Introduction
Neuropathic pain is an unmet medical concern,
affecting individuals globally. The condition has been causally
correlated with functional alterations in the sodium channels of
nociceptors (1). Sodium channels
are integral membrane glycoproteins, which are responsible for the
generation and conduction of action potentials in excitable cells
(2). Previous studies have
revealed that sodium channel blockers, including local anesthetics,
tricyclics and certain anti-convulsants, are able to attenuate pain
in patients with neural injury (3,4). The
voltage-gated sodium channel isoforms, Nav1.8 and
Nav1.9, encoding for slowly-gating
tetrodotoxin-resistant (TTX-R) sodium currents, are particularly
notable with respect to sensory nerve pathophysiology. They are
sensory neuron-specific with Nav1.8 expressed in thinly
unmyelinated (C-fibers) and 10% myelinated (A-fibers) axons. By
contrast, the expression of Nav1.9 is restricted to
small C-fiber dorsal root ganglion (DRG) cells (5). Differential expression of these
sodium channels is coupled with isoform-specific contributions in
neuronal excitability and the transmission of sensory information
(6). Nav1.8 produces
the majority of the depolarizing inward current during an action
potential (7), while
Nav1.9 has been proposed to contribute to maintaining
the resting potential (8). The
absence of these channels in the central nervous system implicates
them as a suitable target for therapeutic intervention in pain
management with few side effects (2).
Peripheral nerve injury, for example axotomy or
nerve transection, causes a downregulation of Nav1.8
expression and a decrease in the electrical current attributed to
this channel in injured neurons (3,9,10).
Additionally, specific knockdown of Nav1.8 with
antisense oligodeoxynucleotides may effectively reverse neuropathic
pain (11). However, it is not
intuitively clear how this contributes to the neuropathic pain
phenotype associated with these models. Previous studies have
observed that upregulation of the expression of Nav1.8
in spared neurons following nerve injury, with the exception of
chronic constriction injury (CCI) or spared nerve injury models,
may provide a reasonable explanation for the contribution of this
channel to the pain phenotype (10,12–14).
In addition, there are specific discrepancies in the evidence among
studies with regards to the involvement of Nav1.9 in
certain neuropathic pain conditions (2,15–17).
The expression of Nav1.9 is downregulated in injured DRG
neurons (15) and is upregulated
in uninjured neurons following peripheral nerve injury (16), while abnormal behavior in the
Nav1.9 knockout mouse remains unchanged in neuropathic
pain models (2). To resolve this
limitation, a CCI model was established in the present study to
evaluate whether Nav1.8 sodium channels mediate
neuropathic pain through a compensatory redistribution to
contralateral uninjured DRG neurons and to elucidate the exact role
of Nav1.9 in neuropathic pain.
Materials and methods
Experimental animals
A total of 72 adult male Sprague-Dawley rats
weighing 150–180 g and aged 6–8 weeks, purchased from the
Experimental Animal Centre of Shenyang Pharmaceutical University
(Shenyang, China), were used in all experiments. Rats were
maintained under controlled environmental conditions at 23±2°C with
a 12-h light/dark cycle and ad libitum access to food and
water. All procedures were performed in accordance with the
guidelines of the International Association for the Study of Pain
(18). The study was approved by
the ethics committee of Shenyang Pharmaceutical University.
CCI model
The CCI model was induced as previously described
(19,20). Briefly, under anesthesia with 3.5%
chloral hydrate (10 ml/kg; Hebei Gaobeidian Chunguang Chemical
Reagent Company, Gaobeidian, China) administered intraperitoneally
(i.p.), the right sciatic nerve was exposed and loosely ligated
with 4–0 chromic catgut (Shanghai Pudong Medical Supplies Co.,
Ltd., Shanghai, China) in four regions separated by ~1 mm, then the
incision was closed with sutures. For the sham group, the right
sciatic nerve was exposed without ligation. Pain-associated
behavioral assessments were performed at the time-points of: −1
(prior to CCI surgery), 1, 3, 5, 7, 10, 14 and 21 days after CCI
surgery.
Behavioral assessment
The abnormal posture of each animal was evaluated
using a subjective pain-associated behavioral grade as described
previously (21) by an
investigator who was blinded to the experimental conditions.
Briefly, grades were determined as: 0, normal; 1, coiling of the
toes; 2, valgus deformity of the paw; 3, partially weight bearing;
4, non-weight bearing and 5, avoidance of any contact with the hind
paw.
Paw withdrawal response to thermal
stimuli
The paw withdrawal response was measured using the
Plantar Test meter (IITC Life Science Inc., Woodland Hills, CA,
USA). Rats were placed individually in a clear, transparent box
(17×11.5×14 cm) and allowed to acclimate for 15 min prior to the
assessment. An infrared beam of a radiant heat source was applied
to irradiate the plantar surface of the hind paw through the glass
plate until the rat withdrew or contracted its paw. The withdrawal
thresholds were measured in each hind paw and the ipsilateral hind
paw was assessed at 15 min intervals, while the contralateral hind
paw was assessed at 5 min intervals. A 20 sec limit was imposed to
prevent tissue damage. Each rat was assessed five times and the
mean value was expressed as the thermal withdrawal latency
(22).
The paw withdrawal response to mechanical stimuli
was measured using the Electronic von Frey Anesthesiometer (IITC
Life Science Inc.). Rats were placed individually into wire
mesh-bottom boxes (20×14×16 cm) and allowed to acclimate for 30 min
prior to assessment. The probe was placed beneath the plantar
surface of the hind paw and the force was increased until the rat
was observed to vellicate its paw. The maximum force was recorded
at the time of paw withdrawal. Withdrawal thresholds were measured
in each hind paw and the ipsilateral hind paw was assessed at 30
sec intervals, while the contralateral hind paw was assessed at 15
sec intervals. Each rat was assessed five times and the average
value was expressed as the mechanical withdrawal threshold
(23).
Immunofluorescence and hematoxylin and
eosin (H&E) staining
L4-6 DRGs, quickly dissected from
recently sacrificed animals, were fixed with 10% formalin overnight
at 4°C. Following embedding the tissue in paraffin, a series of
5-µm sections were cut for immunofluorescence and H&E
staining. Paraffin sections were treated with dimethylbenzene
solution I for 15 min, dimethylbenzene solution II for 15 min,
dimethylbenzene:pure ethanol (1:1) solution for 2 min, 100% ethanol
I for 5 min, 100% ethanol II for 5 min, 95% ethanol solution for 3
min, 90% ethanol solution for 1 min, 85% ethanol solution for 1
min, 75% ethanol solution for 1 min, 50% ethanol solution for 1
min, running water for 2 min, haematoxylin solution for 2 min,
running water for 1 min, 1% hydrochloric acid ethanol solution for
20 sec, running water for 5 min, Eosin solution for 30 sec, running
water for 30 sec, 75% ethanol solution for 30 sec, 85% ethanol
solution for 20 sec, 95% ethanol solution I for 1 min, 95% ethanol
solution II for 1 min, 100% ethanol for 2 min, 100% ethanol II for
2 min, dimethylbenzene solution I for 2 min, dimethylbenzene
solution II for 2 min then dimethylbenzene solution III for 2 min.
The primary antibodies polyclonal rabbit Nav1.8
(ASC-016; 1:200; Alomone Laboratories Ltd., Jerusalem, Israel),
monoclonal mouse neurofilament (NF)200 (N0142; 1:200;
Sigma-Aldrich, St. Louis, MO, USA) and polyclonal rabbit
Nav1.9 (ASC-017; 1:200; Alomone Laboratories Ltd.) were
administered for immunofluorescence staining and used to incubate
the sections overnight at 4°C. Following three washes with
phosphate-buffered saline containing Tween-20, the sections were
incubated with anti-mouse IgG fluorescein isothiocyanate-conjugated
antibody produced in goat (F0257) and anti-rabbit IgG (whole
molecule)-Cy3 antibody produced in sheep (C2306) (1:100;
Sigma-Aldrich) for 1 h at 37°C. Images were captured under an
inverted fluorescence microscope (Olympus BX40; Olympus, Tokyo,
Japan) and imported into Image pro plus 6.0 software (Media
Cybernetics, Silver Spring, MD, USA) for further analysis. H&E
staining was performed according to the manufacturer's instructions
(Sigma-Aldrich). Nuclear material within the nucleus was stained a
deep purple/blue, while the cytoplasmic material, including
connective tissue and collagen appeared orange/pink.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
At 3, 7, 14 and 21 days after CCI, rats were
sacrificed via anesthesia with 3.5% chloral hydrate (10 ml/kg,
i.p.). The ipsilateral and contralateral L4–6 DRGs were
rapidly removed and placed into Eppendorf tubes. Total RNA was
extracted using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) and the cDNA was reverse transcribed using the
PrimeScript® RT reagent kit (Takara Bio Inc., Otsu,
Japan) according to the manufacturer's instructions. PCR was
performed in a 25-µl reaction mixture containing 2 µl
templates, 12.5 µl SYBR® Premix Ex Taq™ (2X), 0.5
µl ROX reference dye II (50X) and 0.4 µM primer for
each gene. The thermal cycling conditions comprised 30 sec
polymerase activation at 95°C, 40 cycles of 15 sec denaturation at
95°C and 1 min at 60°C for annealing and extension. A dissociation
curve was used to determine the amplification specificity. The
primer sequences (24) were as
follows: Nav1.8 (GenBank accession number, U53833)
forward, 5′-GACTCCCGGACAAATCAGAA-3′ and reverse,
5′-AGCAGCGACCTCATCTTCAT-3′; Nav1.9 (GenBank accession
number, AF059030) forward, 5′-TCTCCACCCCTACCTCACTG-3′ and reverse,
5′-CGTTCAGCCAAAAACACAGA-3′; and GAPDH forward,
5′-TGCCAAGTATGATGACATCAAGAAG-3′ and reverse,
5′-AGCCCAGGATGCCCTTTAGT-3′. The threshold cycle values of
Nav1.8 and Nav1.9 mRNA were measured and
normalized to GAPDH, and then expressed as a relative ratio. The
M×3000P qPCR system was used (Agilent Technologies, Inc., Santa
Clara, CA, USA).
Western blot analysis
Bilateral L4–6 DRGs were dissected and
total proteins were extracted via homogenization in ice-cold lysate
buffer (Thermo Fisher Scientific, Waltham, MA, USA). Samples (30–50
µg) were separated on 8% SDS-PAGE separation gels (Amresco,
Boise ID, USA) and subsequently transferred onto polyvinylidene
difluoride membranes (Merck Millipore, Boston, MA, USA). The
membranes were blocked in 5% skimmed milk solution at room
temperature for 2 h and then rabbit polyclonal Nav1.8
(1:500; Alomone Laboratories Ltd.), Nav1.9 (1:500;
Alomone Laboratories Ltd.) and mouse monoclonal β-actin (1:500;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) primary antibodies
were used to incubate the samples overnight at 4°C. The target
bands were detected with secondary horseradish peroxidase
(HRP)-labeled goat anti-rabbit (1:10,000; ZB-2301) or HRP-labeled
goat anti-mouse IgG (1:5,000; ZB2305) (Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China) antibodies for 1 h at room
temperature. The band intensity of Nav1.8 and
Nav1.9 was normalized to that of β-actin and expressed
as a relative ratio.
Patch clamp recording
Rat DRG neurons were acutely dissociated as
previously described (25). The
samples were superfused at a rate of 3 ml/min and all patch clamp
recordings were performed using an Axopatch 200B amplifier
(Molecular Devices, Sunnyvale, CA, USA), filtered at 1 kHz and
digitally sampled at 10 kHz at room temperature (23±2°C). Clampfit
10.0 software (Molecular Devices) was used for data acquisition and
analysis.
The bath solution for the Nav1.8 currents
contained (in mM): 140 NaCl, 1 MgCl2, 3
CaCl2, 5 KCl (Tianjin Bodi Chemical Co. Ltd., Tianjin,
China), 10 tetraethylammonium (TEA)-Cl, 1 4-aminopyridine, 0.2
CdCl2, 10 4-(2-hydroxyethyl)-1-piper-azineethanesulfonic
acid (HEPES), 10 glucose (Tianjin Bodi Chemical Co. Ltd.), 0.001
TTX (Hebei Fisheries Research Institute, Qinhuangdao, China), pH
7.3 with NaOH (Tianjin Bodi Chemical Co. Ltd.). The bath solution
for the Nav1.9 currents contained 30 NaCl, 20 TEA-Cl, 90
choline chloride, 3 KCl, 1 CaCl2, 1 MgCl2, 10
HEPES, 10 glucose, 0.1 CdCl2, 0.001 TTX, pH 7.3 with
Tris base (all purchased from Sigma-Aldrich unless specified). The
pipette solution used for recording the DRG neuron
Nav1.8 and Nav1.9 currents contained (in mM):
140 CsCl, 10 TEA-Cl, 10 ethylene glycol tetraacetic acid (EGTA;
Amresco), 10 HEPES, pH 7.2 with CsOH and 135 CsF, 10 NaCl, 10
HEPES, 5 EGTA, 2 adenosinetriphosphate bisodium, pH 7.2 with CsOH,
respectively. The pipettes, fabricated with a P-97 puller (Sutter
Instruments, Novato, CA, USA), had resistances of 3–5 MΩ when
filled with pipette solution.
Statistical analysis
Data were analysed using SPSS 16.0 (SPSS, Inc.,
Chicago, IL, USA) with a one-way analysis of variance. All
measurements are expressed as the mean ± standard error. P<0.05
was considered to indicate a statistically significant
difference.
Results
CCI-induced neuropathic pain evokes
spontaneous pain, mechanical allodynia and heat hyperalgesia
The CCI model induced the typical features of
neuropathic pain, which were assessed via behavioral grade, the
Plantar Test meter and the Electronic von Frey Anesthesiometer.
Rats with nerve injury (CCI) exhibited curling, eversion, partial
weight bearing or non-weight bearing on the right injured side,
while they exhibited normal behavior on the left uninjured paw. The
score for the right paw increased significantly from the first day
after surgery (data not shown).
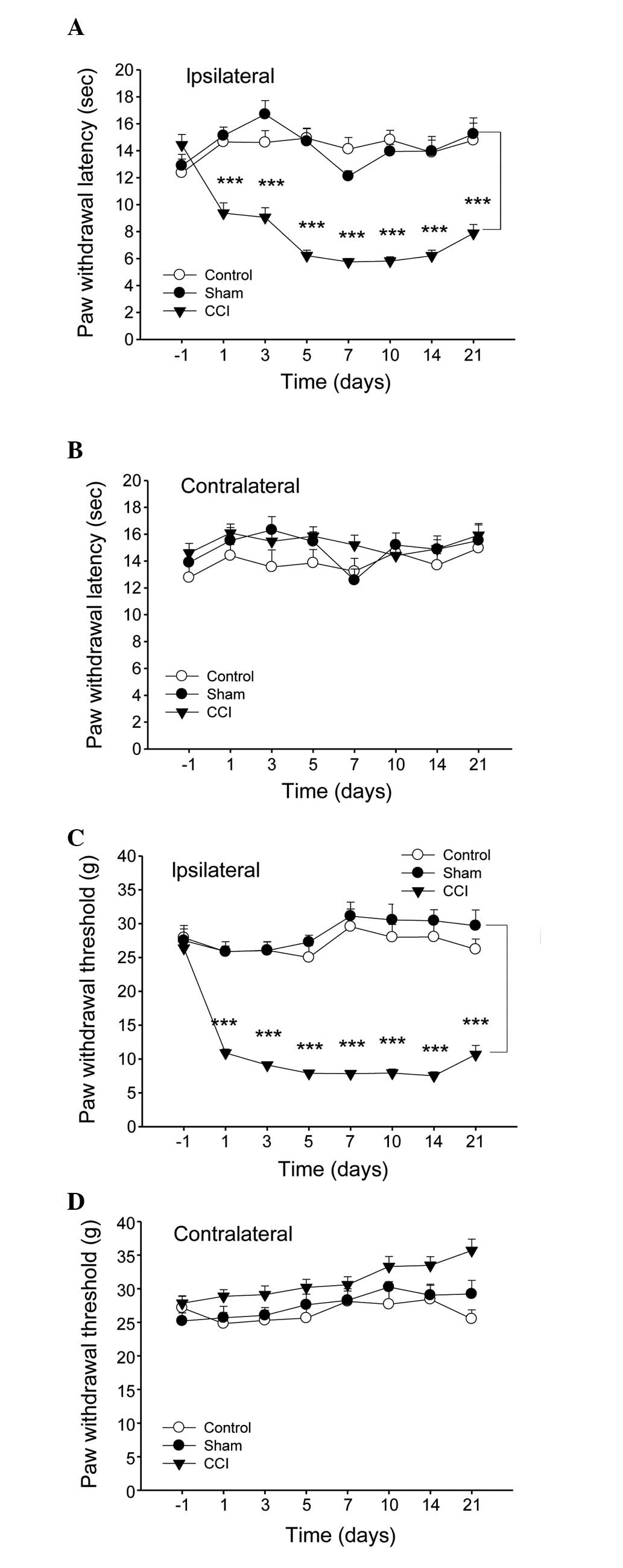
The sensitivity to heat and mechanical stimulus
revealed certain discrepancies among the experimental groups. In
the CCI model group, the thermal withdrawal latency and mechanical
withdrawal threshold of the right injured paw decreased markedly
from day 1 after injury, with the maximal level of hypersensitivity
at 7 days after surgery, while observable changes were not observed
in the sham surgery group (Fig. 1A and
C). By contrast, no differences were observed in the
contralateral paw withdrawal in all groups (Fig. 1B and D).
CCI-induced neuropathic pain impairs DRG
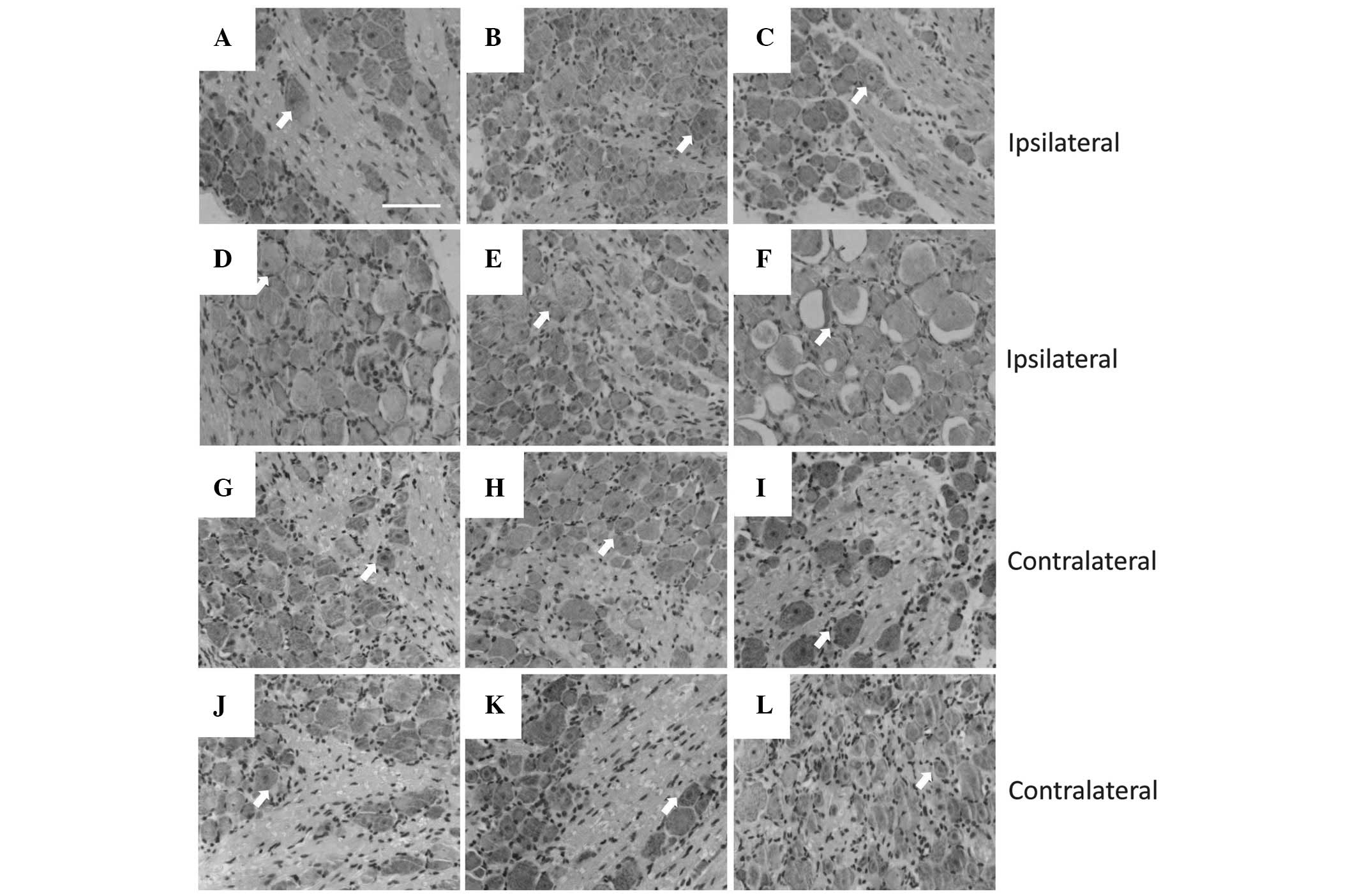
histological morphology
In peripheral nerve injury, the majority of neuronal
cell death is observed in peripheral nerve lesions, which are
considered to be a major factor in the poor clinical outcome
following the injury (26). With
H&E staining, ipsilateral and contralateral DRGs to the injury
(Fig. 2) were analyzed
histologically. In each contralateral DRG group (Fig. 2G–L), the neurons exhibited a normal
cellular appearance, including a large, round nucleus, clear cell
boundaries and even chromatin distribution. However, neurons on the
ipsilateral side to the injury exhibited a range of morphologies at
different time-points (Fig. 2C–F),
although all observed cells had abnormal nuclei exhibiting
pyknosis, membrane irregularities or even vacuolation, with the
maximal magnitude of neuronal death at 21 days after surgery.
Expression profiles of Nav1.8
and Nav1.9 in DRG neurons under CCI-induced neuropathic
pain
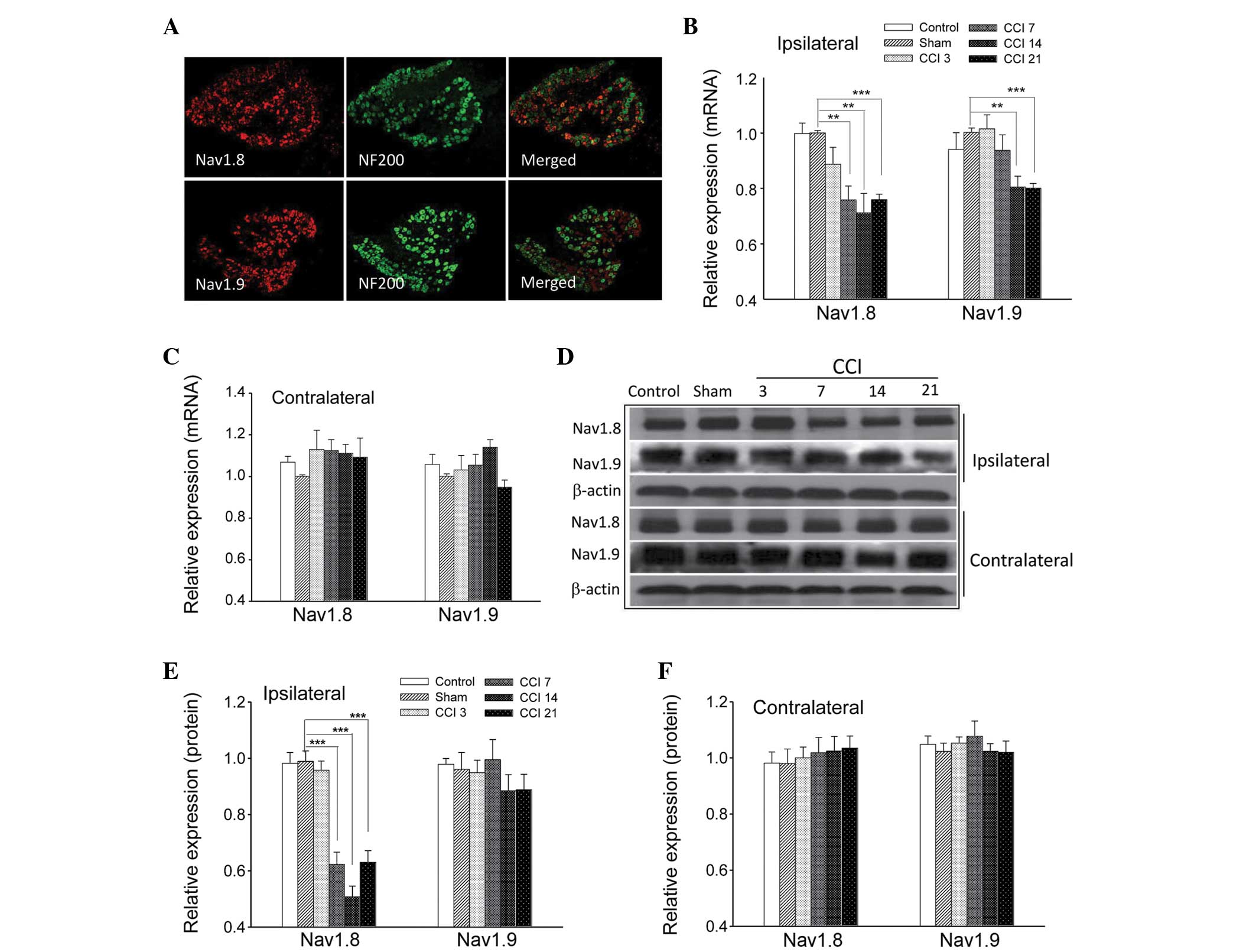
The neurofilament NF200 is preferentially expressed
in large DRG neurons and is a useful marker for this population
(7). When co-incubated with
antibodies against Nav1.8 and Nav1.9 in an
immunofluorescence assay, it was observed that Nav1.8
and Nav1.9 subunits were primarily located in the small
and medium diameter DRG neurons (Fig.
3A).
RT-qPCR and western blot analyses were performed at
3, 7, 14 and 21 days after the CCI surgery instead of at an earlier
point, in order to avoid potential error due to the effects of
post-surgical pain. CCI induced a marked downregulation of the
Nav1.8 transcript at 7 days (0.76±0.05; P<0.01), 14
days (0.71±0.07; P<0.01) and 21 days (0.76±0.02; P<0.001) and
a substantial downregulation of Nav1.9 transcript at 14
days (0.81±0.04; P<0.01) and 21 days (0.806±0.02; P<0.001) in
L4–6 ipsilateral DRGs (Fig.
3B); however, no significant differences were observed among
groups in the contralateral DRGs (Fig.
3C; P>0.05). Representative images of Nav1.8 and
Nav1.9 protein expression within the ipsilateral and
contralateral DRGs are shown in Fig.
3D. Consistent with these findings, RT-qPCR analysis revealed
that the relative ratio of band intensity of Nav1.8
protein in ipsilateral DRGs was significantly reduced at 7 days
(0.62±0.04; P<0.001), 14 days (0.51±0.04; P<0.001) and 21
days (0.63±0.04; P<0.001) after CCI treatment, while
Nav1.9 protein expression following CCI exhibited a
decrease at 14 days (0.88±0.06; P>0.05) and 21 days (0.89±0.06,
P>0.05; Fig. 3E). However, the
differences were not statistically significant when compared with
the sham group. In addition, there were no detectable differences
among contralateral DRG neurons from the rats subjected to CCI
(Fig. 3F). These results markedly
suggested that Nav1.8 and Nav1.9 sodium
channels have distinct roles following CCI treatment.
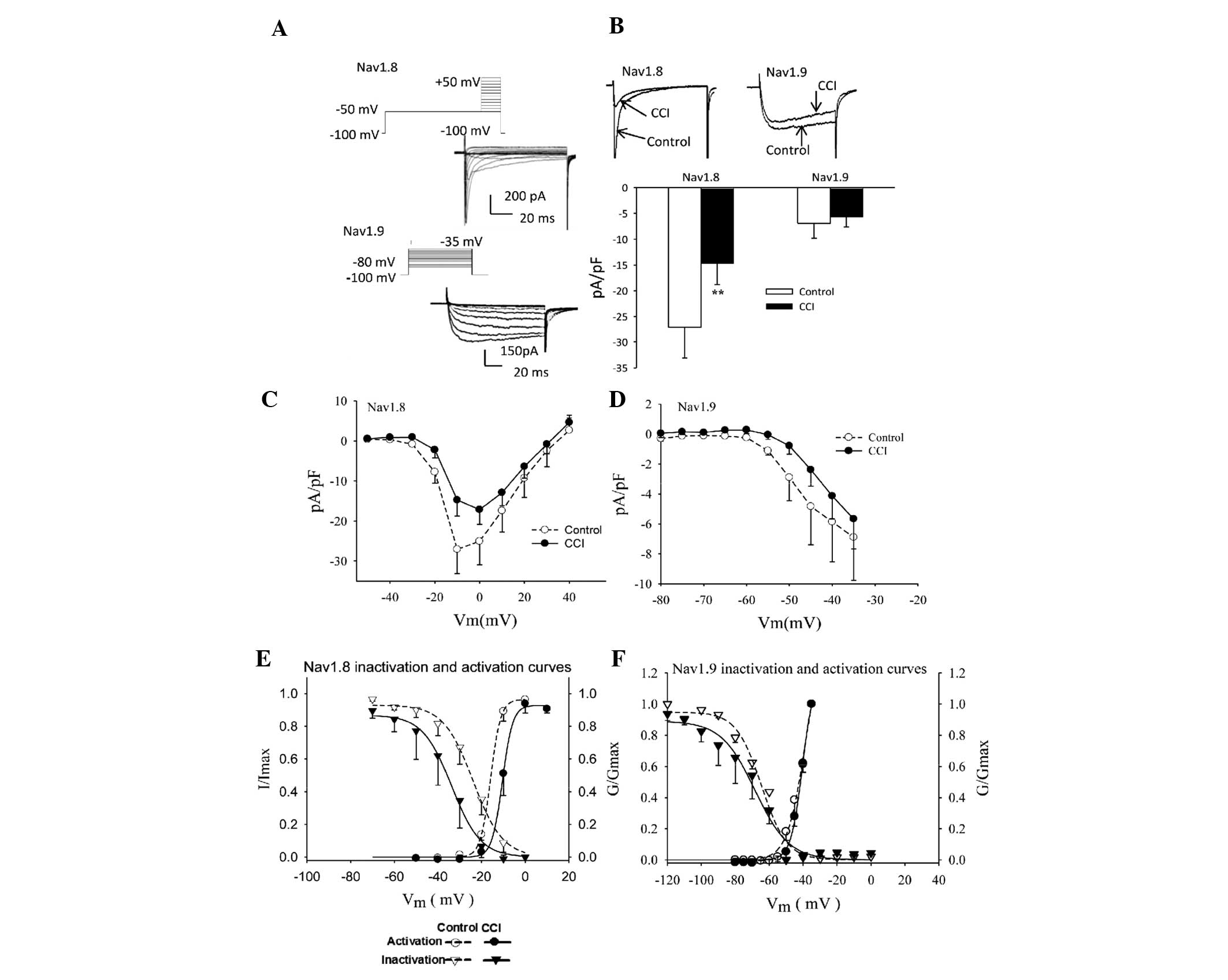
Effect of CCI on TTX-R sodium channels in
rat DRG neurons
The small- and medium-diameter DRG neurons (12–25
µm) were selected as the main focus of the present study.
Differences in the voltage protocols and pharmacological inhibition
by TTX were used to evoke Nav1.8 and Nav1.9
inward currents (Fig. 4A).
Following CCI surgery, the sodium current densities mediated by
Nav1.8 and Nav1.9 decreased ~50% (Fig. 4B; between −27.10±6.03 and
−14.75±4.01 pA/pF; P<0.01) and 18% (Fig. 4B; between −6.90±2.89 and −5.67±2.00
pA/pF; P>0.05), respectively.
Steady-state activation curves were constructed as
described from current-voltage curve experiments. The normalized
activation curves of Nav1.8 and Nav1.9 were
fitted with the Boltzmann function expressed as:
G/Gmax = 1/{1+exp[(V−V1/2)/K,
where V1/2 is the membrane potential at
half-activation and K represents the slope factor.
Characterization of activation curves revealed that CCI treatment
caused a depolarizing shift of 5.3 mV and 1.44 mV in
Nav1.8 (Fig. 4E;
V1/2 between −15.84±0.21 and −10.51±0.24 mV) and
Nav1.9 (Fig. 4F;
between −40.28±1.02 mV and −38.84±2.07 mV) steady-state activation
curves, respectively.
The voltage-dependent inactivation of the
Nav1.8 current is shown in Fig. 4E. The solid lines were fitted with
the Boltzmann equation, I/Imax =
1/{1+exp[(V−V1/2)/K, where
V1/2 is the membrane potential when
I/Imax = 0.5 and K represents the slope
factor. There was a 10-mV depolarizing shift in the midpoint of
inactivation (Fig. 4E; between
−23.85±0.74 and −33.73±0.91 mV). This change reflected a parallel
shift in the availability curve of the current as no change was
observed in the slope factor (between −6.82±0.63 mV and −6.64±0.76
mV). However, no significant differences were identified between
injured and control DRG neurons with respect to the biophysical
properties of Nav1.9 (Fig.
4F).
Discussion
Neuropathic pain originating from pathology within
the nervous system is a serious unmet medical concern. Animal
models of neuropathic pain, although often unrepresentative,
provide important information for understanding the underlying
mechanism of neuropathic pain in humans (27). Peripheral nerve injury may result
in pain-associated behavior characterized by spontaneous pain,
hyperalgesia and allodynia (28).
CCI, as a classical neuropathic pain model, is able to induce
spontaneous pain and hyperalgesia through noxious thermal and
mechanical stimuli (29). In the
present study, it has been demonstrated that CCI is able to stably
induce the typical features of neuropathic pain. This method
exhibited multiple advantages, including a simple surgical
procedure with little tissue damage and a high success rate, with
evident and stable spontaneous pain following surgery.
The present findings confirmed the results of
previous studies investigating the downregulation of the
Nav1.8 current and expression in injured neurons
(3,9,14).
Nav1.8 mRNA transcript and protein levels were
significantly reduced in injured DRGs from 7 days after CCI and the
current density mediated by the Nav1.8 channel was
markedly reduced at 21 days. However, it remains to be elucidated
how this contributes to the neuropathic pain phenotype. Recently,
emerging evidence has revealed that the increase in
Nav1.8 levels and TTX-R current upregulated in adjacent
spared uninjured neurons may provide a reasonable explanation for
the role of the Nav1.8 channel in neuropathic pain
models (12–14). However, in the present study, the
current and expression of Nav1.8 were not affected in
contralateral uninjured DRGs. This indicated that there were no
redistributed compensatory effects of Nav1.8 in the
contralateral uninjured DRGs under CCI-induced neuropathic pain,
contrasting with previous studies (15).
A potential reason for the downregulation of the
Nav1.8 sodium channel is that the surgical procedure
caused neuronal damage. During the present study, it was
demonstrated that, in ipsilateral DRGs, CCI treatment induced
neuronal damage in a time-dependent manner, exhibiting pyknosis and
anachromasis of the nuclei as well as necrosis and shrunken
cavities in the cell bodies. Similarly to behavioral assessments,
the histological morphology of the contralateral DRGs remained
unchanged among groups. It was hypothesized that the quantity of
normal neurons affects the excitability of afferent neurons. Thus,
functional analysis was performed using the patch clamp
electrophysiological technique. It was identified that CCI
treatment caused a depolarizing shift in the Nav1.8
steady-state activation curve. This finding is supported by a
previous study in which Amm VIII, an α-toxin isolated from venom,
is able to induce rapid mechanical and thermal pain
hypersensitivities by negatively shifting the activation curve
(30). The slowly inactivating
Nav1.8 current has been observed to be capable of
generating repeated action potentials (31). In the present study, a positive
shift was also observed in the Nav1.8 activation curve
following the induction of CCI. This may induce the fast
inactivated state and increase action potential firing rates.
Another neuronal TTX-R channel, Nav1.9,
has also been observed to be associated with neuropathic pain. A
previous study revealed that Nav1.9 expression decreased
~3.6 fold under sciatic nerve ligation-induced neuropathic pain
(15). However, during the present
study, CCI-treatment only led to a significant decrease in the
level of Nav1.9 mRNA in DRGs ipsilateral to the injury.
In addition, it is important to acknowledge that no change in the
relative expression of Nav1.9 protein and
Nav1.9 current density and dynamics were observed. This
was conflicting with the normal behavior observed in the
Nav1.9 knockout mouse following CCI treatment (2). The reasons for this discrepancy
remain to be elucidated at present and require further study.
In conclusion, the present study provided evidence
of a role for Nav1.8 in the pathogenesis of neuropathic
pain. Of note, a positive shift of intact Nav1.8 sodium
channels in the DRG neurons ipsilateral to the induced injury was
likely to have promoted ectopic discharge, rather than a
compensatory regulation of contralateral uninjured DRG neurons.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81073081), the
Excellent Talents Plan of Higher Education Institutes in Liaoning
province (grant no. LJQ2013105) and the Key Laboratory of
Cardiovascular Medicine Research (Harbin Medical University),
Ministry of Education.
References
|
1
|
Dib-Hajj SD, Binshtok AM, Cummins TR,
Jarvis MF, Samad T and Zimmermann K: Voltage-gated sodium channels
in pain states: role in pathophysiology and targets for treatment.
Brain Res Rev. 60:65–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leo S, D'Hooge R and Meert T: Exploring
the role of nociceptor-specific sodium channels in pain
transmission using Nav1.8 and Nav1.9 knockout mice. Behav Brain
Res. 208:149–157. 2010. View Article : Google Scholar
|
|
3
|
Amir R, Kocsis JD and Devor M: Multiple
interacting sites of ectopic spike electrogenesis in primary
sensory neurons. J Neurosci. 25:2576–2585. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Joshi SK, Honore P, Hernandez G, et al:
Additive antinociceptive effects of the selective Nav1.8 blocker
A-803467 and selective TRPV1 antagonists in rat inflammatory and
neuropathic pain models. J Pain. 10:306–315. 2009. View Article : Google Scholar
|
|
5
|
Amaya F, Decosterd I, Samad TA, et al:
Diversity of expression of the sensory neuron-specific
TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol
Cell Neurosci. 15:331–342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ho C and O'Leary ME: Single-cell analysis
of sodium channel expression in dorsal root ganglion neurons. Mol
Cell Neurosci. 46:159–166. 2011. View Article : Google Scholar
|
|
7
|
Blair NT and Bean BP: Roles of
tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant
Na+ current, and Ca2+ current in the action
potentials of nociceptive sensory neurons. J Neurosci.
22:10277–10290. 2002.PubMed/NCBI
|
|
8
|
Herzog RI, Cummins TR and Waxman SG:
Persistent TTX-resistant Na+ current affects resting
potential and response to depolarization in simulated spinal
sensory neurons. J Neurophysiol. 86:1351–1364. 2001.PubMed/NCBI
|
|
9
|
Dib-Hajj S, Black JA, Felts P and Waxman
SG: Down-regulation of transcripts for Na channel α-SNS in spinal
sensory neurons following axotomy. Proc Natl Acad Sci USA.
93:14950–14954. 1996. View Article : Google Scholar
|
|
10
|
Chen X, Pang RP, Shen KF, Zimmermann M,
Xin WJ, Li YY and Liu XG: TNF-α enhances the currents of voltage
gated sodium channels in uninjured dorsal root ganglion neurons
following motor nerve injury. Exp Neurol. 227:279–286. 2011.
View Article : Google Scholar
|
|
11
|
Lai J, Gold MS, Kim CS, Bian D, Ossipov
MH, Hunter JC and Porreca F: Inhibition of neuropathic pain by
decreased expression of the tetrodotoxin-resistant sodium channel,
NaV1.8. Pain. 95:143–152. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gold MS, Weinreich D, Kim CS, Wang R,
Treanor J, Porreca F and Lai J: Redistribution of Na(V)1.8 in
uninjured axons enables neuropathic pain. J Neurosci. 23:158–166.
2003.PubMed/NCBI
|
|
13
|
Zhang XF, Zhu CZ, Thimmapaya R, et al:
Differential action potentials and firing patterns in injured and
uninjured small dorsal root ganglion neurons after nerve injury.
Brain Res. 1009:147–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Decosterd I, Ji RR, Abdi S, et al: The
pattern of expression of the voltage-gated sodium channels Na(v)1.8
and Na(v)1.9 does not change in uninjured primary sensory neurons
in experimental neuropathic pain models. Pain. 96:269–277. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Sun H, Della Penna K, et al:
Chronic neuropathic pain is accompanied by global changes in gene
expression and shares pathobiology with neurodegenerative diseases.
Neuroscience. 114:529–546. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berta T, Poirot O, Pertin M, Ji RR,
Kellenberger S and Decosterd I: Transcriptional and functional
profiles of voltage-gated Na(+) channels in injured and
non-injured DRG neurons in the SNI model of neuropathic pain. Mol
Cell Neurosci. 37:196–208. 2008. View Article : Google Scholar
|
|
17
|
Sleeper AA, Cummins TR, Dib-Hajj SD, et
al: Changes in expression of two tetrodotoxin-resistant sodium
channels and their currents in dorsal root ganglion neurons after
sciatic nerve injury but not rhizotomy. J Neurosci. 20:7279–7289.
2000.PubMed/NCBI
|
|
18
|
Yu YQ, Zhao F, Guan SM and Chen J:
Antisense-mediated knockdown of Na(V)1.8, but not Na(V)1.9,
generates inhibitory effects on complete Freund's adjuvant-induced
inflammatory pain in rat. PloS ONE. 6:e198652011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bennett GJ and Xie YK: A peripheral
mononeuropathy in rat that produces disorders of pain sensation
like those seen in man. Pain. 33:87–107. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li X, Kang L, Li G, et al: Intrathecal
leptin inhibits expression of the P2X2/3 receptors and alleviates
neuropathic pain induced by chronic constriction sciatic nerve
injury. Mol Pain. 9:65–73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu Y and Westlund KN: Gabapentin
attenuates nociceptive behaviors in an acute arthritis model in
rats. J Pharmacol Exp Ther. 290:214–219. 1999.PubMed/NCBI
|
|
22
|
Gao YH, Chen SP, Wang JY, Qiao LN, Meng
FY, Xu QL and Liu JL: Differential proteomics analysis of the
analgesic effect of electroacupuncture intervention in the
hippocampus following neuropathic pain in rats. BMC Complement
Altern Med. 12:241–251. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tu WZ, Cheng RD, Cheng B, et al: Analgesic
effect of electroacupuncture on chronic neuropathic pain mediated
by P2X3 receptors in rat dorsal root ganglion neurons. Neurochem
Int. 60:379–386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qiu F, Jiang Y, Zhang H, Liu Y and Mi W:
Increased expression of tetrodotoxin-resistant sodium channels
Nav1.8 and Nav1.9 within dorsal root ganglia in a rat model of bone
cancer pain. Neurosci Lett. 512:61–66. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maingret F, Coste B, Padilla F, Clerc N,
Crest M, Korogod SM and Delmas P: Inflammatory mediators increase
Nav1.9 current and excitability in nociceptors through a coincident
detection mechanism. J Gen Physiol. 131:211–225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McKay Hart A, Brannstrom T, Wiberg M and
Terenghi G: Primary sensory neurons and satellite cells after
peripheral axotomy in the adult rat: timecourse of cell death and
elimination. Exp Brain Res. 142:308–318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ueda H: Molecular mechanisms of
neuropathic pain-phenotypic switch and initiation mechanisms.
Pharmacol Ther. 109:57–77. 2006. View Article : Google Scholar
|
|
28
|
Latremoliere A and Woolf CJ: Central
sensitization: a generator of pain hypersensitivity by central
neural plasticity. J Pain. 10:895–926. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao ZQ: Neural mechanism underlying
acupuncture analgesia. Prog Neurobiol. 85:355–375. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Abbas N, Gaudioso-Tyzra C, Bonnet C, et
al: The scorpion toxin Amm VIII induces pain hypersensitivity
through gain-of-function of TTX-sensitive Na+ channels.
Pain. 154:1204–1215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garrison SR, Weyer AD, Barabas ME, Beutler
BA and Stucky CL: A gain-of-function voltage-gated sodium channel
1.8 mutation drives intense hyperexcitability of A- and C-fiber
neurons. Pain. 155:896–905. 2014. View Article : Google Scholar : PubMed/NCBI
|