Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of cancer, ranked the third most common cause of
cancer-related mortality worldwide, particularly in Africa and Asia
(1). Extracellular
signal-regulated kinase (ERK), a member of the mitogen-activated
protein kinases (MAPK) superfamily, which also includes c-Jun
N-terminal protein kinase (JNK), and p38 family of kinases, has
been implicated in HCC development (1). ERK is activated by a variety of
extracellular stimuli, from growth factors, such as epidermal
growth factor (EGF), to proinflammatory cytokines, including tumor
necrosis factor (TNF)-α (2,3).
Growth factors tend to simultaneously activate the ERK and Akt
pathways (2), whereas TNF-α
activates all three major groups of MAPKs, as well as the IκB
kinase (IKK) pathway (3,4). Extensive studies have shown that
chronic inflammation associated with persistent viral infections
and/or persistent exposure to hepatotoxic agents is clearly the
primary inducer of HCC (1,5). TNF-α and interleukin (IL)-6 are key
proinflammatory cytokines involved in HCC development (6,7). ERK
protects against TNF-α-induced apoptosis and mediates TNF-α-induced
IL-6 expression (8,9). However, the mechanisms underlying the
aberrant activation of the ERK pathway in HCC remain largely
unclear.
Alternative splicing modulates the expression of
various oncogene and tumor-suppressor isoforms (10–12).
Mutations in components of the spliceosome were recently identified
in several types of cancer and are predicted to be driver
mutations, supporting the concept that splicing factors are
important in cancer development (13,14).
Splicing factor 2/alternative splicing factor (SF2/ASF) is a member
of the arginine/serine-rich splicing factor family and has been
identified as a proto-oncogene that is amplified in human tumors
and can transform immortalized mouse fibroblasts, which form
sarcomas in nude mice (15).
Recently, it has been proposed that SF2/ASF is protumorigenic in
HCC through increased alternative splicing and consequent
inactivation of Krüppel-like factor 6 (KLF6), a zinc finger
transcription factor that inhibits cellular growth in part by
transcriptional activation of p21 (16,17).
However, it remains unknown whether SF2/ASF also employs other
mechanism(s) to contribute to HCC development.
The current study was undertaken to investigate the
mechanism(s) other than KLF6 inactivation by which SF2/ASF
contributes to the development of HCC.
Materials and methods
Cell culture and transfection
Cells (SMMC-7721 and BEL-7402) were purchased from
the Shanghai Institutes for Biological Sciences (Shanghai, China)
and were cultured in Dulbecco's modified Eagle's medium
supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA),
100 U/ml penicillin and 100 µg/ml streptomycin
(Sigma-Aldrich, St. Louis, MO, USA) and were maintained at 37°C
with 5% CO2. Small interfering RNAs (siRNAs) against
human SF2/ASF (GCATCTACGTGGGTAACTT, GGAGTTTGTACGGAAAGAA) and
non-targeting control siRNA were synthesized by Shanghai GenePharma
Co., Ltd. (Shanghai, China). Transfection was performed with
Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA,
USA) according to the the manufacturer's instructions.
ELISA
Cells were stimulated with or without 10 ng/ml TNF-α
(R&D Systems, Minneapolis, MN, USA) for 24 h. Then, interleukin
(IL)-6 levels in the supernatants were measured using an ELISA kit
(eBioscience, San Diego, CA, USA) according to the manufacturer's
instructions.
Immunoblotting analysis
Cells were washed twice with ice-cold
phosphate-buffered saline (PBS) and were then lysed with 20 mM
Tris/HCl (pH 7.6), 250 mM NaCl, 3 mM EDTA,3 mM EGTA, 0.5% NP-40, 1
mM DTT, 5 mM NaF, 2 mM Na3VO4 and 0.2
µM Aprotinin. The whole cell extract was clarified at 4°C at
13,800 x g for 15 min. The quantity of protein recovered was
quantified with a Bradford protein assay (Invitrogen Life
Technologies). Equal quantities of protein were resolved by sodium
dodecy1 sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
then transferred to Hybond-P polyvinylidene difluoride (PVDF)
membranes (GE Healthcare Life Sciences, Chalfont, UK). Membranes
were sequentially incubated with primary antibody overnight at 4°C
and horseradish peroxidase-conjugated secondary antibody for 1 h at
room temperature. Bound antibody was detected using an enhanced
chemiluminescence kit (GE Healthcare Life Sciences) and Kodak X-ray
film (Rochester, NY, USA). Antibodies against SF2 (sc-33652), actin
(sc-8432), IKKα/β (sc-7607) and p38 (sc-535) were purchased from
Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Antibodies
against phospho-IKKα/β (2697S), phospho-JNK (9251S), phospho-p38
(9215S), phospho-ERK (9102S) and ERK (4370S) were obtained from
Cell Signaling Technology Inc. (Danvers, MA, USA). Antibodies
against JNK (612541) were obtained from BD Biosciences (Franklin
Lakes, NJ, USA). Antibodies against phospho-Akt (2118-1) were
purchased from Epitomics (Cambridge, MA, USA).
Apoptosis analysis
Cells were adjusted to a density of 2×105
cells/ml, added to 24-well plates in 0.5 ml per well regular
culture medium. Cells were treated with 10 ng/ml TNF-α and 1
µg/ml cycloheximide (CHX, Sigma-Aldrich) for 24 h. Cells
were washed with PBS twice and stained with Annexin V-phycoerythrin
and 7-AAD (Nanjing KeyGen Biotech, Nanjing, China) for 15 min at
room temperature in the dark. The level of apoptosis was determined
by measuring the fluorescence of the cells with a flow cytometer
(FACSCalibur; BD Biosciences).
Statistical analysis
Statistically significant differences between groups
were identified using 2-tailed Student's t-test. Statistical
analysis was conducte using SPSS software, version 13.0 (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
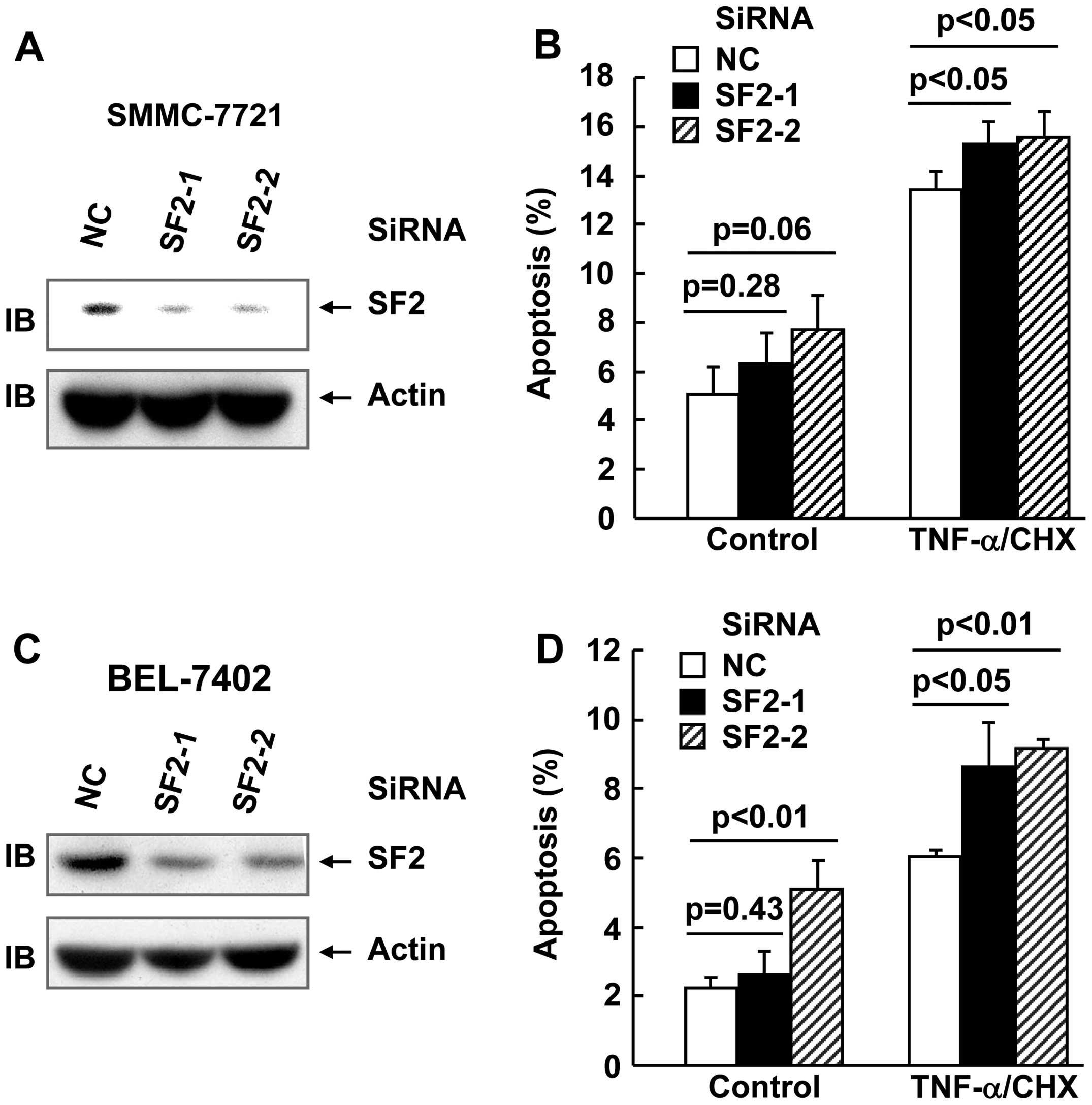
SF2 knockdown marginally enhanced
TNF-α-induced cell death in hepatoma cells
In order to explore whether SF2/ASF affects
TNF-α-induced cell death and TNF-α-induced activation of multiple
signaling pathways in hepatoma cell lines, two siRNAs were designed
against SF2. SMMC-7721 and BEL-7402 hepatoma cells were transfected
with small interfering (si)RNA against SF2 or the non-targeting
control (NC) siRNA. After (48 h) transfection, cell lysates were
obtained and subjected to immunoblotting. As shown in Fig. 1A and B, the SF2 siRNAs designed
significantly inhibited SF2 expression, as compared with the
non-targeting control siRNA. TNF-α usually does not trigger cell
death unless de novo protein synthesis is blocked by
reagents, such as CHX (18). Under
these conditions, hepatoma cells with or without SF2 knockdown were
subjected to TNF-α/CHX treatment for 24 h. Apoptosis analysis
revealed that SF2 knockdown exhibited marginal increase in the
cytotoxicity of TNF-α in the two cell lines (Fig. 1C and D).
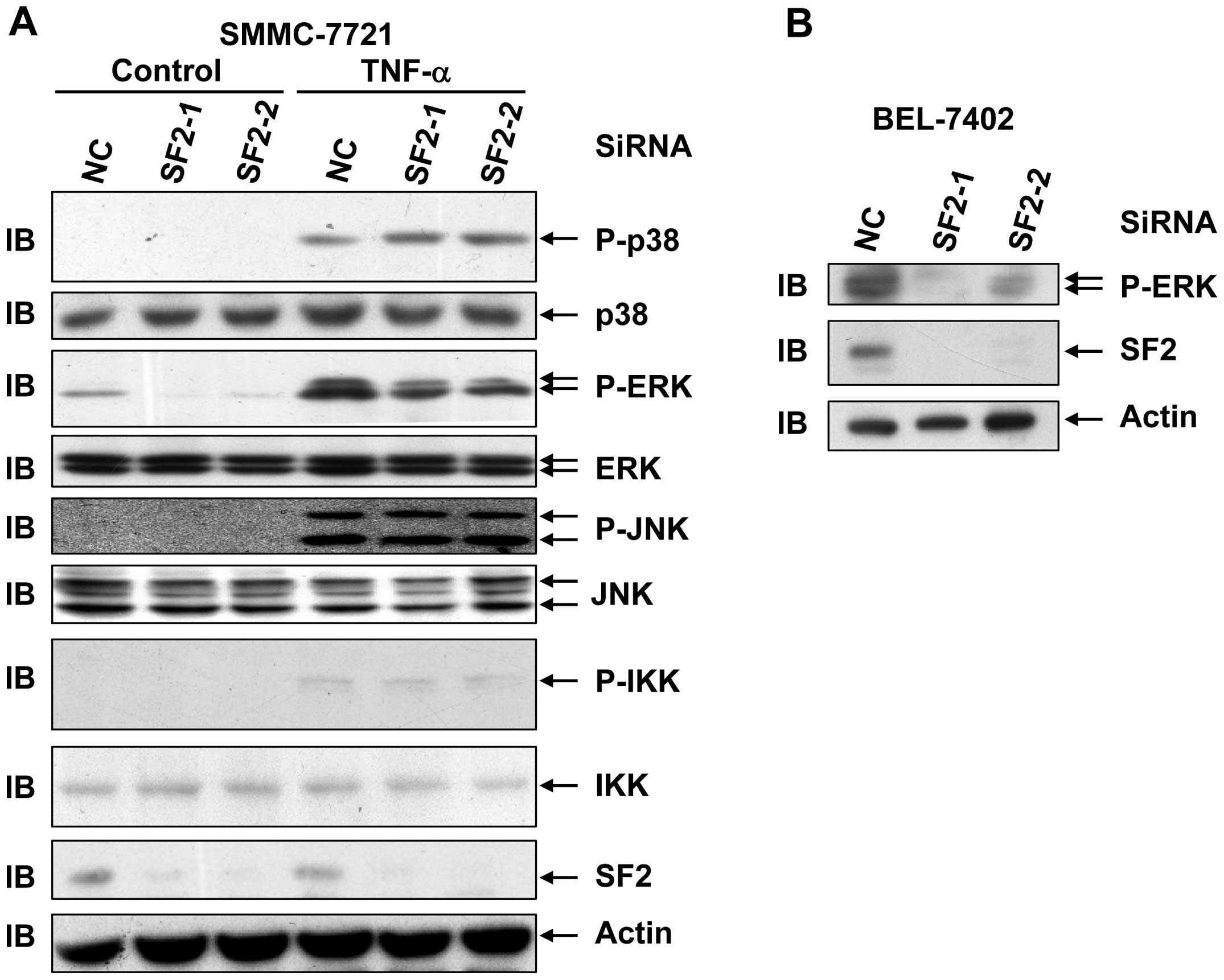
SF2 knockdown leads to reduced levels of
basal ERK activation as well as TNF-α-induced ERK activation in
hepatoma cells
The study also aimed to investigate whether SF2/ASF
affects the cytotoxicity of TNF-α by interfering with TNF-α-induced
activation of signaling pathways in hepatoma cell lines. For this
purpose, SMMC-7721 cells with or without SF2 knockdown were
subjected to TNF-α treatment for 15 min. Immunoblotting analysis
revealed that SF2 knockdown had no effect on TNF-α-induced
activation of the JNK pathway, the p38 pathway or the IKK pathway
in hepatoma cells (Fig. 2A).
However, SF2 knockdown led to reduced levels of basal ERK
activation as well as TNF-α-induced ERK activation without changing
the protein levels of ERK (Fig.
2A). The effect of SF2 knockdown on ERK activation was also
observed in BEL-7402 cells (Fig.
2B). As ERK exhibits a pro-survival role, it is possible that
SF2 antagonizes the cytotoxicity of TNF-α by augmenting ERK
activity.
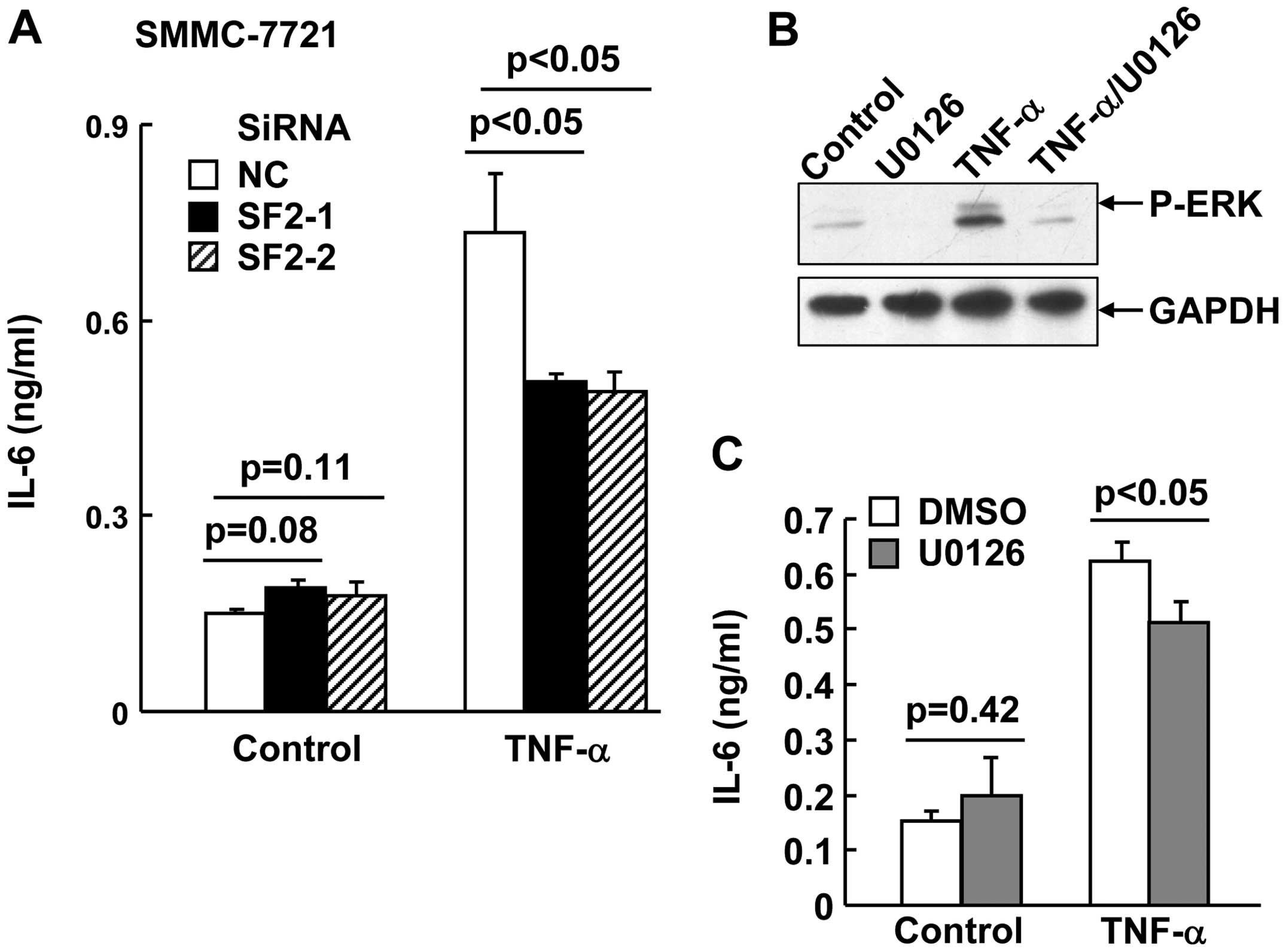
SF2 knockdown and blockade of ERK
activation suppress TNF-α-induced IL-6 production in hepatoma
cells
In addition to TNF-α, IL-6 is also a key
proinflammatory cytokine involved in HCC development (6). ERK has been demonstrated to
contribute to TNF-α-induced IL-6 production (9). In this scenario, it is of interest to
investigate whether SF2 promotes TNF-α-induced IL-6 production.
SMMC-7721 cells with or without SF2 knockdown were subjected to
TNF-α treatment for 24 h. ELISA analysis with the supernatant
revealed that SF2 knockdown resulted in partially reduced IL-6
production in response to TNF-α (Fig.
3A). Consistently, U0126, a specific inhibitor of the ERK
pathway (Fig. 3B), also partially
suppressed TNF-α-induced IL-6 production in SMMC-7721 cells
(Fig. 3C).
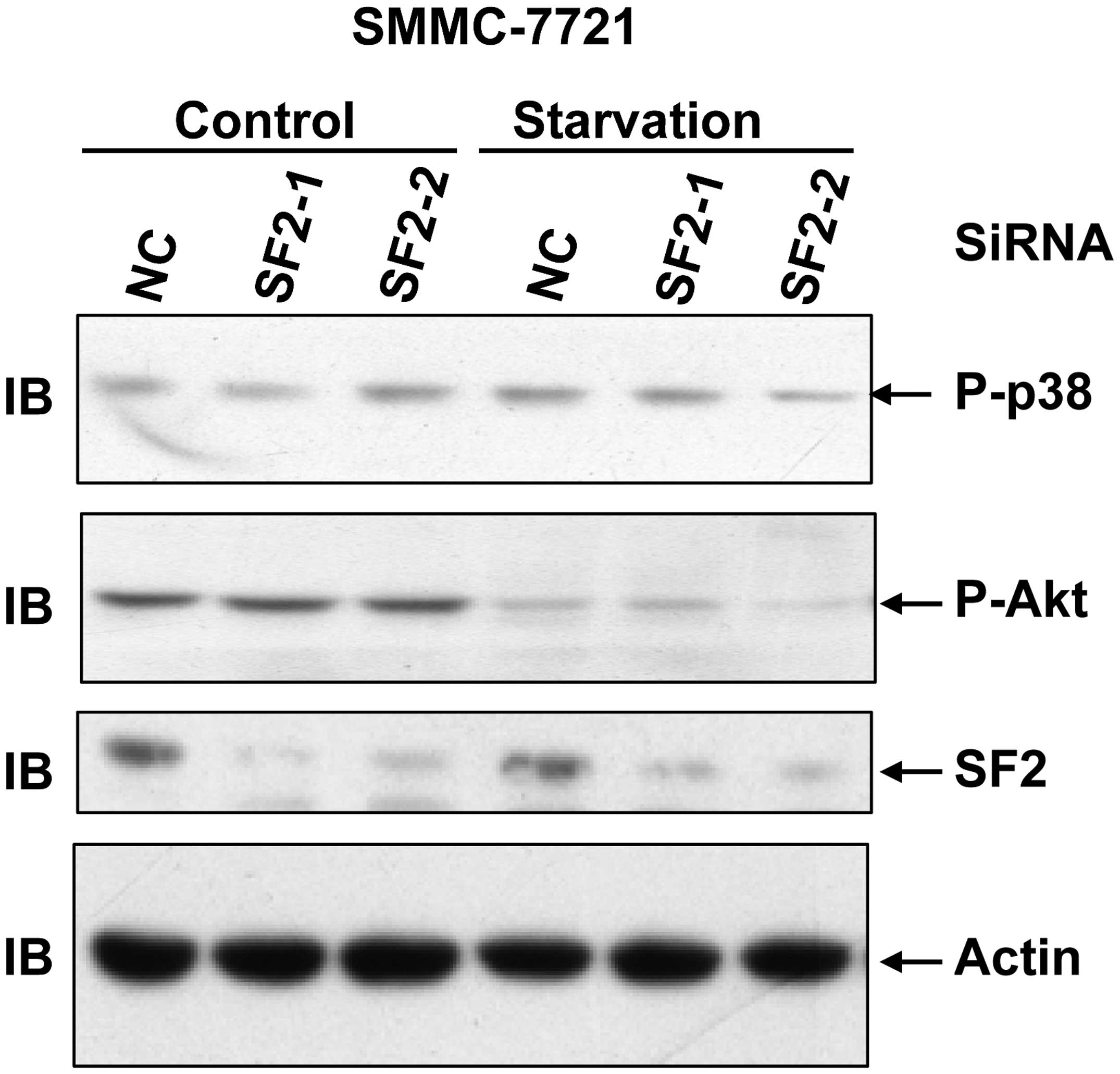
SF2 knockdown is not involved in Akt
activation in hepatoma cells
ERK is activated not only by TNF-α, but also by
growth factors. Growth factors tend to simultaneously activate the
ERK pathway and the Akt pathway (2). To investigate whether SF2 contributes
to ERK activation by affecting the levels of the growth factor
receptors, Akt activation with or without serum starvation was
conducted as it is known that serum contains various growth
factors. As SF2 knockdown exhibited no role in basal Akt activation
and serum-induced Akt activation (Fig.
4), it is unlikely that SF2 affects ERK activation through
modulating the protein levels of certain growth factor
receptors.
Discussion
Recently, splicing factor oncoprotein SRSF1 has been
shown to be a potent proto-oncogene that is upregulated in numerous
types of cancer and can transform immortal mouse and human cells
(15,19). SF2 is a member of the splicing
factor family, which is important in the maintenance of cell
growth, proliferation and inflammation (17,20,21).
However, the underlying mechanisms remain unclear.
Certain studies show that targeting SF2 may be a
strategy for the treatment of leukemia as SF2 silencing promotes
the apoptosis of white blood cells (13,14).
Whether SF2 exhibits the same role in liver cancer cells remains
unclear. In the present study, it was demonstrated that SF2
knockdown leads to reduced levels of basal ERK activation, as well
as TNF-α-induced ERK activation without changing the protein levels
of ERK. As SF2 knockdown exhibited no role in basal Akt activation
and serum-induced Akt activation, SF2 affects ERK activation
through modulating molecular events upstream of ERK, but downstream
of the receptors. Consistently, SF2 knockdown only suppresses ERK
activation, but not p38/JNK activation in response to TNF-α.
ERK may promote HCC development through various
mechanisms, including enhancing cell proliferation, cell survival
and IL-6 production. Since SF2 contributes to ERK activation in
such cells, SF2 knockdown marginally enhanced TNF-α-induced cell
death and partially suppressed TNF-α-induced IL-6 expression. In
conclusion, the present data support the notion that SF2 may be a
therapeutic target for the treatment of hepatocellular
carcinoma.
References
|
1
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Worster DT, Schmelzle T, Solimini NL,
Lightcap ES, Millard B, Mills GB, Brugge JS and Albeck JG: Akt and
ERK control the proliferative response of mammary epithelial cells
to the growth factors IGF-1 and EGF through the cell cycle
inhibitor p57Kip2. Sci Signal. 5:ra192012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Wang Q, Zhu N, Yu M, Shen B,
Xiang J and Lin A: Cyclic AMP inhibits JNK activation by
CREB-mediated induction of c-FLIPL and MKP-1, thereby
antagonizing UV-induced apoptosis. Cell Death Differ. 15:1654–1662.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yan J, Xiang J, Lin Y, Ma J, Zhang J,
Zhang H, Sun J, Danial NN, Liu J and Lin A: Inactivation of BAD by
IKK inhibits TNF-induced apoptosis independently of NF-κB
activation. Cell. 152:304–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He G and Karin M: NF-kappaB and STAT3 –
key players in liver inflammation and cancer. Cell Res. 21:159–168.
2011. View Article : Google Scholar
|
|
6
|
Naugler WE, Sakurai T, Kim S, et al:
Gender disparity in liver cancer due to sex differences in
MyD88-dependent IL-6 production. Science. 317:121–124. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balkwill F and Coussens LM: Cancer, an
inflammatory link. Nature. 431:405–406. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakurai T, Itoh K, Higashisuji H,
Nonoguchi K, Liu Y, Watanabe H, Nakano T, Fukumoto M, Chiba T and
Fujita J: Cirp protects against tumor necrosis factor-α-induced
apoptosis via activation of extracellular signal-regulated kinase.
BBA-Mol Cell Res. 1763:290–295. 2005.
|
|
9
|
Suarez-Cuervo C, Harris KW, Kallman L,
Vaananen HK and Selander KS: Tumor necrosis factor-alpha induces
interleukin-6 production via extracellular-regulated kinase 1
activation in breast cancer cells. Breast Cancer Res Treat.
80:71–78. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Venables JP: Aberrant and alternative
splicing in cancer. Cancer Res. 64:7647–7654. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Srebrow A and Kornblihtt AR: The
connection between splicing and cancer. J Cell Sci. 119:2635–2641.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim E, Goren A and Ast G: Insights into
the connection between cancer and alternative splicing. Trends
Genet. 24:7–10. 2008. View Article : Google Scholar
|
|
13
|
Yoshida K, Sanada M, Shiraishi Y, et al:
Frequent pathway mutations of splicing machinery in myelodysplasia.
Nature. 478:64–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Quesada V, Ramsay AJ and Lopez-Otin C:
Chronic lymphocytic leukemia with SF3B1 mutation. N Engl J Med.
366:25302012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karni R, de Stanchina E, Lowe SW, Sinha R,
Mu D and Krainer AR: The gene encoding the splicing factor SF2/ASF
is a proto-oncogene. Nat Struct Mol Biol. 14:185–193. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yea S, Narla G, Zhao X, Garg R, Tal-Kremer
S, Hod E, Villanueva A, Loke J, Tarocchi M, Akita K, Shirasawa S,
Sasazuki T, Martignetti JA, Llovet JM and Friedman SL: Ras promotes
growth by alternative splicing-mediated inactivation of the KLF6
tumor suppressor in hepatocellular carcinoma. Gastroenterology.
134:1521–1531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Muñoz Ú, Puche JE, Hannivoort R, Lang UE,
Cohen-Naftaly M and Friedman SL: Hepatocyte growth factor enhances
alternative splicing of the Kruppel-like factor 6 (KLF6) tumor
suppressor to promote growth through SRSF1. Mol Cancer Res.
10:1216–1227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang G, Minemoto Y, Dibling B, Purcell NH,
Li Z, Karin M and Lin A: Inhibition of JNK activation through NF-κB
target genes. Nature. 414:313–317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Anczuków O, Rosenberg AZ, Akerman M, Das
S, Zhan L, Karni R, Muthuswamy SK and Krainer AR: The splicing
factor SRSF1 regulates apoptosis and proliferation to promote
mammary epithelial cell transformation. Nat Struct Mol Biol.
19:220–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Geuking MB, Cahenzli J, Lawson MA, Ng DC,
Slack E, Hapfelmeier S, McCoy KD and Macpherson AJ: Intestinal
bacterial colonization induces mutualistic regulatory T cell
responses. Immunity. 34:794–806. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Panasyuk G, Nemazanyy I, Zhyvoloup A,
Filonenko V, Davies D, Robson M, Pedley RB, Waterfield M and Gout
I: mTORbeta splicing isoform promotes cell proliferation and
tumorigenesis. J Biol Chem. 284:30807–30814. 2009. View Article : Google Scholar : PubMed/NCBI
|