Introduction
Congenital heart disease (CHD) is the most prevalent
neonatal disorder in humans; it is the leading cause of
non-infectious infant mortality and affects 4-10 out of every 1,000
live births (1-3). Ventricular septal defect (VSD) is the
most common type of CHD and is present in 30-50% of all cases of
CHD (2,4,5). The
pathogenesis of VSD is multifactorial, with genetic and
environmental factors having important roles (6-7).
Although the embryology and physiology of VSD are widely known, its
etiology and underlying molecular mechanism have remained
elusive.
TWIST1 belongs to the helix-loop-helix (bHLH)
family of transcription factors, is highly conserved and is
involved in embryonic development (8,9). An
animal experiments showed that primary chicken which had lost
TWIST1 gene expression presented with impaired endocardial
cushion development (10). A study
on Twist1-null mice revealed that Twist1 is a novel
bHLH within the cardiac outflow tract (OFT) development (11). In humans, although growing evidence
suggests that TWIST1 is an important tumor biomarker
(12-14), little is known about this gene’s
involvement in human heart development, particularly its
implication in VSD.
Based on previous studies, the present study
hypothesized that TWIST1 may have an important role in the
development of VSD. Mutation or loss of numerous types of single
genes have been predicted to result in VSDs (15–18);
however, the underlying molecular mechanism by which reduced
expression of a certain gene or a transcription factor may cause
defects in cardiac septation, particularly VSDs, has remained
elusive. The aims of the present study were to determine the
differential expression of TWIST1 mRNA and protein levels in
normal and VSD human fetal heart tissues and correlate any observed
changes to the etiology of VSD.
Materials and methods
Tissue samples
All fetal myocardial tissues were obtained from the
Department of Obstetrics and Gynecology at Shengjing Hospital of
China Medical University (Shenyang, China) from October 2011 to
June 2013. All subjects with chromosomal disorders, for example
22q11 deletion syndrome and 21-trisomy syndrome (19,20),
were excluded. The 26 VSD specimens were diagnosed prenatally with
CHDs using three-dimensional echocardiography. The diagnoses were
confirmed by pathological autopsy. Myocardial tissue samples from
12 normal fetuses at 22-28 weeks of gestation were collected during
surgery for pregnancy termination due to spontaneous abortion or
trauma to the pregnant women. All tissue samples were snap frozen
in liquid nitrogen and then stored at -80°C until use in order to
exclude any tissue heterogeneity that may affect results. All fetal
myocardial tissues (with or without VSD) were sampled using the
same protocols. Each pregnant woman or her relatives provided
written informed consent; the Ethics Committee of the National
Research Institute for Family Planning (Shenyang, China) approved
the study. The study protocol followed the principles of the
Declaration of Helsinki. The characteristics of the study subjects
are shown in Tables I and II.
 | Table IClinical classification of VSD in the
subjects. |
Table I
Clinical classification of VSD in the
subjects.
| Group | VSD sub-type of the
sample | Sample number |
|---|
| VSD | Sub-arterial
VSD | S188 S1015 S1000
S211 |
| Perimembranous
VSD | M1005 M1014 M900
M1009 M501 M1001 |
| M198 M1012 M197
M204 M1007 M194 |
| Muscular VSD | A1004 A203 A208
A1006 A1002 |
| Complete
ventricular septal agenesis (functionally univentricular) | CV191 CV183 CV189
CV1013 CV1003 |
| Normal | – | N216 N178 N175 N176
N196 N199 N209 |
| – | N180 N170 N172 N187
N904 |
| Table IIClinical characteristics of the study
population. |
Table II
Clinical characteristics of the study
population.
| Characteristic | VSD (n=26) | Normal (n=12) | Univentricular |
|---|
| Male, n (%) | 19 (73.1) | 7 (58.3) | 5 (100) |
| Female, n (%) | 7 (26.9) | 5 (41.7) | 0 |
RNA isolation and cDNA synthesis
The TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) was used to extract total RNA from 50 mg heart
tissue. A NanoDrop™ 2000 spectrophotometer (Thermo Fisher
Scientific, Waltham, MA, USA) was used to measure the RNA
concentration. The 260/280 nm absorbance ratios of the RNA samples
ranged from 1.91 to 2.03 and they were stored at −80°C until use.
An aliquot (2 μg) of RNA from each sample was reverse
transcribed into cDNA using the PrimeScript™ II 1st strand cDNA
Synthesis kit (Takara, Dalian, China) according to the
manufacturer’s instructions.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
The Sangon Company (Beijing, China) designed the
primers, which were used according to the manufacturer’s
instructions. Details of the primers are listed in Table III. Taq DNA polymerase, 10X Taq
Buffer, desoxyribonucleotide triphosphate mixture and cDNA template
that was reverse transcribed from a previous reaction (all from
Tiangen Biotech, Beijing, China) were used to amplify cDNA (100
ng/l) in vitro. RT-PCR was performed as follows: 94°C for 5
min; 35 cycles of 94°C for 30 sec, 60°C for 30 sec and 72°C for 45
sec; 72°C for 10 min using a Bio-Rad S1000 Thermal Cycler system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). β-actin was used
as an internal standard. PCR products were analyzed by
electrophoresis on 2% agarose gels.
| Table IIIPrimers used in the present
study. |
Table III
Primers used in the present
study.
| Gene | Forward primer | Reverse primer | Product length
(bp) |
|---|
| TWIST1
(human) |
5′-GGAGTCCGCAGTCTTACGAG-3′ |
5′-TCTGGAGGACCTGGTAGAGG-3′ | 211 |
| β-actin
(human) |
5′-TCGTGCGTGACATTAAGGAG-3′ |
5′-ATGCCAGGGTACATGGTGGT-3′ | 178 |
Real-time PCR
Real-time PCR to quantify gene expression was
performed using an ABI Prism 7000 Sequence Detection System
(Applied Biosystems, Life Technologies, Thermo Fisher Scientific)
with SYBR Premix Ex Taq™ II (Takara). The amplification conditions
were: 95°C for 30 sec; 40 cycles of 95°C for 5 sec and 58°C for 34
sec. The levels of β-actin mRNA were used to normalize the
expression of the target genes. The 2−ΔΔCt method was
used to compare differences in relative gene expression between the
controls and VSD samples. Each measurement was repeated three
times. A melting curve of the reaction products yielded a single
peak in each experiment.
Western blot analysis
Frozen heart tissues from all samples were lysed in
radioimmunoprecipitation buffer (C1053; Applygen Technologies Inc,
Beijing, China) together with a protease inhibitor (P1265; Applygen
Technologies). Total soluble protein lysates (80 μg) were
boiled for 10 min and then subjected to 15% gradient SDS-PAGE
(CW2384; CWBiotech, Beijing, China). After electrophoresis, the
semi-dry transfer system (Bio-Rad Laboratories, Inc.) was used to
transfer the proteins onto nitrocellulose membranes (0.45
μm; Bio-Rad Laboratories, Inc.). After blocking in 5%
non-fat dried milk (Wondersun, Inc., Harbin, China), the
nitrocellulose membranes were incubated with mouse monoclonal
anti-Twist1 (1:500; sc-81417; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and mouse monoclonal anti-β-actin (1:2,000;
HC201-01; TransGen Biotech, Beijing, China) primary antibodies.
After washing in phosphate-buffered saline containing Tween 20
(TBST), the membranes were incubated with goat anti-mouse
immunoglobulin G, horseradish peroxidase conjugate (1:3,000; HS201;
TransGen Biotech). The membranes were washed again in TBST. The
enhanced chemiluminescence detection reagents (CW0049; CWBiotech)
were used to visualize the blots. ImageJ software (version 1.4.8;
National Institute of Health, Bethesda, MD, USA) was used to
quantify the protein levels.
Statistical analysis
Statistical analysis was performed using SPSS
(version 13.0; SPSS Inc., Chicago, IL, US) and GraphPad Prism
(version 5.0; GraphPad Software Inc., La Jolla, CA, USA). The
Mann-Whitney test was used for comparisons between the normal
controls and VSD groups. All statistical analyses were two-sided
and P<0.05 was considered to indicate a statistically
significant difference. Values are expressed as the mean ± the
standard error, typically from two to three independent
experiments, each with three replicates.
Results
Clinical characteristics and evaluation
of VSDs
Pregnant women underwent pre-natal screening using
three-dimensional echocardiography to judge whether the heart of
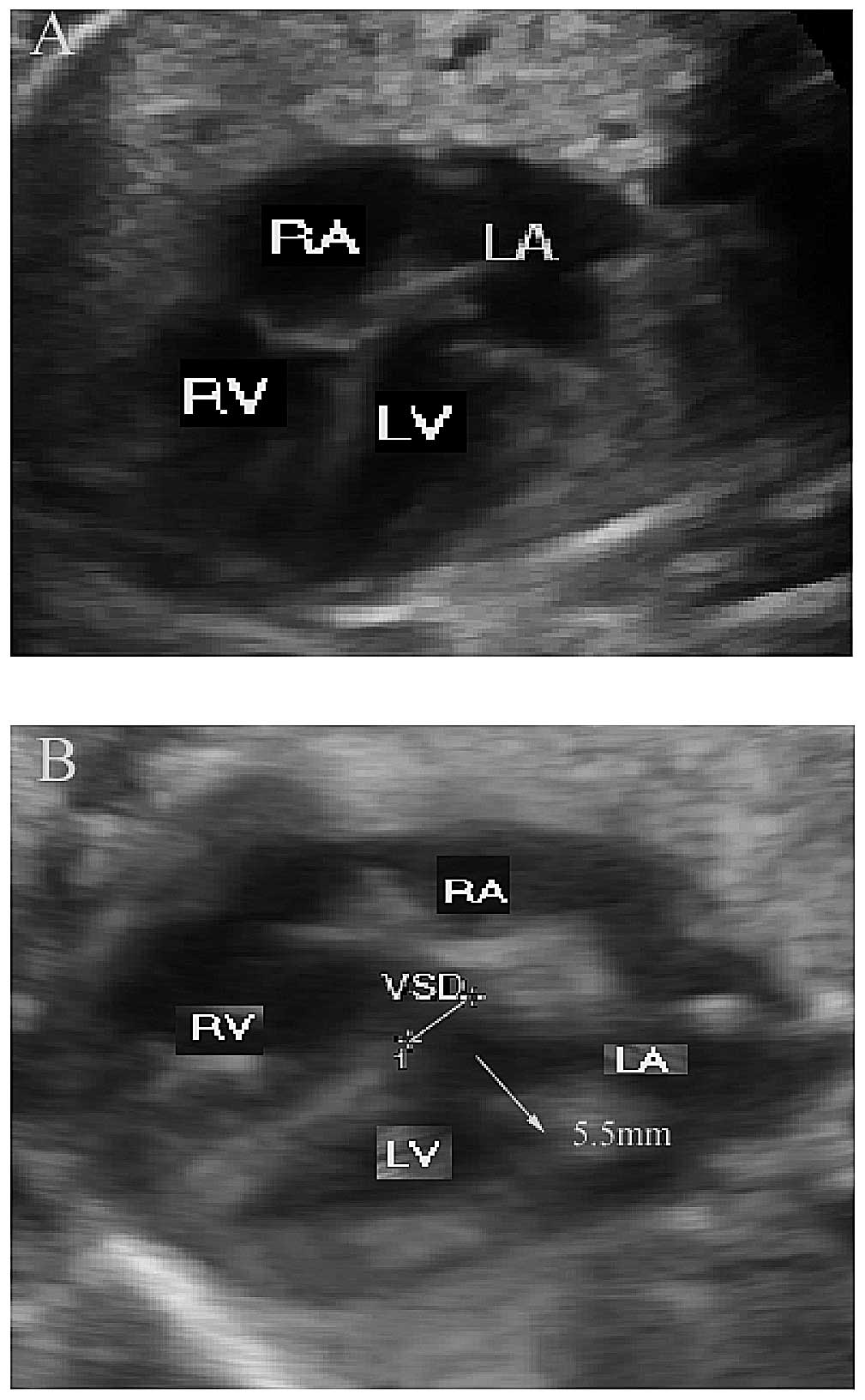
their fetus may be affected by VSD. A normal image of a fetal
four-chambered heart with no VSDs was used as a control (Fig. 1A). The three-dimensional
echocardiography diagnosed 26 fetuses with VSD at 22-28 weeks of
gestation. The sizes of the defects of the 26 VSDs ranged from
4.6-8 mm. The representative image shown in Fig. 1B had a VSD of 5.5 mm. Pathological
autopsy confirmed the diagnosis and gender of the 26 VSD specimens
(73.1% male; 26.9% female). As shown in Table I, 12 cases had defects of the
perimembranous septum, five were muscular VSDs and four were
sub-arterial type VSDs. There were five complete ventricular septal
ageneses among the specimens. As shown in Table II, all five of these complete
ventricular septal ageneses were from male fetuses.
TWIST1 expression in fetal myocardial
tissue
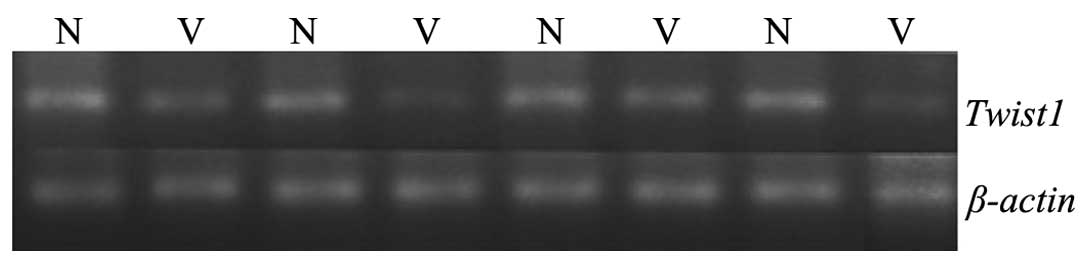
RT-PCR was used to examine the expression of
TWIST1 mRNA in the myocardial tissues of normal fetuses and
those with VSD. Specific primers were designed using the
TWIST1 gene sequence from GenBank and primer specificity was
checked using the basic local alignment search tool (BLAST;
http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome).
TWIST1 mRNA PCR products were not clearly detectable in VSD
samples (n=4), but were clearly present in normal fetal heart
tissues (n=4) (Fig. 2). Details of
the primers are provided in Table
III.
TWIST1 mRNA levels are decreased in
myocardial tissue of fetuses with VSD
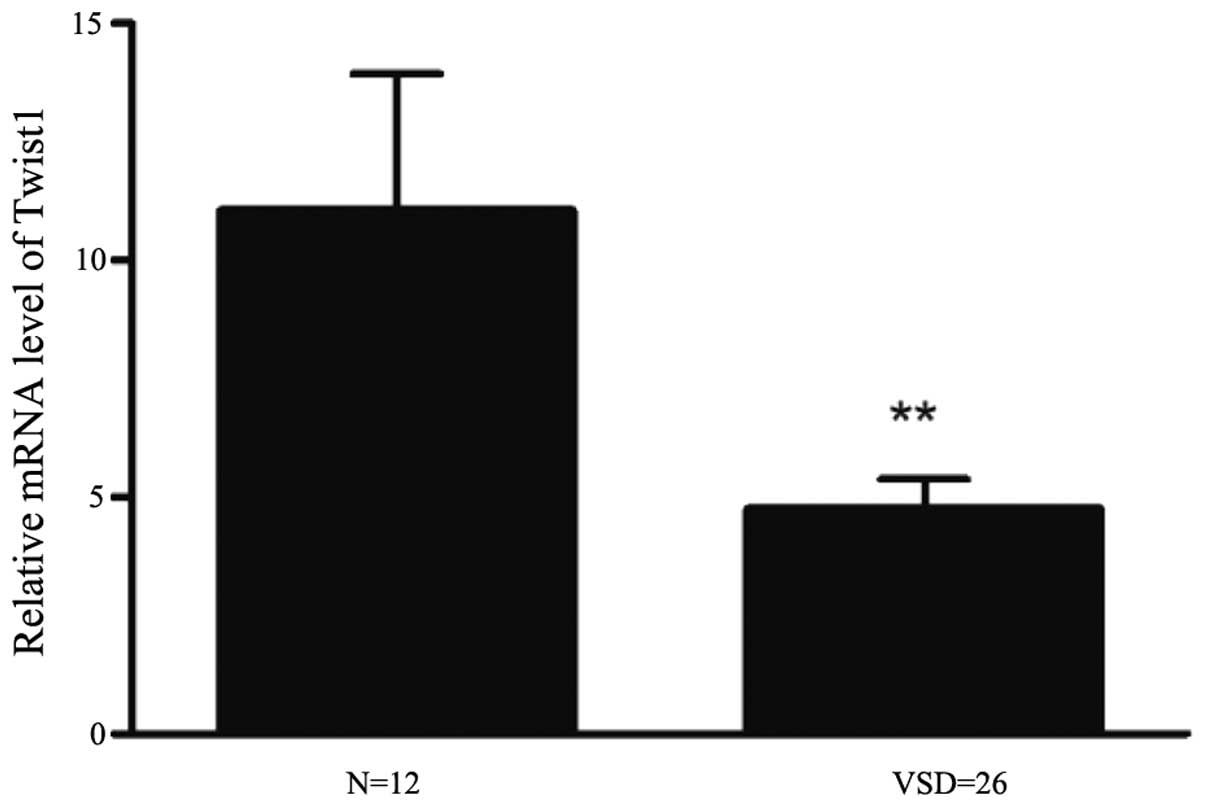
Real-time PCR was used to quantify the relative
differences in expression between TWIST1 mRNA in myocardial
tissues of normal fetuses and those with VSD. The expression of
TWIST1 was two-fold higher in normal tissues compared with
that in VSD tissues (Fig. 3). As
shown in Table IV, TWIST1
mRNA relative expression levels [RQ (2−ΔΔCt) values]
were significantly lower in fetuses with VSD compared with those
from patients with normal fetal myocardial tissues (P<0.01). For
the five complete ventricular septal agenesis samples, similar
results were obtained to those for other VSD fetal myocardial
tissues (P<0.05). These results should be further confirmed
using an increased number of samples.
| Table IVRelative mRNA expression levels of
TWIST1 in samples from controls and patients with VSD or
univentricular septal angenesis. |
Table IV
Relative mRNA expression levels of
TWIST1 in samples from controls and patients with VSD or
univentricular septal angenesis.
| Gene | Control [RQ value
(2−ΔΔCt)a, n=12] | VSD [RQ value
(2−ΔΔCt), n=26] | P-valueb | Univentricular [RQ
value (2−ΔΔCt), n=5] | P-valuec |
|---|
| TWIST1 | 11.04±2.892 | 4.738±0.625 | 0.010 | 3.675±1.126 | 0.039 |
TWIST1 protein levels are decreased in
myocardial tissue of fetuses with VST
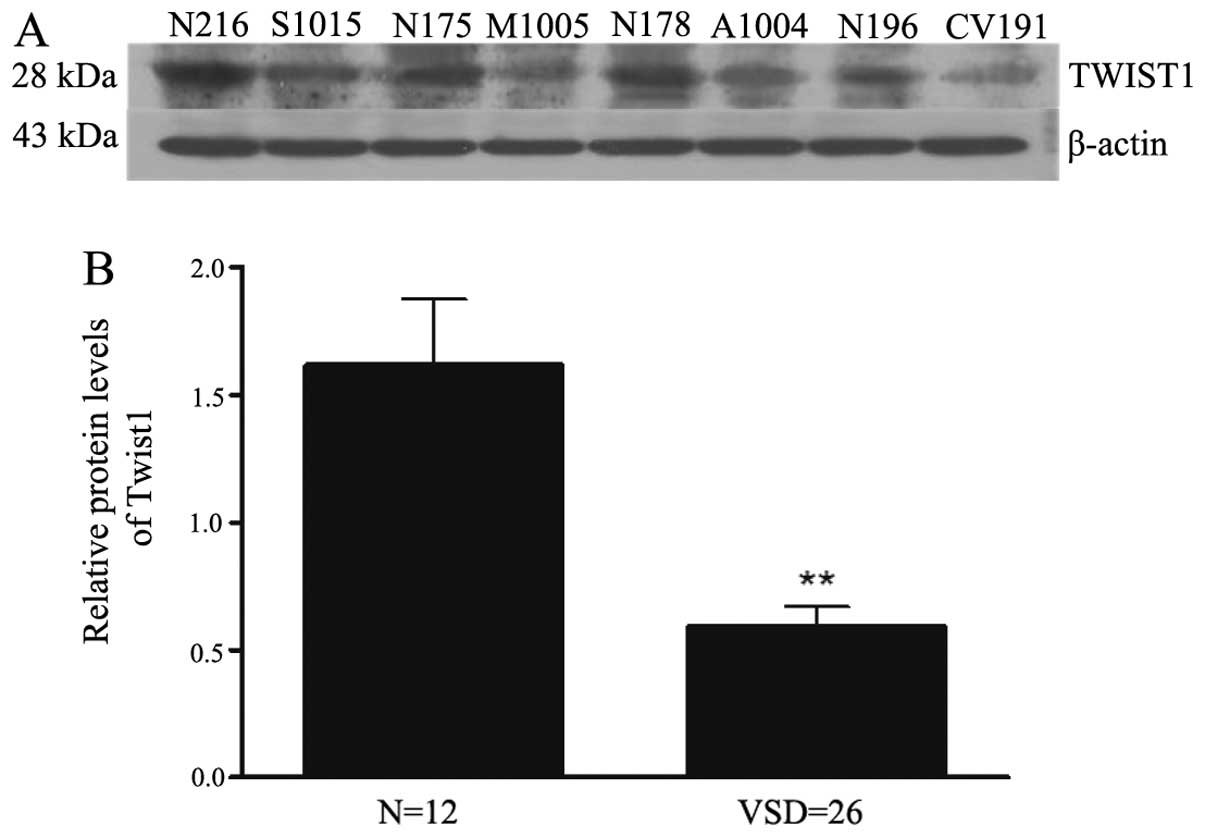
Western blot analysis showed that TWIST1 protein
levels were significantly downregulated in the four fetuses with
different VSD subtypes compared with those from four selected
normal samples (Fig. 4A). To
further confirm these results, an in-depth analysis of the peak
area of the TWIST1 protein band relative to β-actin was performed
using ImageJ software for all samples (12 samples without and 26
samples with VSDs) (Fig. 4B). As
shown in Table V, the results
revealed that TWIST1 expression was decreased by ~three-fold
(P=0.001) in the VSD samples compared with the normal samples.
These data are consistent with the real-time PCR results shown in
Fig. 3 and Table IV.
| Table VRelative protein levels of
TWIST1 in samples from controls and patients with VSD. |
Table V
Relative protein levels of
TWIST1 in samples from controls and patients with VSD.
| Gene | Control (n=12) | VSD (n=26) | P-valuea |
|---|
| TWIST1 | 1.616±0.261 | 0.591±0.078 | 0.001 |
Discussion
With the increased incidence of congenital heart
disease, neonatal disorders have placed a heavy economic burden on
patients and their families, with multiple surgeries required to
correct the numerous anatomical defects (21,22).
VSDs form a large proportion of CHDs and have attracted increasing
attention from researchers (23).
VSDs can be classified into four broad categories: Sub-arterial,
perimem-branous and muscular VSD, as well as complete ventricular
septal agenesis (functionally univentricular) (24,25).
Of note, it was found that the VSD samples were predominantly from
males (73.1% male; 26.9% female), particularly in the complete
ventricular septal agenesis sub-group (100% male). This finding is
consistent with the observations by Zhao et al (25), who found female predominance in
mild CHD [small VSD/patent ductus arteriosus/atrial septal defect
(ASD)] and male predominance in severe CHD (large VSD, functionally
univentricular heart and tetralogy of Fallot). The results of the
present study are in contrast with those of Sands et al
(26), who reported that there
were significantly more females in the group with VSDs compared
with the control group (P=0.004). This discrepancy is likely caused
by the differences in sample selection: Live birth neonates were
used by Sands et al (26),
while fetal myocardial tissue with lethal embryonic defects was
used in the present study. Thus, males with severe CHDs, including
a large VSD or a functionally univentricular heart, would not
survive to birth and would be missing from the analysis in the
study by Sands et al (26).
Previous studies have shown that TWIST1, as a
tumor biomarker, is overexpressed in different types of cancer,
including gastric carcinomas (27), breast cancer (28), pancreatic cancer (29), esophageal squamous cell carcinoma
(30) and colorectal cancer
(31). During embryonic
development, TWIST1 has an essential role in specification
of the mesoderm and differentiation of the mesoderm-derived
tissues. It also regulates morphogenesis in a variety of developing
organ systems, including the heart, in animal models (32). In the primary chicken endocardial
cushion, Twist1 promoted cell proliferation (10). In Twist1 (−/−) mice, the
cardiac OFT displayed defective maturation and tracking (11). The cardiac OFT and endocardial
cushion are important in primordial heart development, suggesting
that Twist1 is essential for normal heart development. The
cardiac cushions separate into the atrioventricular septum;
however, a relevant developmental disorder would lead to complete
ventricular septal agenesis or subarterial VSD. The cardiac OFT
functionally separates the aortic arch and pulmonary trunk; a
relevant developmental disorder may result in a perimembranous VSD
(21,24). Previous studies have shown that
mice overexpressing TWIST1 mutants in cardiomyocytes
developed pathological cardiac remodeling, including ASD and VSD,
induced by abnormal Akt signaling (enhancement or reduction)
(33). TWIST1, also a valve
progenitor marker (34), is
expressed in newly formed mesenchymal cells of the
atrio-ventricular canal and outflow tract and has been shown to
have roles in cell self-renewal, proliferation, migration and
differentiation (35,36). Persistent TWIST1 expression was
shown to increase cell proliferation (37,38);
however, decreased expression of TWIST1 may cause VSD.
In the present study, the expression levels of
TWIST1 mRNA and protein in fetal myocardial tissue samples
were measured using RT-PCR, real-time PCR and western blotting. The
results suggested that TWIST1 was not only involved in fetal
heart tissue development, but was also downregulated in myocardial
cells of fetuses with VSD compared with that in normal fetuses. To
the best of our knowledge, the present study was the first to show
differences in TWIST1 mRNA as well as protein levels between
myocardial tissues of fetuses with VSD and normal samples,
suggesting that TWIST1 is a candidate target gene for VSD
prevention as well as a prospective marker for VSD.
The strengths of the present study include the
strict sample collection process. Data were obtained from freshly
frozen cardiac tissue and may be more accurate than those from
other studies using cardiac tissue fixed with formalin, which can
cause random DNA base damage, thereby affecting PCR fidelity
(39). The present study focused
on an early time interval of cardiac development, namely embryonic
hearts at 22-28 weeks of gestation while the embryo was in utero.
This was earlier than the stages examined by other studies on
cardiac development of newborns or children and it therefore
provided more comprehensive and original information for the study
of cardiac development in humans.
Several limitations of the present study should be
considered. First, due to the difficulty of obtaining fetal
myocardial tissue samples, it was not possible to obtain a
sufficient number of complete gestational age-matched samples from
fetuses with VSD and healthy controls. Second, it was not possible
to obtain heart tissue samples at a variety of developmental stages
during pregnancy due to the limitations of pre-natal diagnostic
three-dimensional echocardiography techniques. According to
previous studies, the earliest time-point at which a diagnosis of
congenital heart disease in a fetus can be made is at 14 weeks of
gestation; however, the diagnostic specificity is poor (40). Further studies using a larger
number of samples are warranted to verify the findings of the
present study. In addition, based on the results of the present
study obtained from clinical samples, associated in-depth studies
will continue to investigate the molecular mechanisms responsible
for downregulated gene expression in VSD.
In conclusion, the present study showed that
TWIST1 gene expression in fetal myocardial tissue was
significantly decreased in VSD samples compared with that in normal
control subjects. These findings made a significant contribution to
the understanding of the development of human VSD and provided
novel knowledge regarding the etiology of CHD.
Acknowledgments
The authors are grateful to Miss. Chaofan Zhang,
Miss. Shuang Song, Miss. Weiju Jiang and Mr. Tianchu Huang from the
Department of Obstetrics and Gynecology at Shengjing Hospital of
China Medical University (Shenyang, China) for collecting the
samples. In particular, the authors would like to thank all of the
subjects for participating in this study. The present study was
supported by the Science and Technology Program of Liaoning
Province (no. 2011225017) and the Natural Science Foundation of
Liaoning province (no. 2014021004).
References
|
1
|
Go AS, Mozaffarian D, Roger VL, et al:
Heart disease and stroke statistics-2013 update: a report from the
American heart association. Circulation. 127:e6–e245. 2013.
View Article : Google Scholar
|
|
2
|
Hoffman JI: Incidence of congenital heart
disease: I. Postnatal incidence. Pediatr Cardiol. 16:103–113. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hoffman JI and Kaplan S: The incidence of
congenital heart disease. J Am Coll Cardiol. 39:1890–1900. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Misra C, Sachan N, McNally CR, et al:
Congenital heart disease-causing Gata4 mutation displays functional
deficits in vivo. PLoS Genet. 8:e10026902012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xuan C, Jia KG, Wang BB, et al:
Identification of two novel mutations of the HOMEZ gene in Chinese
patients with isolated ventricular septal defect. Genet Test Mol
Biomarkers. 17:390–394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jenkins KJ, Correa A, Feinstein JA, et al:
Noninherited risk factors and congenital cardiovascular defects:
current knowledge: a scientific statement from the American heart
association council on cardiovascular disease in the young:
endorsed by the American academy of pediatrics. Circulation.
115:2995–3014. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pierpont ME, Basson CT, Benson DW Jr, et
al: Genetic basis for congenital heart defects: current knowledge:
a scientific statement from the American heart association
congenital cardiac defects committee, council on cardiovascular
disease in the young: endorsed by the american academy of
pediatrics. Circulation. 115:3015–3038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Conway SJ, Firulli B and Firulli AB: A
bHLH code for cardiac morphogenesis. Pediatr Cardiol. 31:318–324.
2010. View Article : Google Scholar :
|
|
9
|
Castanon I and Baylies MK: A Twist in
fate: evolutionary comparison of Twist structure and function.
Gene. 287:11–22. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shelton EL and Yutzey KE: Twist1 function
in endocardial cushion cell proliferation, migration and
differentiation during heart valve development. Dev Biol.
317:282–295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vincentz JW, Barnes RM, Rodgers R, Firulli
BA, Conway SJ and Firulli AB: An absence of Twist1 results in
aberrant cardiac neural crest morphogenesis. Dev Biol. 320:131–139.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X, Ling MT, Guan XY, et al:
Identification of a novel function of TWIST, a bHLH protein, in the
development of acquired taxol resistance in human cancer cells.
Oncogene. 23:474–482. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang J, Mani SA, Donaher JL, et al: Twist,
a master regulator of morphogenesis, plays an essential role in
tumor metastasis. Cell. 117:927–939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang J, Mani SA and Weinberg RA: Exploring
a new twist on tumor metastasis. Cancer Res. 66:4549–4552. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rajagopal SK, Ma Q, Obler D, et al:
Spectrum of heart disease associated with murine and human GATA4
mutation. J Mol Cell Cardiol. 43:677–685. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Garg V: Insights into the genetic basis of
congenital heart disease. Cell Mol Life Sci. 63:1141–1148. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mori AD and Bruneau BG: TBX5 mutations and
congenital heart disease: Holt-Oram syndrome revealed. Curr Opin
Cardiol. 19:211–215. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Biben C, Weber R, Kesteven S, et al:
Cardiac septal and valvular dysmorphogenesis in mice heterozygous
for mutations in the homeobox gene Nkx2–5. Circ Res. 87:888–895.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huber J, Peres VC, de Castro AL, et al:
Molecular screening for 22Q11.2 deletion syndrome in patients with
congenital heart disease. Pediatr Cardiol. 35:1356–1362. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tan M, Xu C, Sim SK, Seow AL, Tan TH and
Quek SC: Types and distribution of congenital heart defects
associated with trisomy 21 in Singapore. J Paediatr Child Health.
49:223–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bruneau BG: The developmental genetics of
congenital heart disease. Nature. 451:943–948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Verheugt CL, Uiterwaal CS, van der Velde
ET, et al: The emerging burden of hospital admissions of adults
with congenital heart disease. Heart. 96:872–878. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu C, Yu ZB, Chen XH, et al: Screening
for differential methylation status in fetal myocardial tissue
samples with ventricular septal defects by promoter methylation
microarrays. Mol Med Rep. 4:137–143. 2011.PubMed/NCBI
|
|
24
|
Soto B, Becker AE, Moulaert AJ, Lie JT and
Anderson RH: Classification of ventricular septal defects. Br Heart
J. 43:332–343. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao QM, Ma XJ, Jia B and Huang GY:
Prevalence of congenital heart disease at live birth: an accurate
assessment by echocardio-graphic screening. Acta Paediatr.
102:397–402. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sands AJ, Casey FA, Craig BG, Dornan JC,
Rogers J and Mulholland HC: Incidence and risk factors for
ventricular septal defect in ‘low risk’ neonates. Arch Dis Child
Fetal Neonatal Ed. 81:F61–F63. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Z, Li XJ, Luo T and Wu XT: Expression
of TWIST in gastric carcinoma and its correlation with clinical
significance. Sichuan Da Xue Xue Bao Yi Xue Ban. 42:625–629.
2011.In Chinese. PubMed/NCBI
|
|
28
|
Gort EH, Suijkerbuijk KP, Roothaan SM, et
al: Methylation of the TWIST1 promoter, TWIST1 mRNA levels and
immunohistochemical expression of TWIST1 in breast cancer. Cancer
Epidemiol Biomarkers Prev. 17:3325–3330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ohuchida K, Mizumoto K, Ohhashi S, et al:
Twist, a novel oncogene, is upregulated in pancreatic cancer:
clinical implication of Twist expression in pancreatic juice. Int J
Cancer. 120:1634–1640. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuen HF, Chan YP, Wong ML, et al:
Upregulation of Twist in oesophageal squamous cell carcinoma is
associated with neoplastic transformation and distant metastasis. J
Clin Pathol. 60:510–514. 2007. View Article : Google Scholar
|
|
31
|
Valdés-Mora F, Gómez del Pulgar T, Bandrés
E, et al: TWIST1 overexpression is associated with nodal invasion
and male sex in primary colorectal cancer. Ann Surg Oncol.
16:78–87. 2009. View Article : Google Scholar
|
|
32
|
Barnes RM and Firulli AB: A twist of
insight-the role of Twist-family bHLH factors in development. Int J
Dev Biol. 53:909–924. 2009. View Article : Google Scholar :
|
|
33
|
Lu S, Nie J, Luan Q, et al:
Phosphorylation of the Twist1-family basic helix-loop-helix
transcription factors is involved in pathological cardiac
remodeling. PLoS One. 6:e192512011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wirrig EE and Yutzey KE: Conserved
transcriptional regulatory mechanisms in aortic valve development
and disease. Arterioscler Thromb Vasc Biol. 34:737–741. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vrljicak P, Cullum R, Xu E, et al: Twist1
transcriptional targets in the developing atrio-ventricular canal
of the mouse. PLoS One. 7:e408152012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee MP and Yutzey KE: Twist1 directly
regulates genes that promote cell proliferation and migration in
developing heart valves. PLoS One. 6:e297582011. View Article : Google Scholar
|
|
37
|
Chakraborty S, Wirrig EE, Hinton RB,
Merrill WH, Spicer DB and Yutzey KE: Twist1 promotes heart valve
cell proliferation and extracellular matrix gene expression during
development in vivo and is expressed in human diseased aortic
valves. Dev Biol. 347:167–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Firulli BA, McConville DP, Byers JS III,
Vincentz JW, Barnes RM and Firulli AB: Analysis of a Hand1
hypomorphic allele reveals a critical threshold for embryonic
viability. Dev Dyn. 239:2748–2760. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Quach N, Goodman MF and Shibata D: In
vitro mutation artifacts after formalin fixation and error prone
translesion synthesis during PCR. BMC Clin Pathol. 4:12004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Johnson B and Simpson LL: Screening for
congenital heart disease: a move toward earlier echocardiography.
Am J Perinatol. 24:449–456. 2007. View Article : Google Scholar : PubMed/NCBI
|