under permissive conditions [33°C, 5% (v/v)
CO2, RPMI-1640 (Gibco Life Technologies, Carlsbad, CA,
USA), 10% (v/v) fetal bovine serum (Gibco Life Technologies), 50–10
U/ml γ-interferon (ProSpec-Tany TechnoGene Ltd., Ness Ziona,
Israel)] and under formulary conditions (37°C without γ-interferon)
for external differentiation. Podocytes, which were between
passages 5 and 20 were used. Differentiated cells were identified
by their large arborized shape and by cell expression of
synaptopodin mRNA, a known marker for differentiation. An inverted
microscope (TS100; Nikon, Tokyo, Japan) was used to observe the
growth of podocytes.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Synaptopodin mRNA expression was identified using
synaptopodin DNA. Total RNA from differentiated and
non-differentiated podocyte cells was isolated using the RNAiso
Plus extraction reagent and cDNA was produced using PrimeScript
reverse transcriptase (Takara Bio, Inc., Otsu, Japan) according to
the manufacturer’s instructions. RT-qPCR was then conducted using
SYBR Premix Ex Taq II (Takara Bio, Inc.) in 25 μl reactions
with 2 μl cDNA and 0.4 μM each of the forward and
reverse primers using the following thermocycling conditions: 95°C
for 30 sec, 95°C for 5 sec and 60°C for 30 sec for 40 cycles. Data
was collected and all expression levels were normalized to β-actin.
The ABI7900 thermocycler (Life Technologies, Carlsbad, CA, USA) was
used. The following primers were used: Synaptopodin, forward
5′-CCTGCCCGTAACTTCCGTG-3′, and reverse 5′-GAGCGGCGGTAGGGAAAAG-3′;
β-actin, forward 5′-CATCCGTAAAGACCTCTATGCCAAC-3′, and reverse
5′-ATGGAGCCACCGATCCACA-3′. The primers were synthesized by Jie Li
Biology (Shanghai, China).
Subsequently, the cells were treated with TNF-α (15
ng/ml; Sigma-Aldrich, St. Louis, MO, USA), IL-1β (15 ng/ml;
Sigma-Aldrich) and lipopolysaccharide (LPS; 1 μg/ml;
Sigma-Aldrich) and ATS (50 μl/ml) + human serum (30
μl/ml) for the indicated durations. Pyrrolidine
dithiocarbamate (PDTC; Sigma-Aldrich) was used to treat podocytes
at various concentrations for 2 h before the cells were treated
with TNF-α, IL-1β, LPS and ATS + human serum for 24 h.
In the podocyte-mesangial cell co-culturing system,
podocytes at 80% confluence were co-cultured with mesangial cells
[prepared from the kidney cortex of male Sprague-Dawley rats as
described previously (26) and
planted in nested transwell plates (Corning Inc., Corning, NY, USA)
at 30% confluence] in six-well plates. Sprague-Dawley rats were
obtained from the Animal Center of the College of Basic Medicine of
Fudan University). A total of 10 rats at 2 months-old were used in
the present study. The protocol was approved by the Ethics
Committee from the College of Basic Medicine, Fudan University.
Subsequently, the dual culture system was treated with ATS
>1:1,000 for different time periods.
The ATS was produced in our laboratory (27). It is able to combine with antigens
of rat mesangial cells and activate complement in the serum, and
the resulting immune complex is capable of stimulating podocytes
directly in the co-culture system. The ATS was verified to be
efficient at a dilution of 1:1,000 using the agarose
immunodiffusion method as described previously (27).
Western blot analysis
Gel electrophoresis and western blotting were
performed as described previously (19). Samples (15 μl) of the
podocyte lysates, including soluble cell proteins, were separated
by SDS-PAGE [10% (w/v) polyacrylamide gel; Bio-Rad Laboratories,
Inc., Hercules, CA, USA] and then transferred electrophoretically
onto polyvinylidene difluoride membranes (Millipore). The membranes
were blocked with 5% (v/v) nonfat milk and probed with primary
antibodies against UCH-L1 (Millipore), p65/p-p65 (1:2,000; cat. no.
4764; Cell Signaling Technology) and β-actin (as a loading control)
overnight at 4°C following incubation with horseradish
peroxidase-conjugated secondary antibody (Proteintech Group,
Chicago, IL, USA). All washing steps were performed in
Tris-buffered saline containing 0.1% (v/v) Tween 20 (Sangon Biotech
Co., Ltd.). The immune reaction was visualized via an enhanced
chemiluminescence detection kit according to the manufacturer’s
instructions (Pierce Biotechnology, Rockford, IL, USA) and exposure
to X-ray film (Eastman Kodak, Rochester, NY, USA). The relative
band intensities in the blots were determined using Adobe Photoshop
software (Adobe Systems Inc., San Jose, CA, USA). Each experiment
was performed at least three times.
Statistical analysis
All statistical analyses were performed with SPSS
software (SPSS, Inc., Chicago, IL, USA) and the results are
presented as the mean ± standard error of the mean. Paired means
were analyzed using Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference and P<0.01 was
considered to indicate a highly statistically significant
difference.
Results
NF-κB and UCH-L1 distribution in diseased
podocytes in glomerulonephritis
A previous study by our group identified that UCH-L1
expression was significantly increased in several types of immune
complex-mediated glomerulonephritis (2). Therefore, the distribution of p65 was
further examined using immunohistochemistry and it was compared
with UCH-L1 expression in podocytes in a number of types of immune
complex-mediated glomerulonephritis, including LN, IgAN and MGN.
WT1 was also used as a marker of podocytes in successive sections
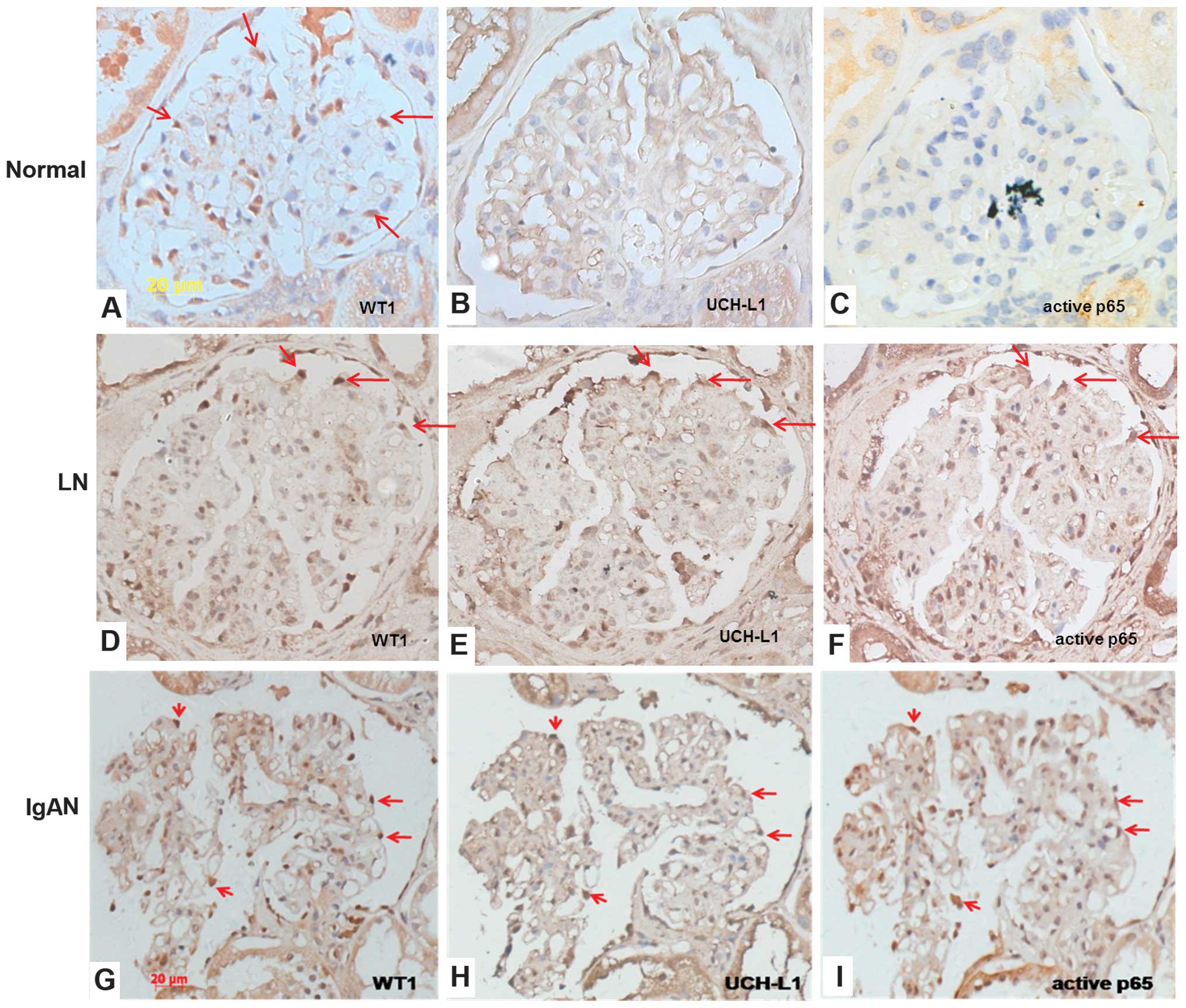
of renal biopsy tissues (Fig.
1A–O).
In the normal kidney, WT1 is expressed in the
peripheral cells of the capillary tufts and was therefore used to
indicate the location of podocytes (Fig. 1A). No marked expression of UCH-L1
and active p65 was observed in the corresponding areas (Fig. 1B–C). LN is one of the typical forms
of immune complex-mediated glomerulonephritis. As shown in Fig. 1E, positive staining for UCH-L1 was
increased in the glomeruli of patients with LN and was located
mainly at the peripheral area of the capillary tufts in accordance
with the distribution of WT1 (Fig.
1D). Activation of p65 was also increased in the glomeruli of
patients with LN, with more extensive cells, including mesangial
cells, endothelial cells and infiltrating leukocytes in addition to
podocytes (Fig. 1F). In podocytes,
the expression of UCH-L1 paralleled the activation of NF-κB in the
glomeruli and appeared to be positively correlated in patients with
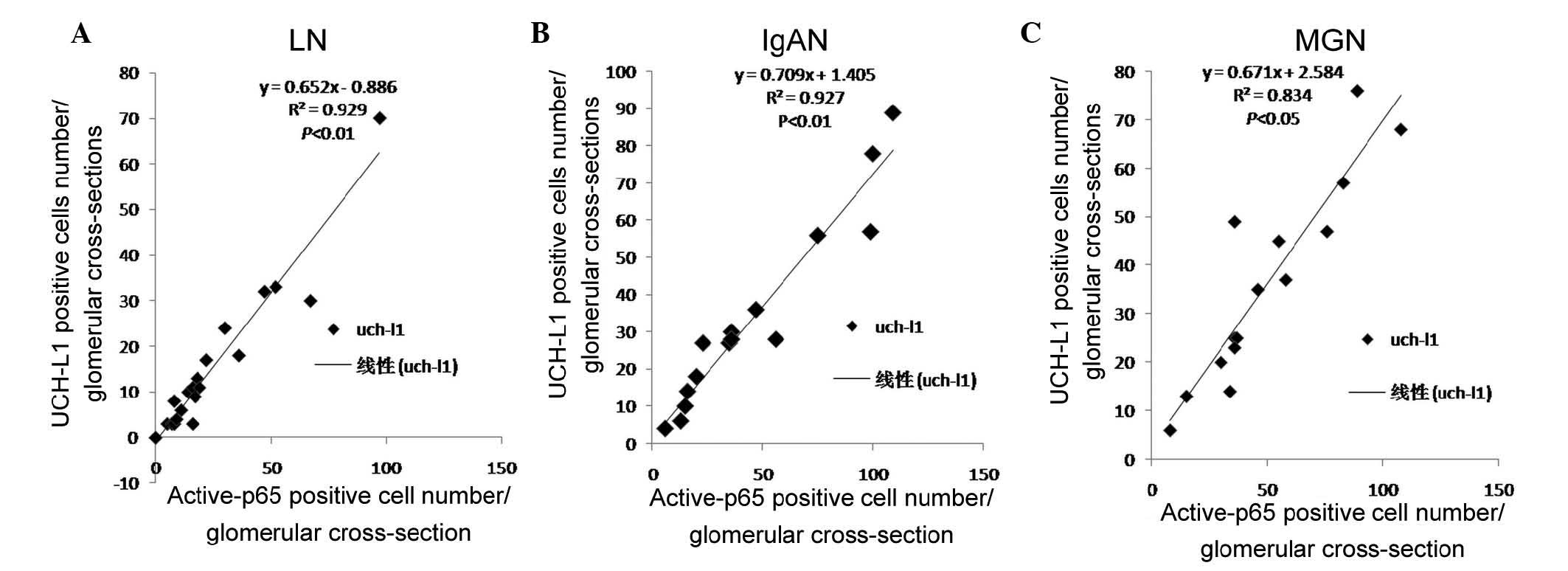
LN (R=0.925, P<0.01; Fig. 2A).
As shown in Fig. 1G–L, the
localization of WT1, UCH-L1 and activated p65 in patients with IgAN
and MGN were concordant with those in patients with LN. All of
these proteins were expressed in the respective podocytes. The
expression of UCH-L1 also paralleled the activation of NF-κB in the
glomeruli and also appeared positively correlated in IgAN (R=0.927,
P<0.01; Fig. 2B), and in MGN
(R=0.834, P<0.05; Fig. 2C).
Furthermore, the expression levels of UCH-L1 and active p65 were
examined in MCD, a non-immune complex-mediated type of
glomerulonephritis, in which no distinct inflammatory changes were
observed in the glomeruli. Additionally, no clear expression of
UCH-L1 and activated p65 was observed in podocytes in MCD (Fig. 1M and O). These results suggested
that there was a close association between UCH-L1 and the
activation of NF-κB in immune complex-mediated
glomerulonephritis.
Upregulation of UCH-L1 expression via
inflammatory stimulation with phosphorylation of p65
The cultured podocytes were divided into four
groups, each with or without treatment with TNF-α, IL-1β, LPS and
ATS. Western blot analysis revealed the expression of UCH-L1 in the
stimulated groups of TNF-α, and IL-1β was markedly increased in a
time-dependent manner at 12 and 24 h, reaching a maximum at 24 h
(Fig. 3A and B). In the LPS and
ATS groups, UCH-L1 expression also increased with the duration of
treatment; however, the maximum level was achieved later than in
the TNF-α and IL-1β groups mentioned above, namely at 48 h. Among
the stimulation groups, the highest level of UCH-L1 expression was
observed in podocytes following ATS stimulation (Fig. 3C and D). These results confirmed
that UCH-L1 expression in podocytes may be upregulated by
pro-inflammatory mediators as well as immune stimulation.
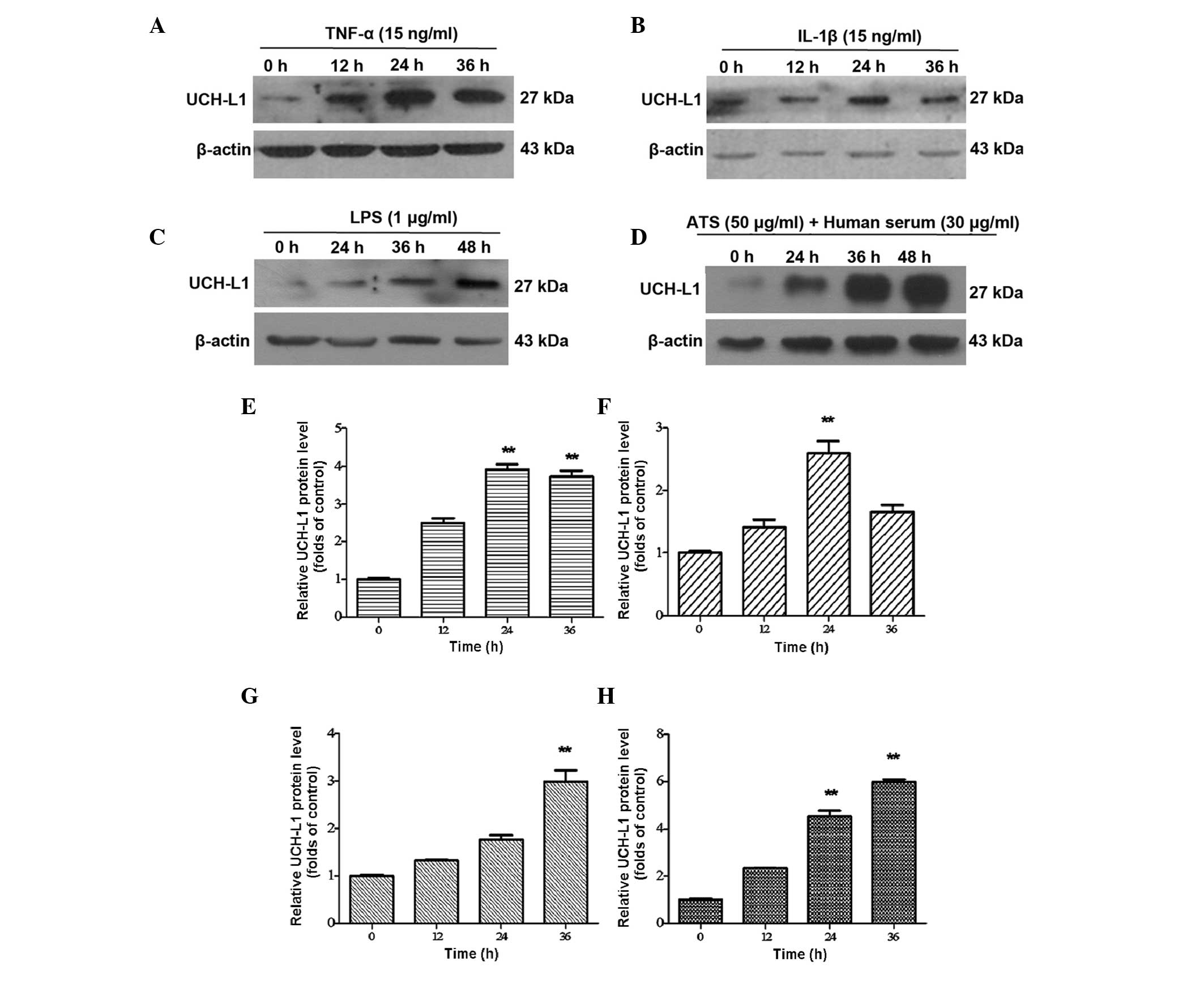 | Figure 3UCH-L1 expression in podocytes
treated with TNF-α, IL-1β, LPS and ATS. Lysates from the podoctyes
were assessed by western blot analysis using murine anti-UCH-L1
antibody. β-Actin was used as a protein loading control. Murine
podocytes treated with (A) TNF-α (15 ng/ml), (B) IL-1β (15 ng/ml),
(C) LPS (1 μg/ml) and (D) ATS (50 μl/ml) + human
serum (30 μl/ml) for the indicated durations. (E–H) Relative
expression of UCH-L1 quantified from the western blot analysis of
A–D, respectively. Data are representative of three independent
experiments. **P<0.005 vs. 0 h. LPS,
lipopolysaccharide; UCH-L1, ubiquitin carboxy-terminal hydrolase
L1; TNF, tumor necrosis factor; IL, interleukin; ATS, anti-rat
thymocyte serum. |
NF-κB is a common transcription factor family
consisting of different subunits, which may function as homo- or
heterodimers. p65 is a well-known subunit, which is involved in the
NF-κB family in regulating the transcription of a number of genes.
Phosphorylation of p65 is a typical feature in the activation of
NF-κB. Therefore, the phosphorylation of p65 in the cell nucleus
was further examined following treatment with TNF-α, IL-1β, LPS and
ATS. It was identified that following treatment for 15 min, the p65
subunit was clearly phosphorylated in cultured podocytes and the
phosphorylation lasted for several hours (Fig. 4A–D). Of note, after 6 h, the
phosphorylation of p65 was greater than that in the control group.
It was confirmed that the NF-κB signaling pathway was activated
following stimulation with the four different treatments. Among the
four treated groups, immune stimulation with ATS was able to cause
the most persistent phosphorylation of p65, which lasted for at
least 12 h and was maintained at an elevated level (Fig. 4D).
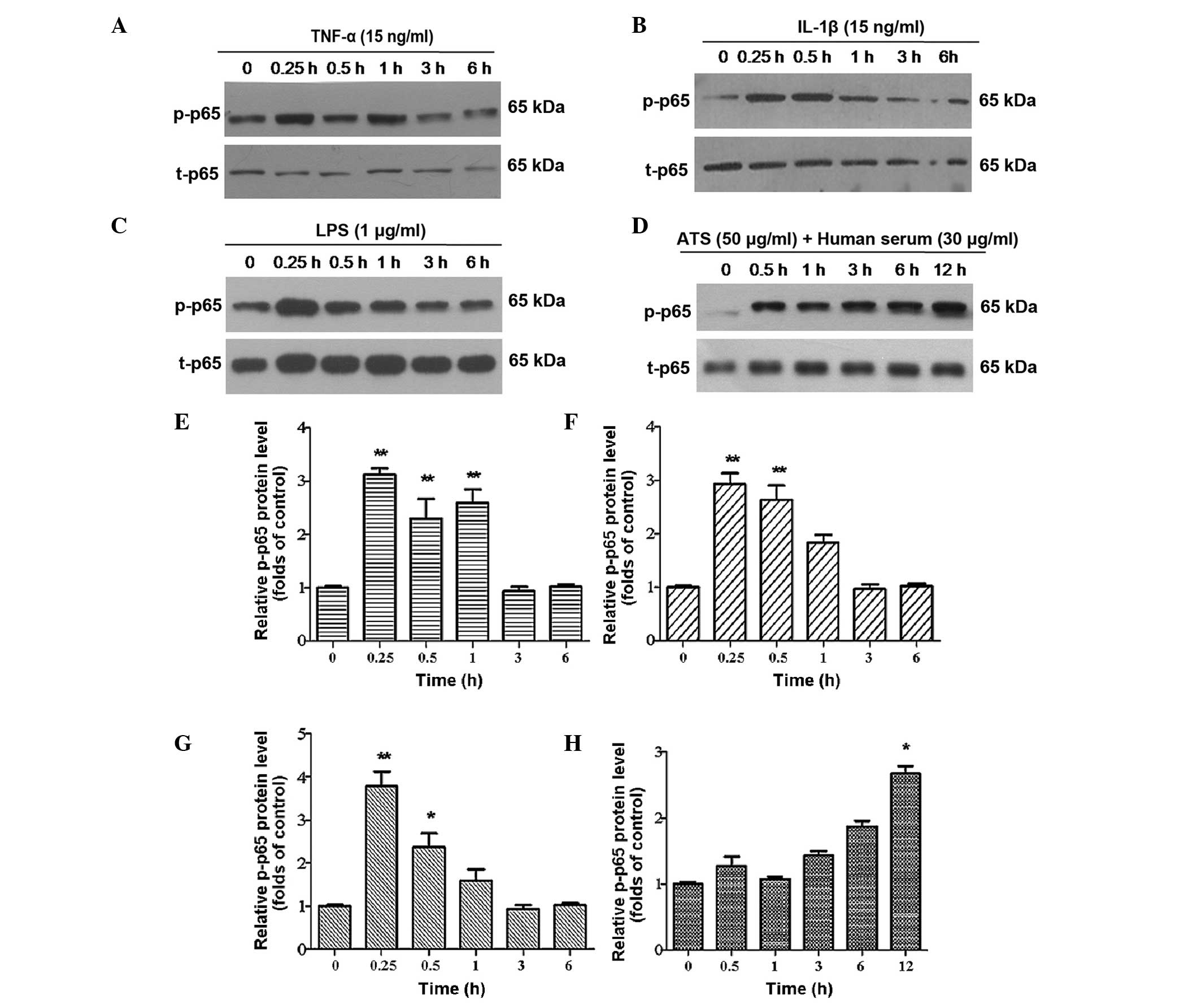 | Figure 4p-p65 expression in podocytes treated
with TNF-α, IL-1β, LPS and ATS. Lysates from the podocytes were
assessed by western blot analysis using rabbit anti-p-p65 antibody,
and total-p65 was used as control for protein loading. Murine
podocytes were treated with (A) TNF-α (15 ng/ml), (B) IL-1β (15
ng/ml), (C) LPS (1 μg/ml) and (D) ATS (50 μl/ml) +
human serum (30 μl/ml) for the indicated durations. (E–H)
Relative expression of p-p65 quantified from the western blot
analysis of A–D, respectively. Data are representative of three
independent experiments. *P <0.05,
**P<0.005 vs. 0 h. LPS, lipopolysaccharide; TNF,
tumor necrosis factor; IL, interleukin; ATS, anti-rat thymocyte
serum; p, phosphorylated; t, total. |
Effect of NF-κB inhibition on the
expression of UCH-L1
Subsequently, an inhibitor of the NF-κB pathway was
used to assess the specificity of NF-κB phosphorylation and
regulation. PDTC, a specific inhibitor of NF-κB, is capable of
preventing the degradation of inhibitor of κB and the subsequent
translocation of NF-κB from the cytoplasm into the nucleus without
inhibiting other nuclear factors, including specificity protein-1,
Oct and cyclic adenosine monophosphate response element-binding
protein (4). In the present study,
podocytes were pre-treated with PDTC at various concentrations for
2 h and then subjected to treatment with TNF-α, IL-1β, LPS and ATS
for 24 h. UCH-L1 expression significantly decreased in podocytes
treated with PDTC in a dose-dependent manner (Fig. 5A–D). This result demonstrated that
the inhibition of NF-κB activation blocked the expression of UCH-L1
in response to stimulation by TNF-α, IL-1β, LPS and ATS.
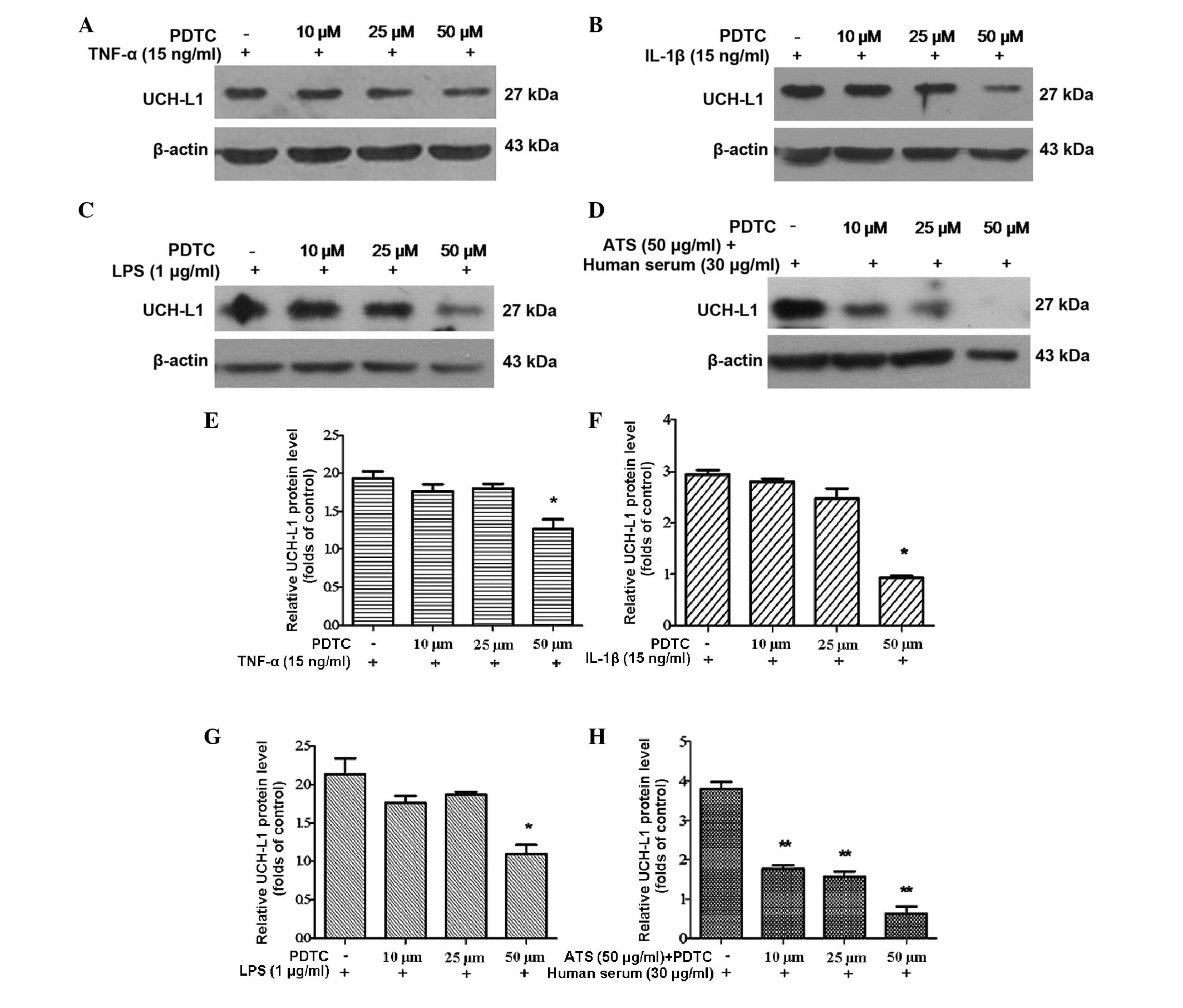 | Figure 5Inhibition of NF-κB with PDTC reduced
UCH-L1 expression in podocytes treated with TNF-α, IL-1β, LPS and
ATS. Lysates from the stimulated cells were assessed by western
blot analysis using murine anti-UCH-L1 antibody, and β-actin was
used as a control for protein loading. Murine podocytes were
pre-treated with PDTC at the stated concentrations for 2 h,
following stimulation with (A) TNF-α (15 ng/ml), (B) IL-1β (15
ng/ml), (C) LPS (1 μg/ml) and (D) ATS (50 μl/ml) +
human serum (30 μl/ml) for 24 h. (E–H) Relative expression
of UCH-L1 quantified from the western blot analysis of A–D,
respectively. Data are representative of three independent
experiments. *P<0.05, **P<0.005, vs.
PDTC. LPS, lipopolysaccharide; TNF, tumor necrosis factor; IL,
interleukin; ATS, anti-rat thymocyte serum; UCH-L1, ubiquitin
C-terminal hydrolase 1; PDTC, pyrrolidine dithiocarbamate. |
Discussion
The present study provided further evidence
supporting that UCH-L1 expression is upregulated through the
activation of NF-κB in diseased podocytes in human
glomerulonephritis and in cultured mouse podocytes in vitro.
It has been suggested that the NF-κB signaling pathway is important
in the underlying mechanism of podocyte injury in
glomerulonephritis (28).
A study has indicated that the podocyte is a primary
vulnerable cell in glomerular diseases (2). The pathological features of injured
podocytes may be observed as effacement of the processes,
pseudocystic changes and microvilli formation, which are closely
associated with the development of proteinuria and
glomerulosclerosis (29–31). In general, these podocyte changes
are recognized as prominent characteristics in MCD and focal
segmental glomerulosclerosis (FSGS) amongst others, which are known
as non-immune complex-mediated glomerular diseases. However, it
should not be ignored that severe damage to podocytes also occurs
in a number of other types of glomerulonephritis, including IgAN,
MGN and LN, via the immune injury mechanism (1,2,25,32,33).
Studies have demonstrated that the changes to podocyte morphology
and function following injury were closely associated with the
abnormalities of numerous podocyte proteins, including UCH-L1,
nephrin and synaptopondin (1,34,35).
A previous study by our group demonstrated that UCH-L1 is
upregulated in podocytes in a number of types of immune
complex-mediated glomerulonephritis, which was detected by
immunoelectron microscopy, accompanied by foot process fusion
(2). In addition, it was reported
that the expression of UCH-L1 is associated with podocyte
differentiation (25). In immature
podocytes, UCH-L1 expression was higher, whereas when the cell was
differentiating into a mature cell, UCH-L1 expression was
significantly reduced and then eliminated, accompanied with the
formation of foot processes. When the glomerular lesions formed,
UCH-L1 expression increased again with foot process fusion. These
results implied that elevated UCH-L1 expression is an indication of
podocyte injury, which may appear in non-immune complex-mediated
and immune complex-mediated glomerular diseases. However, the
regulatory mechanisms of UCH-L1 or other podocyte-specific proteins
are complex and remain to be elucidated.
It has been demonstrated that inflammation leads to
podocyte injury involving multiple signaling pathways, including
the samds, mitogen-activated protein kinase, NF-κB, Wnt/β-catenin
and TGF-β1 signaling pathways (12,29,35,36).
NF-κB signaling is considered to be the most prominent activation
pathway in the pathogenesis of human kidney diseases and numerous
associated animal models (11,37).
In the present study, renal biopsy sections were
analyzed in numerous types of immune complex-mediated
glomerulonephritis, including LN, IgAN and MGN, and the expression
of active NF-κB p65 and UCH-L1 were identified in the podocytes. In
addition, NF-κB p65 over-activation was correlated with the
expression of UCH-L1 in podocytes. By contrast, in non-immune
complex-mediated glomerulonephritis, such as MCD, no marked
expression of active p65 and UCH-L1 was identified in podocytes.
These results indicated that immune injurious stimulation is able
to increase the expression of UCH-L1 through the NF-κB signaling
pathway, which may be vital in the pathogenesis of podocyte injury
in immune complex-mediated glomerulonephritis. Furthermore,
although podocyte injury and foot process effacement appeared in
non-immune complex-mediated glomerulonephritis, there were no
evident inflammatory changes in these glomeruli. Correspondingly,
there was also no marked activation of NF-κB and no increase of
UCH-L1 in MCD. It is therefore suggested that there may be another
mechanism for podocyte injury in non-immune complex-mediated
glomerulonephritis.
NF-κB is known to be activated by the exposure of
cells to pro-inflammatory mediators, including TNF-α and IL-1β
(38,39). It is also activated by the binding
of Toll-like receptors with their cognate ligands, such as LPS
(4). In the present study,
podocytes were therefore treated with TNF-α, IL-1β and LPS in
vitro, which all caused translocation of phosphorylated p65 to
the nucleus and then upregulated the expression of the UCH-L1
protein. Thus, the present study further verified that UCH-L1 is
one downstream target protein of NF-κB in podocytes. Subsequently,
ATS was used in combination with human serum to treat podocytes
co-cultured with rat mesangial cells, duplicating a model of immune
injury. Persistent phosphorylation of p65 and a significant
upregulation of UCH-L1 were identified, which were more marked than
those in the groups treated with TNF-α, IL-1β and LPS. This was
consistent with the results of a previous study by our group
(2), which demonstrated that the
expression of UCH-L1 in podocytes was not elevated in non-immune
complex-mediated nephritis, including MCD and FSGS. By contrast,
UCH-L1 was markedly increased in immune complex-mediated
glomerulonephritis (2). It has
been reported that ATS is capable of interacting with antigens on
the cell membrane of mesangial cells to form immune complexes and
subsequently activate complements in fresh serum to assemble
sublytic C5b-9, leading to immune injury to cells in vitro
(40,41). As podocytes present Fc and C3
receptors on the cell surface, it is possible that immune complexes
and sublytic complement compounds are stimulatory factors affecting
podocytes in glomeruli (41,42).
Therefore, the deposition of immunity compounds may be a key cause
of the activation of NF-κB to upregulate the expression of UCH-L1
in podocytes in immune complex-mediated glomerulonephritis.
In conclusion, the present study demonstrated that
in human renal biopsy samples of several forms of immune
complex-mediated glomerulonephritis, the increase of NF-κB and
UCH-L1 was positively correlated with diseased podocytes. However,
in non-immune complex-mediated glomerulonephritis, no clear
activation of NF-κB and no increase of UCH-L1 expression was
observed. In vitro, immune stimulation also upregulated
UCH-L1 through the NF-κB signaling pathway in mouse podocytes.
These results suggested that the activation of NF-κB and
upregulation of UCH-L1 in podocytes are vital for podocyte injury
in immune complex-mediated glomerulonephritis.
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no. 81070566),
a grant from the General Project of Weifang Medical University
(grant no. K1302012) and a grant from the Science and Technology
Development Plan of Medicine and Healthcare in Shandong province
(grant no. 2013WS0279). The authors would like to thank Professor
Hong Xu (Affiliated Children’s Hospital of the Medical College of
Fudan University, Shanghai, China) for her gift of the podocyte
line.
References
|
1
|
Meyer-Schwesinger C, Meyer TN, Sievert H,
Hoxha E, Sachs M, Klupp EM, Münster S, Balabanov S, Carrier L,
Helmchen U, et al: Ubiquitin c-terminal hydrolase-l1 activity
induces polyubiquitin accumulation in podocytes and increases
proteinuria in rat membranous nephropathy. Am J Pathol.
178:2044–2057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu Y, Wu J, Wu H, Wang T, Gan H, Zhang X,
Liu Y, Li RX, Zhao Z, Chen Q, Guo MY and Zhang Z: Uch-l1 expression
of podocytes in diseased glomeruli and in vitro. J Pathol.
217:642–653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greka A and Mundel P: Cell biology and
pathology of podocytes. Annu Rev Physiol. 74:299–323. 2012.
View Article : Google Scholar
|
|
4
|
Liu X, Ye L, Christianson G, Yang JQ,
Roopenian DC and Zhu X: Nf-kappab signaling regulates functional
expression of the MHC class I-related neonatal FC receptor for IgG
via intronic binding sequences. J Immunol. 179:2999–3011. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guijarro C and Egido J: Transcription
factor-kB (NF-kB) and renal disease. Kidney Int. 59:415–424. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baeuerle PA: Pro-inflammatory signaling:
last pieces in the NF-kappaB puzzle? Curr Biol. 8:R19–R22. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sakurai H, Shigemori N, Hisada Y, Ishizuka
T, Kawashima K and Sugita T: Suppression of NF-kappa B and AP-1
activation by glucocorticoids in experimental glomerulonephritis in
rats: molecular mechanisms of anti-nephritic action. Biochim Biophs
Acta. 1362:252–262. 1997. View Article : Google Scholar
|
|
8
|
Massy ZA, Guijarro C, O’Donnell MP, Kim Y,
Kashtan CE, Egido J, Kasiske BL and Keane WF: The central role of
nuclear factor-kappa B in mesangial cell activation. Kidney Int.
(Suppl 71): 76–79. 1999. View Article : Google Scholar
|
|
9
|
Rovin BH, Dickerson JA, Tan LC and Hebert
CA: Activation of nuclear factor-kB correlates with MCP-1
expression by human mesangial cells. Kidney Int. 48:1263–1271.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ashizawa M, Miyazaki M, Abe K, Furusu A,
Isomoto H, Harada T, Ozono Y, Sakai H, Koji T and Kohno S:
Detection of nuclear factor-kappaB in IgA nephropathy using
southwestern histochemistry. Am J Kidney Dis. 42:76–86. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng L, Sinniah R and Hsu SI: In situ
glomerular expression of activated NF-kappaB in human lupus
nephritis and other non-proliferative proteinuric glomerulopathy.
Virchows Arch. 448:172–183. 2006. View Article : Google Scholar
|
|
12
|
Bruggeman LA, Drawz PE, Kahoud N, Lin K,
Barisoni L and Nelson PJ: TNFR2 interposes the proliferative and
NF-kappaB-mediated inflammatory response by podocytes to TNF-alpha.
Lab Invest. 91:413–425. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Palanisamy N, Kannappan S and Anuradha CV:
Genistein modulates NF-kappaB-associated renal inflammation,
fibrosis and podocyte abnormalities in fructose-fed rats. Eur J
Pharmacol. 667:355–364. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mudge SJ, Paizis K, Auwardt RB, Thomas RJ
and Power DA: Activation of nuclear factor-kappa B by podocytes in
the autologous phase of passive heymann nephritis. Kidney Int.
59:923–931. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morigi M, Buelli S, Angioletti S, Zanchi
C, Longaretti L, Zoja C, Galbusera M, Gastoldi S, Mundel P, Remuzzi
G and Benigni A: In response to protein load podocytes reorganize
cytoskeleton and modulate endothelin-1 gene: implication for
permselective dysfunction of chronic nephropathies. Am J Pathol.
166:1309–1320. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morigi M, Buelli S, Zanchi C, Longaretti
L, Macconi D, Benigni A, Moioli D, Remuzzi G and Zoja C:
Shigatoxin-induced endothelin-1 expression in cultured podocytes
autocrinally mediates actin remodeling. Am J Pathol. 169:1966–1975.
2006. View Article : Google Scholar
|
|
17
|
Bondeva T, Roger T and Wolf G:
Differential regulation of toll-like receptor 4 gene expression in
renal cells by angiotensin II: dependency on AP1 and PU.1
transcriptional sites. Am J Nephrol. 27:308–314. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu LX, Lin QX, Liao H, Feng JH, Dong XH
and Ye JM: Tgf-beta1 induces podocyte injury through
SMAD3-ERK-NF-kappaB pathway and FYN-dependent TRPC6
phosphorylation. Cell Physiol Biochem. 26:869–878. 2010. View Article : Google Scholar
|
|
19
|
Zhang H, Sun Y, Hu R, Luo W, Mao X, Zhao
Z, Chen Q and Zhang Z: The regulation of the UCH-l1 gene by
transcription factor NF-kB in podocytes. Cell Signal. 25:1574–1585.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fukasawa H: The role of the
ubiquitin-proteasome system in kidney diseases. Clin Exp Nephrol.
16:507–517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Amerik AY and Hochstrasser M: Mechanism
and function of deubiquitinating enzymes. Biochim Biophys Acta.
1695:189–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wilson PO, Barber PC, Day IN, Thompson RJ
and Polak JM: The immunolocalization of protein gene product 9.5
using rabbit polyclonal and mouse monoclonal antibodies. Br J Exp
Pathol. 69:91–104. 1988.PubMed/NCBI
|
|
23
|
D’Andrea V, Malinovsky L, Berni A,
Biancari F, Biassoni L, Di Matteo FM, Corbellini L, Falvo L,
Santoni F, Spyrou M and De Antoni E: The immunolocalization of PGP
9.5 in normal human kidney and renal cell carcinoma. G Chir.
18:521–524. 1997.
|
|
24
|
Shirato I, Asanuma K and Takeda Y: Protein
gene product 9.5 is selectively localized in parietal epithelial
cells of Bowman’s capsule in the rat kidney. J Am Soc Nephrol.
11:2381–2386. 2000.PubMed/NCBI
|
|
25
|
Meyer-Schwesinger C, Meyer TN, Munster S,
Klug P, Saleem M, Helmchen U and Stahl RA: A new role for the
neuronal ubiquitin C-terminal hydrolase-l1 (UCH-l1) in podocyte
process formation and podocyte injury in human glomerulopathies. J
Pathol. 217:452–464. 2009. View Article : Google Scholar
|
|
26
|
Zhang M, Guo MY, Chen Q and M JH: The
culture of rat glomerular mesangial cells. J Shanghai Med Univ.
207–209. 1995.
|
|
27
|
Chen GP, Guo MY and Zhang YE: Preparation
of anti-thy1 serum and establishment of mesangioproliferative
glomerulonephritis model in rat. J Clin Exp Pathol. 241–243.
1996.
|
|
28
|
Chiang ML, Hawkins EP, Berry PL, Barrish J
and Hill LL: Diagnostic and prognostic significance of glomerular
epithelial cell vacuolization and podocyte effacement in children
with minimal lesion nephrotic syndrome and focal segmental
glomerulosclerosis: An ultrastructural study. Clin Nephrol.
30:8–14. 1988.PubMed/NCBI
|
|
29
|
Wang D, Dai C, Li Y and Liu Y: Canonical
WNT/β-catenin signaling mediates transforming growth
factor-β1-driven podocyte injury and proteinuria. Kidney Int.
80:1159–1169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghayur A, Liu L, Kolb M, Chawla A, Lambe
S, Kapoor A and Margetts PJ: Adenovirus-mediated gene transfer of
TGF-β1 to the renal glomeruli leads to proteinuria. Am J Pathol.
180:940–951. 2012. View Article : Google Scholar
|
|
31
|
Shankland SJ: The podocyte’s response to
injury: role in proteinuria and glomerulosclerosis. Kidney Int.
69:2131–2147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Borza DB, Zhang JJ, Beck LH Jr, C M and
Luo W: Mouse models of membranous nephropathy: the road less
travelled by. Am J Clin Exp Immunol. 2:135–145. 2013.PubMed/NCBI
|
|
33
|
Meyer-Schwesinger C, Dehde S, Sachs M,
Mathey S, Arefi K, Gatzemeier S, Balabanov S, Becker JU, Thaiss F
and Meyer TN: Rho-kinase inhibition prevents proteinuria in
immune-complex-mediated antipodocyte nephritis. Am J Physiol Renal
Physiol. 303:F1015–F1025. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kato T, Mizuno S and Kamimoto M: The
decreases of nephrin and nuclear WT1 in podocytes may cause
albuminuria during the experimental sepsis in mice. Biomed Res.
31:363–369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Greka A and Mundel P: Cell biology and
pathology of podocytes. Annu Rev Physiol. 74:299–323. 2012.
View Article : Google Scholar
|
|
36
|
Hirota M, Watanabe K, Hamada S, Sun Y,
Strizzi L, Mancino M, Nagaoka T, Gonzales M, Seno M, Bianco C and
Salomon D: SMAD2 functions as a co-activator of canonical
WNT/beta-catenin signaling pathway independent of SMAD4 through
histone acetyltransferase activity of p300. Cell Signal.
20:1632–1641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rangan G, Wang Y and Harris D: NF-kappaB
signalling in chronic kidney disease. Front Biosci (Landmark Ed).
14:3496–3522. 2009. View
Article : Google Scholar
|
|
38
|
Li QI and Verma M: NF-κB regulation in the
immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schjerven H, Brandtzaeg P and Johansen FE:
A novel NF-κB/Rel site in intron 1 cooperates with proximal
promoter elements to mediate TNF-α-induced transcription of the
human polymeric Ig receptor. J Immunol. 167:6412–6420. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nangaku M, Shankland SJ and Couser WG:
Cellular response to injury in membranous nephropathy. J Am Soc
Nephrol. 16:1195–1204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rus HG, Niculescu FI and Shin ML: Role of
the c5b-9 complement complex in cell cycle and apoptosis. Immunol
Rev. 180:49–55. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kazatchkine MD, Fearon DT, Appay MD,
Mandet C and Bariety J: Immunohistochemical study of the human
glomerular c3b receptor in normal kidney and in seventy-five cases
of renal diseases: loss of c3b receptor antigen in focal hyalinosis
and in proliferative nephritis of systemic lupus erythematosus. J
Clin Invest. 69:900–912. 1982. View Article : Google Scholar : PubMed/NCBI
|