Introduction
Spinal cord injury (SCI) occurs as a result of a
multifactorrial process, involving primary injury subsequently
followed by secondary effects. Primary injury usually occurs due to
mechanical trauma, whereas secondary injury occurs as a result of
the primary injury; in addition, the severity of secondary injury
is proportional to that of the primary mechanical damage (1). Secondary injury is characterized by
apoptotic or necrotic events, however the predominant causes of
such events are mediated through oxidative stress (2,3). The
central mediator in SCI injury is the generation of reactive oxygen
species (ROS), which have various effects at the cellular level.
Previous studies have suggested that the initial formation of ROS
and reactive nitrogen species (RNS) molecules initiate lipid
peroxidation and protein carbonylation (4). Lipid peroxidation results in the
disorganization of phospholipids in membranes; in addition, the end
products of lipid peroxidation damage the structure and function of
proteins, thereby exacerbating oxidative stress conditions. In
addition, various signaling events mediated through the
deregulation of ion homeostasis contribute to oxidative stress and
tissue damage (5). It has been
widely reported and accepted that the end results of free
radical-mediated oxidative stress and subsequent inflammatory
mechanisms serve a key role in SCI. Oxidative stress and
deregulation of ion homeostasis initiate and promote cellular
damage; therefore, protection against these effects using
pharmacological intervention may be an effective therapeutic
strategy. Thus, the present study aimed to investigate the
protective effects of gallic acid against SCI injury in the context
of oxidative stress and ion homeostasis.
Gallic acid is a phenolic compound with clear
antioxidant activity (6). This
compound is widely present in gallnuts, tea leaves, green tea,
apples, grapes, strawberries and pineapples (7). Gallic acid has been reported to exert
chemopreventive activities through ameliorating oxidative stress
and enhancing the antioxidant status (8). In addition to its free radical
scavenging abilities and cytoprotective effects, gallic acid has
been well reported to act as an anticancer and anti-inflammatory
agent (9–12). The present study was designed to
analyze the effects of gallic acid against SCI-induced oxidative
stress and ion imbalance by evaluating lipid peroxide levels,
protein carbonylation, ROS levels, antioxidant status and the
enzymatic activities of the Na+/K+,
Ca2+ and Mg2+ Adenosine triphophatase
(ATPase) in Wistar rats.
Materials and methods
Reagents
Gallic acid (97%), ketamine, xylazine, Tris-HCl
buffer, 2′,7′-dichlorofluorescein diacetate (DCF-DA),
phosphate-buffered saline (PBS), Griess,
H2O2, nitrocellulose membranes and
Tris-buffered saline with Tween 20 (TBST) were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Pierce ECL Western Blotting
Substrate was purchased from Life Technologies (Rockford, IL,
USA).
Animals and treatment schedule
All experiments performed in the present study were
approved by the Institutional Animal Care Committee at the
117th Hospital of People’s Liberation Army (Hangzhou,
China). A total of 30 male Wistar rats (160–190g) were obtained
from the Animal Center of the Hospital of People’s Liberation Army
(Beijing, China), were housed under controlled conditions (21±2°C;
relative humidity, 75%) and were fed with a Food and Drug
Association-approved diet and water ad libitum. Subsequent
to acclimatization, the animals were allocated into the following 3
groups with 10 animals in each: Group I, sham operated animals,
laminectomy alone; group II, laminectomy followed by SCI injury;
group III, laminectomy followed by SCI and intraperitoneal (i.p.)
injection of gallic acid (10 mg/kg) following 1 h of SCI on day 1,
which continued until day 10. The laminectomy was performed at the
T9 vertebra and the weight drop technique was adopted for the
induction of conducive SCI. During this technique, weight (15 g)
was dropped at a height of 2.5 cm onto the spinal cord for 2 min.
Subsequent to the appropriate treatment schedule for the group, the
rats were sacrificed with ketamine (75 mg/kg; i.p.) and xylazine
(10 mg/kg; i.p.). Blood was collected through cardiac puncture and
the serum was separated and stored at ‒80°C. The spinal cord
tissues (2 cm) were obtained. Tissue samples were homogenized using
ice-cold Tris-HCl buffer (50 mM, pH 7.4) and centrifuged at 800 × g
for 15 min at 4°C. The supernatant was aliquoted and stored at
‒20°C until used for the measurement of oxidative stress parameters
and western blot analysis. Protein estimation was conducted as
previously described (13).
Estimation of serum total antioxidant
capacity (TAC) and total oxidant status (TOS)
The TAC of serum was determined as described
previously (14). This method
involved quantifying hydroxyl radical formation during reactions
between antioxidants in the sample and free radicals. The results
are expressed as mmol of Trolox equivalent/l. TOS of serum was
determined as described by Erel (15). The results are expressed as
μmol H2O2 equivalent/l. The assay
measures oxidation of ferrous ion-o-dianisidine complex to the
ferric ion by the oxidants in the serum sample. The formation of
the colored complex was measured spectrophotometrically (AquaMate
8000 UV-Vis Spectrophotometer; Thermo Fisher Scientific, Waltham,
MA, USA), which is proportional to the quantity of oxidant
molecules present. The quantity of oxidants was then compared with
that of H2O2. The results are expressed as
μmol H2O2 equivalent/l.
Determination of tissue oxidative stress
and antioxidant status
The tissue homogenate was prepared using ice-cold
Tris-HCl buffer (50 mM, pH 7.4) at 4°C and the Ultra-Turrax T 25
Basic Homogenizer (IKA-Werke GmbH & Co., Staufen, Germany). The
homogenate was centrifuged at 800 × g for 15 min. The supernatant
was isolated and aliquoted into separate vials and stored at ‒20°C
until further use in various biochemical studies.
Oxidative stress
Estimation of tissue protein carbonyl
content
The protein carbonyls formed were determined as
described previously (16). The
formation of the Schiffs base during a reaction between the
carbonyl group and 2,4-dinitrophenylhydrazone (DNPH) resulted in
the formation of carbonyl contents, the levels of which were
measured spectrophotometrically (AquaMate 8000 UV-Vis
Spectrophotometer) at 370 nm. The results were expressed as nmol
carbonyl formed/mg protein.
Estimation of lipid peroxidation
The lipid peroxide levels (thiobarbituric acid
reactive substances; TBARS) were determined, as described
previously (17). The estimation
involved a reaction between malondialdehyde (MDA) and
thiobarbituric acid (TBA), which resulted in the formation of
TBARS. The pink colored chromogen formed was measured
spectrophometrically (AquaMate 8000 UV-Vis Spectrophotometer) at
532 nm. Results were expressed as nmol TBA reactants formed/g wet
tissue.
ROS generation
Levels of ROS generated were determined as described
by Hashimoto et al (18).
The homogenate was incubated with DCF-DA at 37°C for 15 min. At the
end of the incubation time, centrifugation at 9,000 × g was
conducted for 15 min. The resultant pellet was re-suspended in PBS
and incubated for 60 min at 37°C. ROS levels were measured
spectrofluorimetrically (AquaMate 8000 UV-Vis Spectrophotometer) at
excitation (485 nm) and emission (528 nm) wavelengths. The results
were expressed as the percentage of ROS generation when the control
group represented 100%.
Estimation of nitrite levels
The conversion of nitrates to nitrites by nitrate
reductase was determined by the addition of Griess reagent. The
reaction results in the formation of purple azo compound;
therefore, absorbance, which is proportional to the nitrite levels
(Nitrate/Nitrite Colorimetric Assay kit; Cayman Chemical Company,
Ann Arbor, MI, USA) in the sample, was then measured
spectrophotometrically (AquaMate 8000 UV-Vis
Spectrophotometer).
Antioxidant status
Levels of the non-enzymatic
antioxidant glutathione (GSH)
GSH levels were measured based on the principle
reaction between 5,5′-dithiobis(2-nitrobenzoic acid) and reduced
GSH. The resultant yellow color formed was measured
spectrophotometrically (AquaMate 8000 UV-Vis Spectrophotometer) at
405 nm. The concentration of GSH was the calculated from standard
GSH levels (Glutathione Assay kit; Trevigen, Inc., Gaithersburg,
MD, USA).
Superoxide dismutase (SOD)
activity
SOD activity was determined as described previously
by Sun et al (19). The
assay was based on the reduction of nitrobluetetrazolium (NBT). A
total of 1 Unit SOD activity = the amount required for 50%
inhibition of NBT reduction. SOD activity is expressed as U/mg
protein.
Catalase (CAT) activity
CAT activity was determined according to the method
described by Aebi (20). The
reaction mixture contained tissue homogenate (50 μg) and 30
mM H2O2 in 50 mM PBS, pH 7.0. The activity
was estimated by the reduced absorbance of
H2O2 at 240 nm.
GSH-S-transferase (GST) activity
The reaction between 1-chloro-2,4-dinitro benzene
(CDNB) and reduced GSH resulted in formation of dinitrophenyl
thioether, which was measured at 340 nm, as previously described
(21). A total of 1 Unit GST =
amount of enzyme producing 1 mmol CDNB-GSH conjugate/min.
GSH peroxidase (GPx) activity
The GPx activity was measured as described by Paglia
and Valentine (22). Oxidized GSH
is reduced by GSH reductase and NADPH. The oxidation of NADPH to
NADP+ was measured by the reduction in absorbance at 340
nm. GPx activity is expressed as U/mg protein.
Na+/K+,
Ca2+ and Mg2+ ATPase enzymatic
activities
Tissue Na+/K+ ATPase (Bonting)
(23), Mg2+ ATPase
(Ohnishi et al) (24) and
Ca2+-ATPase (Hjertén and Pan) (25) activities were measured by
estimating the inorganic free phosphate. Phosphate reacts with
ammonium molybdate to form phosphomolybdate. The reaction between
1-amino, 2-naphthol4-sulfonic acid and phosphomolybdate results in
the formation of a blue colored complex measured at a wavelength of
620 nm. Results are expressed as nmole Pi-released/min/mg
protein.
Western blot analysis
Tissue homogenates (50 μg) from different
treatment groups were analyzed for nuclear factor (NF)-κB and
cycloxygenase (COX)-2 expression. Western blot analysis was
conducted as described by Towbin et al (26). Briefly, the proteins were separated
using 12% SDS-PAGE (Mini-Protean Tetra Cell systems; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and transferred to
nitrocellulose membranes. The blots were blocked with non-fat milk
(5%) at room temperature for 1 h. Subsequent to washing with TBST,
the blots were incubated with the following primary antibodies
overnight at 4°C: Goat polyclonal COX-2 (C-20; sc-1745) and rabbit
polyclonal IgG NFκB p65 (C-20; sc-372). Following another wash with
TBST, the blots were probed with the goat anti-rabbit COX-2 and
rabbit anti-mouse NFκB secondary antibodies for 1 h at room
temperature. Specific primary and secondary antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Danvers, TX, USA).
Protein expression levels were visualized using the Enhanced
Chemiluminescence Detection System and the band intensities were
measured using ImageJ software, version 1.41 (http://imagej.nih.gov/ij/).
Statistical analysis
Data were analyzed using a one-way analysis of
variance followed by Tukey’s multiple comparison test. All
biochemical experiments were performed in triplicate to ensure
reproducibility. SPSS software, version 22.0 (IBM SPSS, Armonk, NY,
USA) was used for the statistical analysis.
Results
Effect of gallic acid on serum TAC and
TOS levels
As shown in Table
I, the results of the present study demonstrated a
statistically significant increase in TOS levels (P<0.001) in
rats with SCI when compared with control rats. By contrast, the
levels of TAC were significantly reduced (P<0.001) in SCI rats
when compared with the control. Treatment with gallic acid resulted
in a significant reduction in TOS levels (P<0.001) with an
increase in TAC levels (P<0.001) when compared with SCI rats
(Table I).
 | Table IEffect of gallic acid on TAC and
TOS. |
Table I
Effect of gallic acid on TAC and
TOS.
| Parameter | Group I | Group II | Group III |
|---|
| TAC | 4.01±0.01 | 2.19±0.01a | 3.53±0.01b |
| TOS | 21±1.9 | 69±3.12a | 39±1.81b |
Gallic acid ameliorates SCI-induced
oxidative stress in Wistar rats
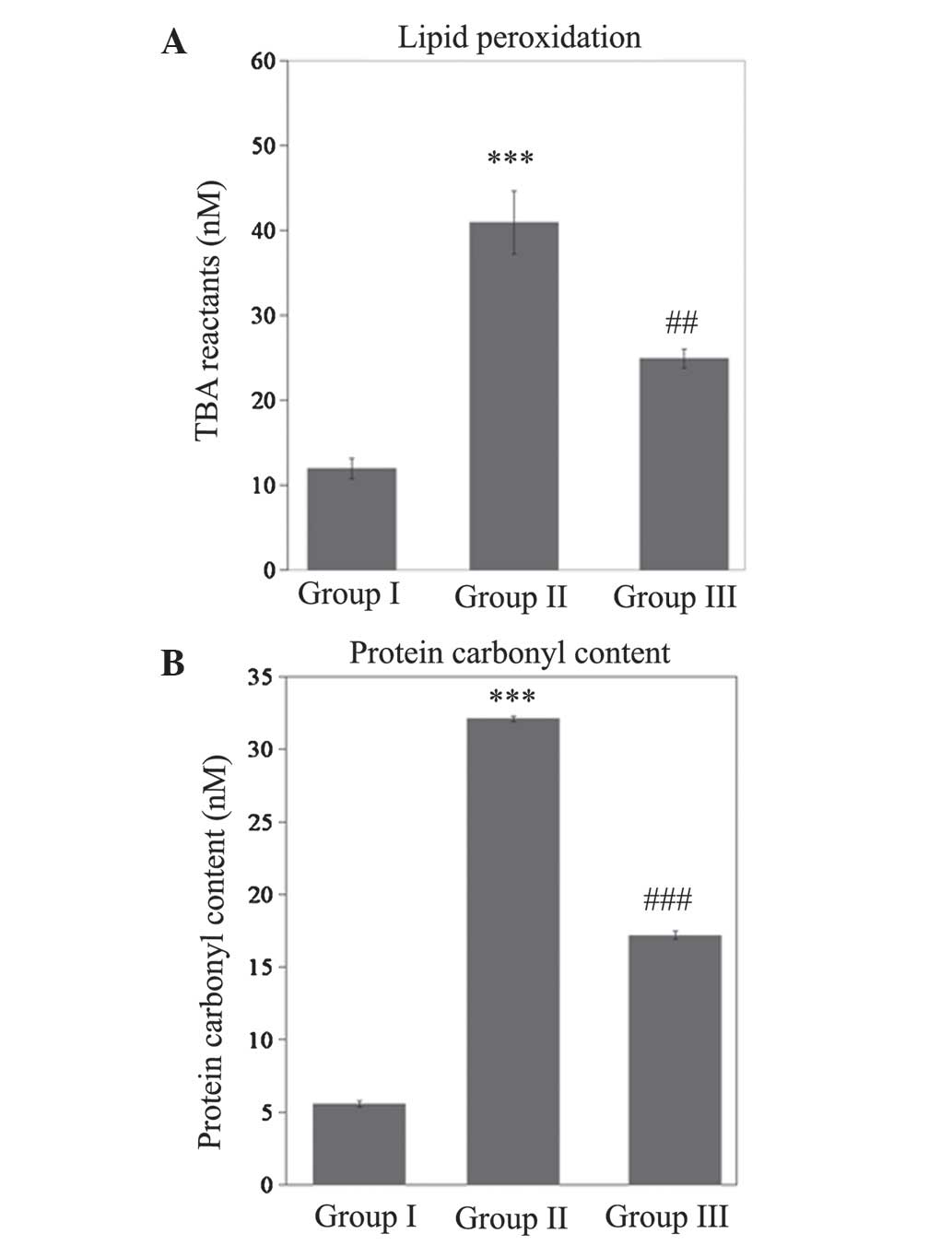
Significant increases in lipid peroxide levels
(P<0.001) and protein carbonyl content (P<0.001) were
observed in rats with SCI injury, when compared with the control
(Fig. 1). In addition, there was a
significant reduction (P<0.01) in these levels when compared
with SCI rats. This indicated that treatment with gallic acid
attenuated the rise in lipid peroxidation (P<0.01) and protein
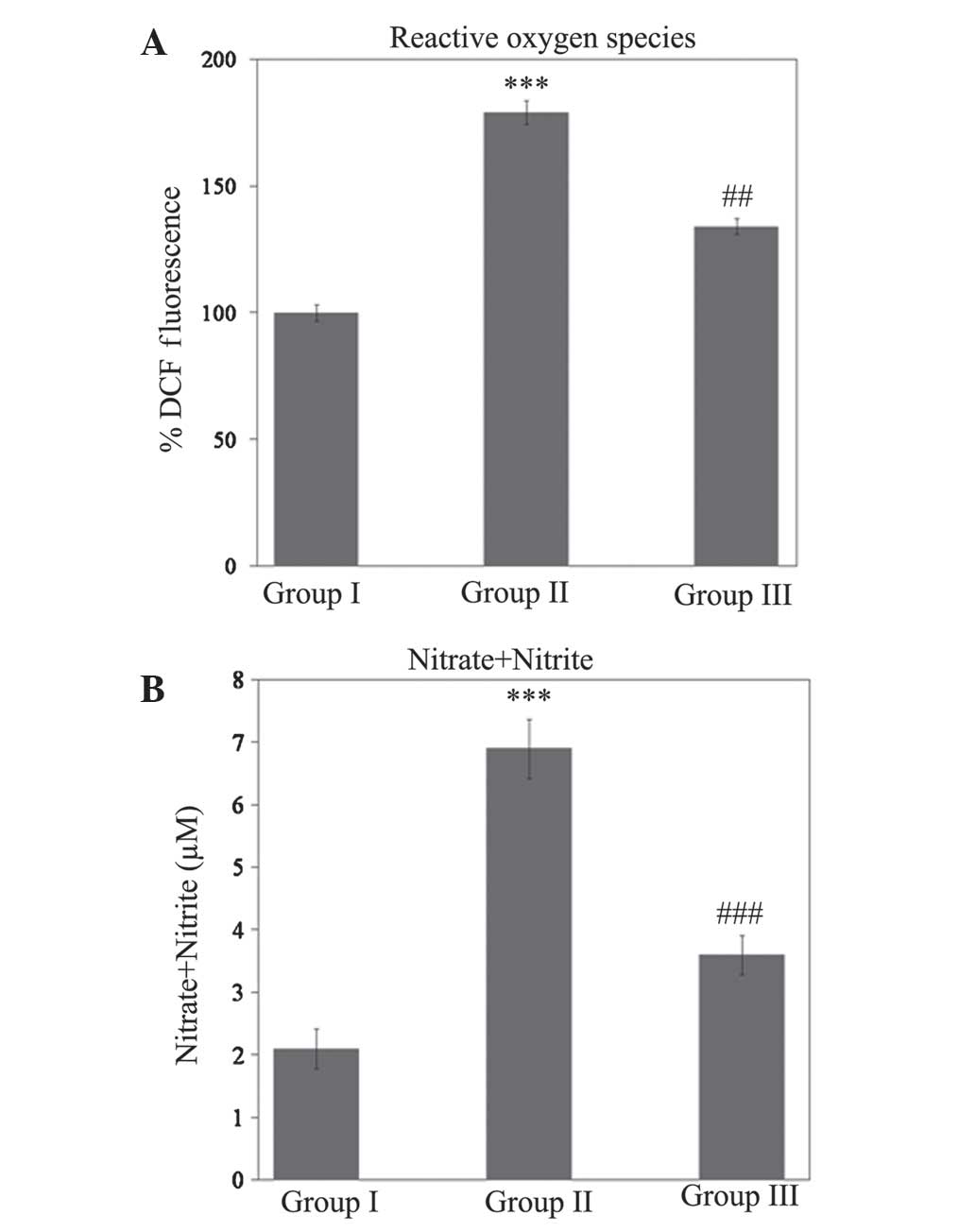
carbonylation (P<0.001). As shown in Fig. 2, a significant increase
(P<0.001) in ROS and nitrite levels during SCI injury was
ameliorated (P<0.01 and P<0.001 for ROS and nitrite levels,
respectively) during treatment with gallic acid (Fig. 2).
Gallic acid enhances antioxidant status
and prevents SCI in Wistar rats
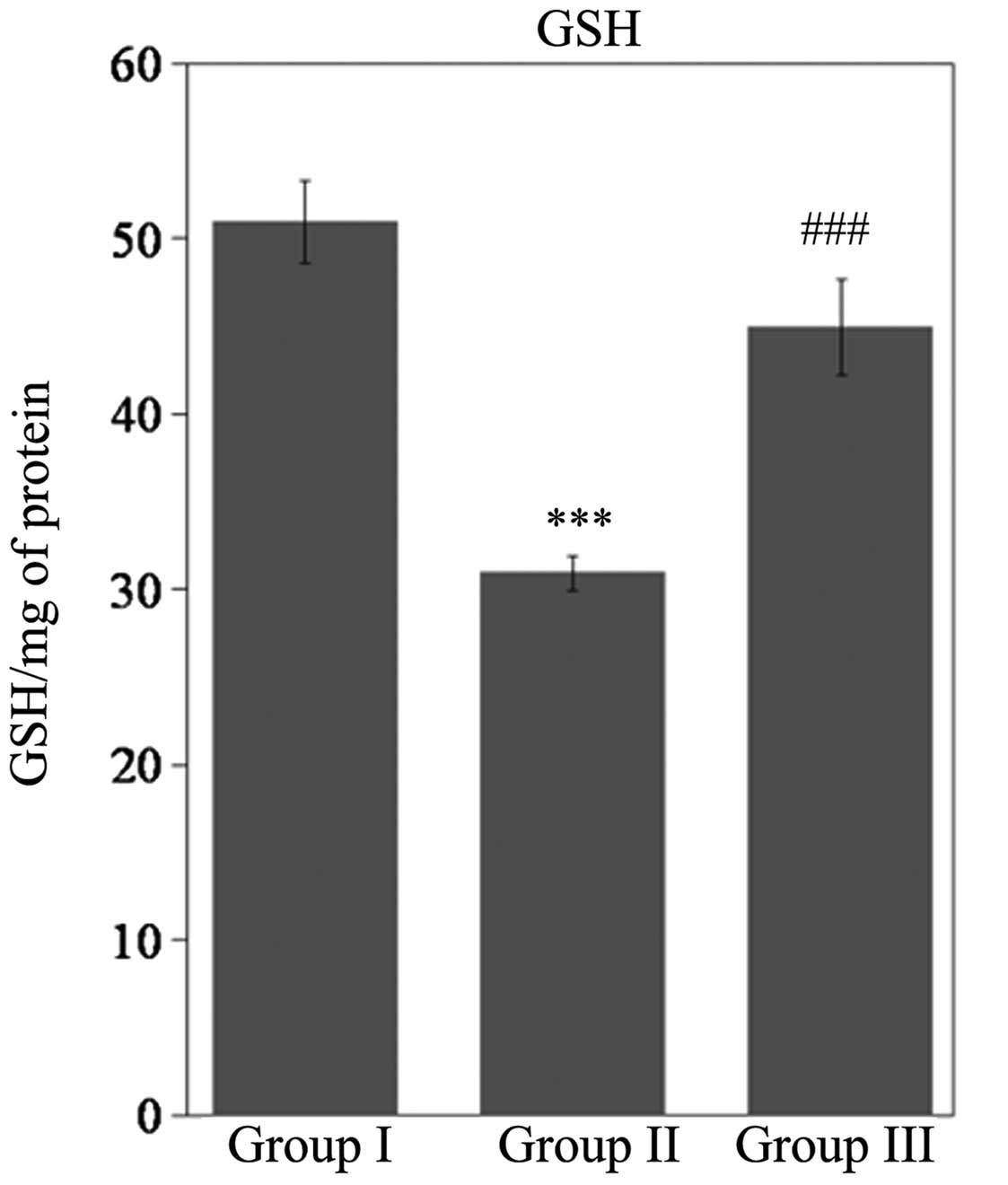
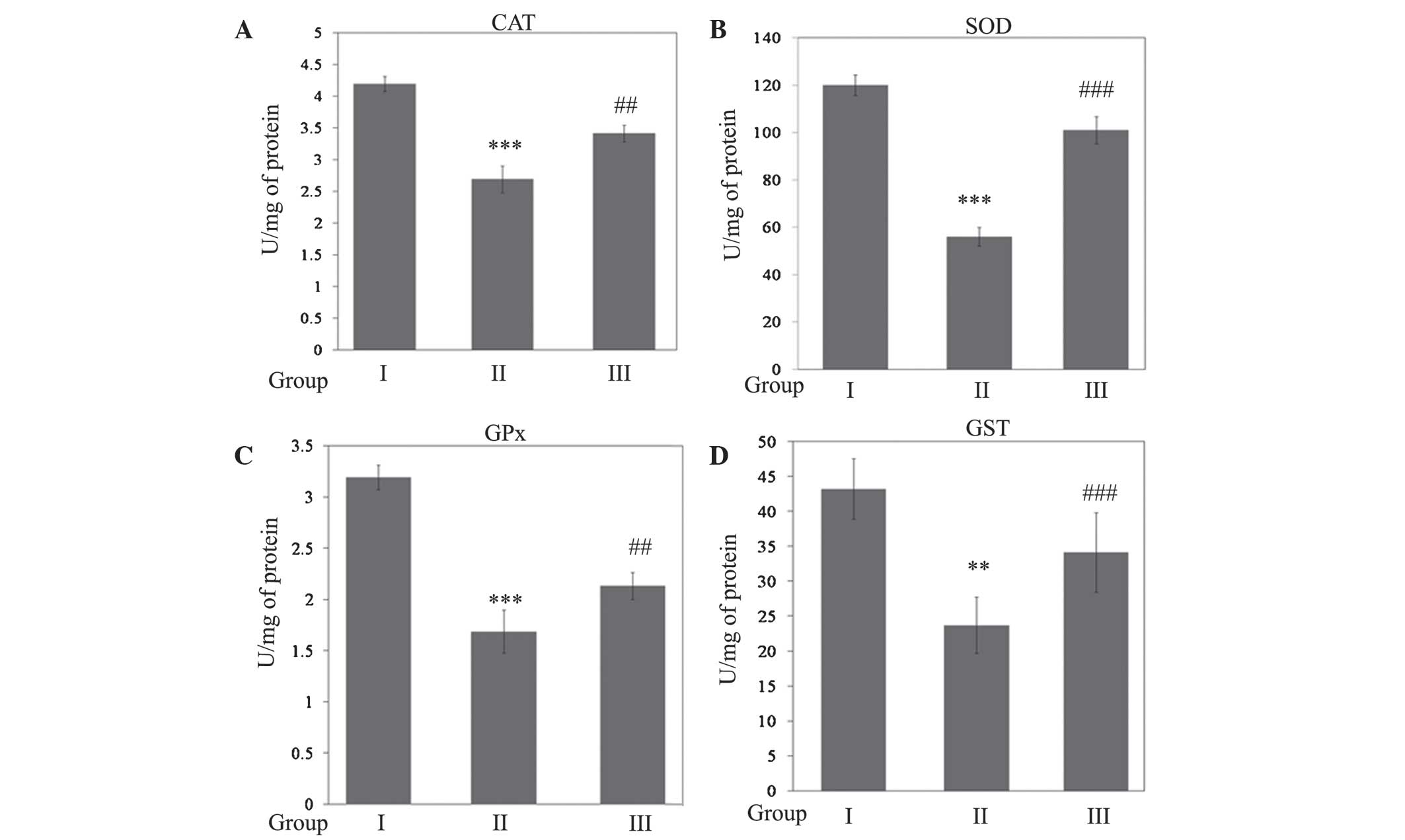
Activity levels of GSH, CAT, SOD, GPx (P<0.001)
and GST (P<0.01) were significantly reduced during SCI in Wistar
rats (Figs. 3 and 4). However, rats treated with gallic acid
followed by SCI injury exhibited a statistically significant
increase (P<0.001) when compared with SCI rats. In response to
this decline in antioxidant status, the levels of GSH (P<0.001),
SOD (P<0.001), CAT (P<0.01), GPx (P<0.01) and GST
(P<0.001) were significantly increased in rats treated with
gallic acid followed by SCI when compared with SCI rats (Fig. 4).
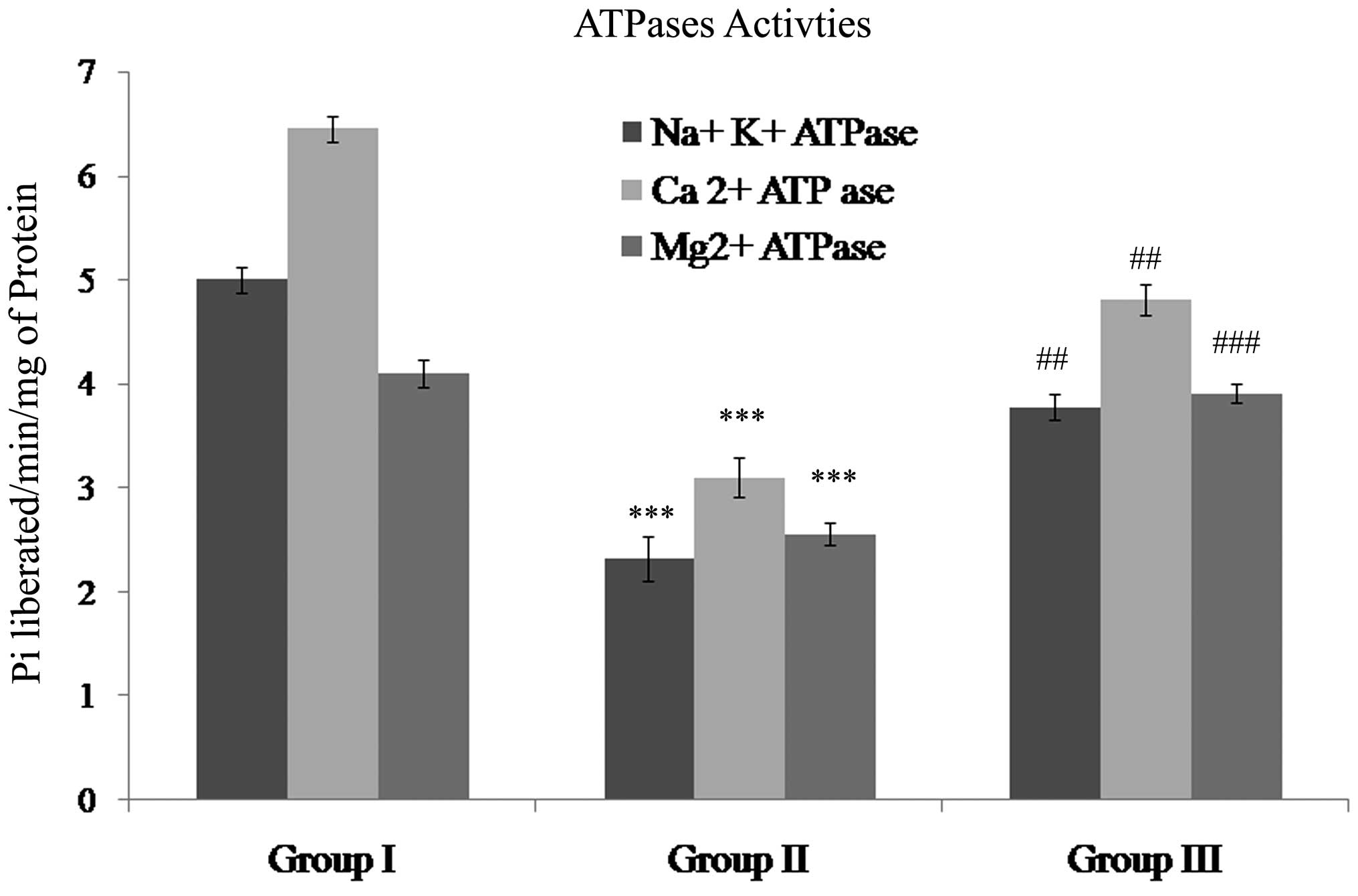
Gallic acid restored the activity of the
Na+/K+, Ca2+ and Mg2+
ATPases during SCI
The activities of the Na+/K+,
Ca2+ and Mg2+ ATPases in SCI were identified
to be significantly reduced (P<0.001) when compared with control
rats. Treatment with gallic acid showed statistically significant
increases in the activities of the Na+/K+
(P<0.01), Ca2+ (P<0.01) and Mg2+
(P<0.001) ATPases compared with those in the SCI group (Fig. 5).
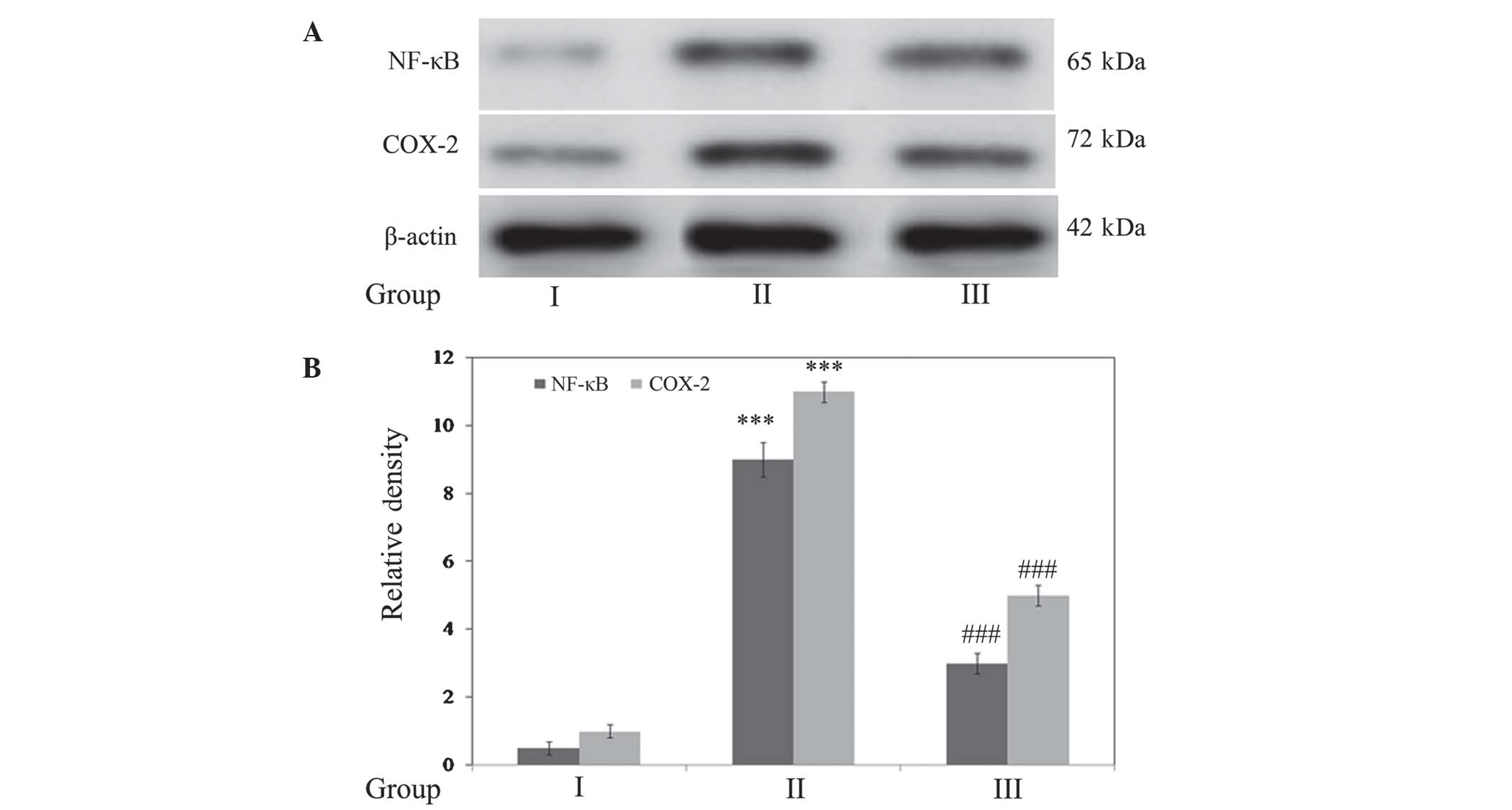
Gallic acid inhibits SCI-induced
inflammatory responses
The expression levels of the inflammatory markers
NF-κB and COX-2 were determined. Densitometric analysis
demonstrated that expression of these markers was significantly
upregulated (P<0.001) in SCI rats compared with the control. In
addition, treatment with gallic acid significantly downregulated
(P<0.001) the expression of NF-κB and COX-2 when compared with
SCI rats (Fig. 6).
Discussion
The key mediator of persistent SCI is secondary
injury, which occurs as a result of physical and conducive damage
to the spinal cord. Within minutes to hours of the initiation of
primary mechanical injury, the secondary effect is initiated and
the adverse effects which occur are proportional to that of the
primary injury (5). The
degenerative events which occur as a result of secondary injury are
mediated through the activation of various signaling cascades,
disturbances in ion homeostasis and the production of ROS.
Oxidative stress is key in the production of tissue damage
(27–30). Thus, spinal cord damage and
subsequent loss of neurological function occurs via
apoptosis/necrosis. As secondary damage has been reported to be
mediated by oxidative stress and its downstream events,
pharmacological intervention with free radical scavengers is
suggested to be able to efficiently block this oxidation-induced
tissue damage. The present study demonstrated promising results,
identifying that gallic acid, a potential antioxidant compound,
prevented SCI via pharmacological intervention.
The measurement of TAC and TOS is a widely used
marker for the evaluation of the total oxidative stress status
during injury. The current study observed that treatment with the
antioxidant gallic acid was able to significantly prevent the total
oxidant levels in serum and the associated increase in total
antioxidant capacity during SCI. Previous studies have reported
that free radical-mediated loss of neurological function is a key
event in SCI (31,32). Thus, the present study evaluated
various markers of oxidative stress, which resulted in the
observation that SCI led to a significant rise in ROS generation.
The reaction between various reactive oxygen and nitrogen species
formed during stress conditions results in the formation of
peroxynitrite radicals. Reports suggest that these peroxy radicals
have critical roles in the initiation of mechanisms mediating lipid
peroxidation (31).
Lipid peroxidation is a complex process resulting in
the damage of lipids, which alters the cellular membranes and
ultimately cellular function. These oxidative alterations in the
membrane lipids are irreversible. Lipid peroxidation and the
resulting oxidative damage to lipids occurs as a result of the
insertion of an oxygen molecule through enzymatic or non-enzymatic
mechanisms (33). The impact of
lipid peroxi-dation leads to alterations in membrane fluidity,
permeability, in addition to the loss of cell-cell contacts.
Alterations to the membrane lipids result in an aggravation of
oxidative stress and pro-inflammatory mechanisms (34). The toxic byproducts of lipid
peroxidation have been reported to mediate carcinogenic and
mutagenic effects (35). Thus,
complex interactions between lipid peroxides, ROS and RNS molecules
are suggested to result in the loss of neurological function. In
addition, these peroxides react with specific amino acids in
proteins such as arginine, cysteine and lysine to form
carbonylation proteins, resulting in the loss of protein function
(33). Various studies have
demonstrated that lipid peroxidation serves a crucial role in SCI
(32,36,37).
The present study identified that tissue levels of ROS, lipid
peroxides, protein carbonylation products and nitrites were
significantly increased during SCI, and that these effects were
ameliorated by gallic acid treatment, which resulted in the
decreased expression of these oxidative stress markers.
Normal cellular functions are controlled by cell
through maintaining the oxidant and antioxidant balance. However,
in cases of extreme oxidative stress, cells lose control over this
balance, which results in increased oxidative stress with depletion
of the antioxidant status (4).
Several non-enzymatic and enzymatic antioxidants are involved in
the regulation of oxidative stress. GSH is an endogenous
antioxidant, which commonly acts as a first line of defense against
stress conditions. The reaction between ROS and GSH during stress
oxidizes and inactivates GSH (38); oxidative damage overwhelms cellular
antioxidant levels, which results in tissue injury. In addition to
GSH, cells also contain various enzymatic antioxidants, including
CAT, SOD, GPx and GST, which protect against cellular damage. The
present study observed a significant reduction in the activities of
the antioxidant status during SCI. However, gallic acid treatment
enhanced antioxidant levels through increasing the activities of
non-enzymatic and enzymatic antioxidant activities. Kim et
al (39) demonstrated that
curcumin treatment enhanced the plasma antioxidant status of cells
and reduced lipid peroxidation levels during in acute SCI. Enhanced
GSH levels and inhibition of MDA levels previously were reported in
rats treated with oleuropein during SCI (40). The protective effects of gallic
acid mediated by the upregulation of antioxidant mechanisms have
been previously reported in various oxidative stress conditions
(41–43).
Oxidative stress is closely associated with the
activation of inflammatory genes, as ROS is a key mediator in these
events (44). Initiation of
inflammation largely occurs with activation of NF-κB followed by
downstream genes, including those for COX-2, inducible nitric oxide
synthase and various interleukins and cytokines (45). The present study demonstrated the
significant upregulation of inflammatory proteins, such as NF-κB
and COX-2, during SCI injury in rats; in addition, gallic acid
exhibited anti-inflammatory effects by downregulating these
proteins. These results are consistent with previous studies, where
SCI induced inflammatory mechanisms via regulation of COX-2, nitric
oxide levels and the release of prostaglandins (46,47).
Similar protection by gallic acid through inhibition of the NF-κB
protein was demonstrated in the prevention of cancer (48) and lipopolysaccharide-induced
inflammation (49).
Previous studies have identified that SCI resulted
in the deregulation of ion homeostasis, including calcium ion
influx and subsequent calcium overload (50,51).
Calcium overload results in the activation of various biochemical
cascades and signaling mechanisms, which have been demonstrated to
lead to the adverse effects of oxidative stress and inflammatory
responses, resulting in tissue damage and loss of normal cell
function (50). Previous studies
have also indicated that early events in SCI induce depolarization
of the membranes, which results in the opening of various ion
channels (52). These ion
concentrations are maintained by membrane bound ATPase enzymes;
however, as these enzymes are inactivated under oxidative stress,
alterations in lipid composition occur (53). The present study observed extensive
oxidative stress during SCI; therefore, it was investigated whether
SCI results in any alterations in the activity of ATPase enzymes.
This was achieved by evaluating the activities of the
Na+/K+, Ca2+ and Mg2+
ATPases during SCI. The results demonstrated a significant
reduction in ATPase activity; in addition, treatment with gallic
acid restored the enzyme activities and maintained normal ion
homeostasis. This was consistent with a previous study by Vijaya
Padma et al (54), which
identified that gallic acid treatment significantly maintained the
antioxidant status and ATPase activity, thereby preventing
cardiotoxicty in Wistar rats.
In conclusion, the results of the present study
provided evidence that pharmacological intervention with gallic
acid prevented and restored SCI-induced oxidative stress and ion
homeostasis.
References
|
1
|
Amar AP and Levy ML: Pathogenesis and
pharmacological strategies for mitigating secondary damage in acute
spinal cord injury. Neurosurgery. 44:1027–1039. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hall ED: The role of oxygen radicals in
traumatic injury: clinical implications. J Emerg Med. 11(Suppl 1):
31–36. 1993.PubMed/NCBI
|
|
3
|
Hall ED: Lipid antioxidants in acute
central nervous system injury. Ann Emerg Med. 22:1022–1027. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hall ED: Antioxidant therapies for acute
spinal cord injury. Neurotherapeutics. 8:152–167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li L, Ng TB, Gao W, Li W, Fu M, Niu SM,
Zhao L, Chen RR and Liu F: Antioxidant activity of gallic acid from
rose flowers in senescence accelerated mice. Life Sci. 77:230–240.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ng TB, He JS, Niu SM, Zhao L, Pi ZF, Shao
W and Liu F: A gallic acid derivative and polysaccharides with
antioxidative activity from rose (Rosa rugosa) flowers. J Pharm
Pharmacol. 56:537–545. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giftson JS, Jayanthi S and Nalini N:
Chemopreventive efficacy of gallic acid, an antioxidant and
anticarcinogenic polyphenol, against 1,2-dimethyl hydrazine induced
rat colon carcinogenesis. Invest New Drugs. 28:251–259. 2010.
View Article : Google Scholar
|
|
8
|
Kroes BH, van den Berg AJ, Quarles van
Ufford HC, van Dijk H and Labadie RP: Anti-inflammatory activity of
gallic acid. Planta Med. 58:499–504. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pellegrina CD, Padovani G, Mainente F,
Zoccatelli G, Bissoli G, Mosconi S, Veneri G, Peruffo A,
Andrighetto G, Rizzi C and Chignola R: Anti-tumour potential of a
gallic acid-containing phenolic fraction from Oenothera biennis.
Cancer Lett. 226:17–25. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hsu CL, Lo WH and Yen GC: Gallic acid
induces apoptosis in 3T3-1 pre-adipocytes via a Fas- and
mitochondrial-mediated pathway. J Agric Food Chem. 55:7359–7365.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Inoue M, Suzuki R, Sakaguchi N, Li Z,
Takeda T, Ogihara Y, Jiang BY and Chen Y: Selective induction of
cell death in cancer cells by gallic acid. Biol Pharm Bull.
18:1526–1530. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Patel SS and Goyal RK: Cardioprotective
effects of gallic acid in diabetes-induced myocardial dysfunction
in rats. Pharmacognosy Res. 3:239–245. 2011. View Article : Google Scholar
|
|
13
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
14
|
Erel O: A novel automated method to
measure total antioxidant response against potent free radical
reactions. Clin Biochem. 37:112–119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Erel O: A new automated colorimetric
method for measuring total oxidant status. Clin Biochem.
38:1103–1111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levine RL, Garland D, Oliver CN, Amici A,
Climent I, Lenz AG, Ahn BW, Shaltiel S and Stantman ER:
Determination of carbonyl content in oxidatively modified proteins.
Methods Enzymol. 186:464–478. 1990.PubMed/NCBI
|
|
17
|
Esterbauer H and Cheeseman KH:
Determination of aldehydic lipid peroxidation products:
malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 186:407–421.
1990.PubMed/NCBI
|
|
18
|
Hashimoto M, Tanabe Y, Fujii Y, Kikuta T,
Shibata H and Shido O: Chronic administration of docosahexaenoic
acid ameliorates the impairment of spatial cognition learning
ability in amyloid beta-infused rats. J Nutr. 135:549–555.
2005.PubMed/NCBI
|
|
19
|
Sun Y, Oberley LW and Li Y: A simple
method for clinical assay of superoxide dismutase. Clin Chem.
34:497–500. 1988.PubMed/NCBI
|
|
20
|
Aebi H: Catalase. Methods of Enzymatic
Analysis. Bergmeyer U: Academic Press; New York: pp. 673–677.
1974
|
|
21
|
Habig WH, Pabst MJ and Jakoby WB:
Glutathione S-transferases. The first enzymatic step in mercapturic
acid formation. J Biol Chem. 249:7130–7139. 1974.PubMed/NCBI
|
|
22
|
Paglia DE and Valentine WN: Studies on the
quantitative and qualitative characterisation of erythrocyte
glutathione peroxidase. J Lab Clin Med. 70:158–169. 1967.PubMed/NCBI
|
|
23
|
Bonting SL: Sodium-potassium activated
adenosine triphosphatase and cation transport. Membranes and Ion
Transport. Bittar EE: Wiley-Interscience; London: pp. 257–263.
1970
|
|
24
|
Ohnishi T, Suzuki T, Suzuki Y and Ozawa K:
A comparative study of plasma membrane Mg2+ -ATPase activities in
normal, regenerating and malignant cells. Biochim Biophys Acta.
684:67–74. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hjertén S and Pan H: Purification and
characterization of two forms of a low-affinity Ca2+ -ATPase from
erythrocyte membranes. Biochim Biophys Acta. 728:281–288. 1983.
View Article : Google Scholar
|
|
26
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: procedure and some applications. Proc Natl
Acad Sci USA. 76:4350–4354. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hall ED and Braughler JM: Central nervous
system trauma and stroke. II Physiological and pharmacological
evidence for involvement of oxygen radicals and lipid peroxidation.
Free Radic Biol Med. 6:303–313. 1989. View Article : Google Scholar
|
|
28
|
Tator CH and Fehlings MG: Review of the
secondary injury theory of acute spinal cord trauma with emphasis
on vascular mechanisms. J Neurosurg. 75:15–26. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Faden AI: Therapeutic approaches to spinal
cord injury. Adv Neurol. 72:377–386. 1997.PubMed/NCBI
|
|
30
|
Faden AI and Salzman S: Pharmacological
strategies in CNS trauma. Trends Pharmacol Sci. 13:29–35. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hall ED and Braughler JM: Free radicals in
CNS injury. Res Publ Assoc Res Nerv Ment Dis. 71:81–105.
1993.PubMed/NCBI
|
|
32
|
Koc RK, Akdemir H, Karakücük EI, Oktem IS
and Menkü A: Effect of methylprednisolone, tirilazad mesylate and
vitamin E on lipid peroxidation after experimental spinal cord
injury. Spinal Cord. 37:29–32. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Halliwell B and Gutteridge JM: Oxidative
stress. Free Radicals in Biology and Medicine. 3rd. Oxford
University Press; New York: pp. 246–350. 1999
|
|
34
|
Greenberg ME, Li XM, Gugiu BG, Gu X, Qin
J, Salomon RG and Hazen SL: The lipid Whisker model of the
structure of oxidized cell membranes. J Biol Chem. 283:2385–2396.
2008. View Article : Google Scholar
|
|
35
|
West JD and Marnett LJ: Endogenous
reactive intermediates as modulators of cell signaling and cell
death. Chem Res Toxicol. 19:173–194. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Christie SD, Comeau B, Myers T, Sadi D,
Purdy M and Mendez I: Duration of lipid peroxidation after acute
spinal cord injury in rats and the effect of methylprednisolone.
Neurosurg Focus. 25:E52008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Diaz-Ruiz A, Rios C, Duarte I, Correa D,
Guizar-Sahagun G, Grijalva I, Madrazo I and Ibarra A: Lipid
peroxidation inhibition in spinal cord injury: cyclosporin-A vs.
methylprednisolone. Neuroreport. 11:1765–1767. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Price A, Lucas PW and Lea PJ: Age
dependent damage and glutathione metabolism in ozone fumigated
barley: a leaf section approach. J Exp Bot. 41:1309–1317. 1990.
View Article : Google Scholar
|
|
39
|
Kim KT, Kim MJ, Cho DC, Park SH, Hwang JH,
Sung JK, Cho HJ and Jeon Y: The neuroprotective effect of treatment
with curcumin in acute spinal cord injury: laboratory
investigation. Neurol Med Chir (Tokyo). 54:387–394. 2014.
View Article : Google Scholar
|
|
40
|
Khalatbary AR and Ahmadvand H:
Neuroprotective effect of oleuropein following spinal cord injury
in rats. Neurol Res. 34:44–51. 2012. View Article : Google Scholar
|
|
41
|
Nabavi SF, Habtemariam S, Jafari M, Sureda
A and Nabavi SM: Protective role of gallic acid on sodium fluoride
induced oxidative stress in rat brain. Bull Environ Contam Toxicol.
89:73–77. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ramkumar K, Vijayakumar R, Vanitha P,
Suganya N, Manjula C, Rajaguru P, Sivasubramanian S and Gunasekaran
P: Protective effect of gallic acid on alloxan-induced oxidative
stress and osmotic fragility in rats. Hum Exp Toxicol. 33:638–649.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mansouri MT, Farbood Y, Sameri MJ, Sarkaki
A, Naghizadeh B and Rafeirad M: Neuroprotective effects of oral
gallic acid against oxidative stress induced by 6-hydroxydopamine
in rats. Food Chem. 138:1028–1033. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kurtoglu T, Basoglu H, Ozkisacik EA, Cetin
NK, Tataroglu C, Yenisey C and Discigil B: Effects of cilostazol on
oxidative stress, systemic cytokine release, and spinal cord injury
in a rat model of transient aortic occlusion. Ann Vasc Surg.
28:479–488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Carroll JE, Hess DC, Howard EF and Hill
WD: Is nuclear factor-kappaB a good treatment target in brain
ischemia/reperfusion injury? Neuroreport. 11:R1–4. 2000.PubMed/NCBI
|
|
46
|
Maihöfner C, Schlötzer-Schrehardt U,
Gühring H, Zeilhofer HU, Naumann GO, Pahl A, Mardin C, Tamm ER and
Brune K: Expression of cyclooxygenase-1 and -2 in normal and
glauco-matous human eyes. Invest Ophthalmol Vis Sci. 42:2616–2624.
2001.
|
|
47
|
Yamamoto T and Nozaki-Taguchi N: Analysis
of the effects of cyclooxygenase (COX)-1 and COX-2 in spinal
nociceptive transmission using indomethacin, a non-selective COX
inhibitor and NS-398, a COX-2 selective inhibitor. Brain Res.
739:104–110. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morais MC, Luqman S, Kondratyuk TP,
Petronio MS, Regasini LO, Silva DH, Bolzani VS, Soares CP and
Pezzuto JM: Suppression of TNF-α induced NFκB activity by gallic
acid and its semi-synthetic esters: possible role in cancer
chemoprevention. Nat Prod Res. 24:1758–1765. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Choi KC, Lee YH, Jung MG, Kwon SH, Kim MJ,
Jun WJ, Lee J, Lee JM and Yoon HG: Gallic acid suppresses
lipopolysaccharide-induced nuclear factor-kappaB signaling by
preventing RelA acetylation in A549 lung cancer cells. Mol Cancer
Res. 7:2011–2021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bains M and Hall ED: Antioxidant therapies
in traumatic brain and spinal cord injury. Biochim Biophys Acta.
1822:675–684. 2012. View Article : Google Scholar
|
|
51
|
Fleming JC, Norenberg MD, Ramsay DA,
Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD
and Weaver LC: The cellular inflammatory response in human spinal
cords after injury. Brain. 129:3249–3269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Oyinbo CA: Secondary injury mechanisms in
traumatic spinal cord injury: A nugget of this multiply cascade.
Acta Neurobiol Exp (Wars). 71:281–299. 2011.
|
|
53
|
Carageorgiou H, Tzotzes V, Pantos C,
Mourouzis C, Zarros A and Tsakiris S: In vivo and in vitro effects
of cadmium on adult rat brain total antioxidant status,
acetylcholinesterase, (Na+, K+)-ATPase and Mg2+-ATPase activities:
protection by L-cysteine. Basic Clin Pharmacol Toxicol. 94:112–118.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Vijaya Padma V, Poornima P, Prakash C and
Bhavani R: Oral treatment with gallic acid and quercetin alleviates
lindane-induced cardiotoxicity in rats. Can J Physiol Pharmacol.
91:134–140. 2013. View Article : Google Scholar : PubMed/NCBI
|