Introduction
Developing treatment strategies for diabetes, a
metabolic disease characterized by a chronic increase in blood
glucose levels resulting from defects in insulin secretion, is an
acknowledged challenge. As a potential therapeutic drug,
glucagon-like peptide 1 (GLP-1) has several physiological benefits,
including improving the synthesis and secretion of insulin,
inhibiting the secretion of glucagon, promoting the proliferation
and genesis of β-cells, inhibiting the apoptosis of β-cells,
reducing food intake, slowing gastric emptying, improving insulin
utilization in peripheral tissues and reducing the output of
glycogen (1). Despite its numerous
attributes, natural GLP-1 has a half-life of ~1–2 min and is
rapidly degraded by dipeptidyl peptidase IV. As a result of these
disadvantages, natural GLP-1 cannot be used directly to treat type
2 diabetes mellitus. Liraglutide (LRG) is a long-acting GLP-1
analogue, which is not degraded as easily as natural GLP-1 and
retains the pleiotropic effects of GLP-1, has been reported to
prevent multiple risk factors of diabetes by reducing blood
glucose, protecting β-cells, reducing body weight, reducing blood
pressure and regulating blood lipids (2).
Previous studies have confirmed the safety of LRG
and its advantages in treating type 2 diabetes mellitus and
preventing cardiovascular diseases (3,4).
However, the exact mechanisms involved remain to be elucidated.
Palmitic acid may activate an endoplasmic reticulum (ER) stress
response in β-cells and induce cellular dysfunction, while
autophagy effectively reduces ER stress (5,6). It
has been confirmed that autophagy, which caused many physiological
accommodations, was closely with pancreatic β-cells in previous
studies (7–10). Autophagy may not only protect
pancreatic β-cells from apoptosis induced by external stimuli, but
it may also maintain the structure, number and functionality of
β-cells, and internal homeostasis (11,12).
Autophagy is associated with the autophagy-lysosome
pathway, which has been suggested to remove excess or damaged
organelles and maintain internal hemostasis, thereby regulating the
growth, development and aging of cells, and correcting
hypermetabolism (13). Adverse
conditions, including oxidative stress and intracellular lipid
deposition, may induce autophagy (14). Persistent adverse conditions may
even induce metabolic diseases, including type 2 diabetes mellitus,
lipid metabolism disorders and metabolic syndrome (14). Previous studies have demonstrated
that several factors, including nutrient deficiencies, insulin
resistance, energy deficiency, hypoxia, injury, pathogen infection
and ER stress, may induce autophagy (15). Autophagy remains at a low level
under normal conditions and effective induction of autophagy is
critical for stress adaptation (16).
The universally accepted gold standard for the
detection of autophagy is the identification of autophagosomes
(double-membrane vesicles) and associated subcellular structures.
The most commonly used methods in previous studies include
detecting the transformation of light chain 3 (LC3) (LC3B/lC3A), a
biomarker of autophagy, using western blot analysis, and detecting
aggregation of the LC3 expression plasmid, which contains green
fluorescent protein (GFP), under a fluorescence microscope. The
formation of LC3B and an increase in green fluorescence are
objective evidence of the presence of autophagy (17). Knockout of autophagy-associated
genes (ATGs) and the use of autophagy inhibitors have been widely
used in autophagy inhibition experiments (18). As an autophagy inhibitor with a low
toxicity and the ability to inhibit autophagy in vitro and
in vivo, chloroquine has been widely used in investigations
associated with autophagy (19).
Another autophagic activity marker protein is
nucleoporin p62 (p62). When autophagy occurs, p62 combines with
ubiquitinated proteins and binds to LC3B to form a complex, which
degrades in autophagolysosomes, resulting in a decrease in the
expression of p62 (20). When the
activity of autophagy is inhibited, p62 accumulates in the
cytoplasm and its expression increases (20). Autophagy protein 5 (ATG5) is
another autophagy-associated gene, which is important in the
formation and extension of the membrane of autophagosomes (21). The Beclin1 gene, which is a homolog
of ATG6, is also a specific gene that is involved in autophagy in
mammals. The Beclin1 gene combines with phosphatidylinositol
3-kinase (PI3K) to form a complex, which regulates the activity of
autophagy through the regulation of other ATG proteins (22,23).
Previous studies have demonstrated that upregulation of Beclin1 may
effectively promote autophagy in cells from mammals (24), and reports have indicated that the
Beclini1-GFP complex is widely distributed in the cytoplasm
throughout the whole body, not restricted to several specific cells
(25–28). These results contradict previous
observations that Beclin1 is restricted to the trans-Golgi network,
suggesting that Beclin1 may have more extensive effects than
expected (29). In embryonic
experiments, excessive apoptosis was observed in Beclin1-deprived
embryos, suggesting that Beclin1 is a potential autophagy inhibitor
(30).
ER is inactivated under normal conditions. The three
trans-membrane proteins on the ER membrane, inosito1-requiring
transmembrane kinase (IRE-1), pancreatic eukaryotic initiation
factor-2α kinase-like ER kinase (PERK) and activation transcription
factor 6 (ATF6) may bind to GRP78, a molecular chaperone, under
normal conditions, exerting no biological activity. PERK, ATF6 and
IRE-1 receive ER stress signals and transmit those signals into the
nuclear membrane and cytoplasm to assist the adaptation of cells to
the initial stress state. In addition, unfolded proteins induce the
detachment of GRP78 from the three transmembrane proteins. The
interaction of these three transmembrane proteins activates
different stress pathways to reduce the accumulation of unfolded
and misfolded proteins, to ensure the correct folding of exported
proteins (31). The PERK, ATF6 and
IRE-1 signaling pathways may activate the survival pathway of ER
stress under mild stimulation, while persistent or prolonged ER
stress may activate the downstream pro-apoptotic signaling
molecule, CHOP/GADD153 (32,33).
CHOP/GADD153 is a growth inhibitor which induces DNA damage at a
genetic level. Previous studies have demonstrated that increased
expression of CHOP significantly reduce the expression of the
B-cell lymphoma 2 (Bcl-2) anti-apoptotic protein, reduce the
synthesis of glutathione and increase the level of reactive oxygen
species in cells, which may in turn induce apoptosis of the cells
(34).
Previous studies have demonstrated that GLP-1
activates the cyclic adenosine monophosphate and PI3K pathways to
increase the levels of Bcl-2 and Bcl-extra large (Bcl-x1), and
reduce the level of caspase-3, thereby inhibiting apoptosis and
promoting the proliferation of β-cells (35). It has also been hypothesized that
LRG upregulates the production of nitric oxide to improve
anti-inflammatory effects, which may protect pancreatic β-cells
from apoptosis (36). LRG may also
inhibit the expression of adhesion factors and reduce the function
of endothelial cells in apolipoprotein E (ApoE)−/− mice
(37). Therefore, the protective
effects of LRG on pancreatic β-cells have been widely accepted,
however, the mechanisms underlying these effects remain to be fully
elucidated.
The present study aimed to investigate the
protective effects of LRG on pancreatic β-cells. Free fatty acids
(FFA) and LRG were administered to pancreatic β-cells, apoptosis
was determined and the mechanisms underlying the activation of
autophagy by LRG were identified. The effects of autophagy
inhibitors on the protective effects of LRG were also investigated.
In an ApoE−/− mice diabetic model, the effects of LRG on
body weight, blood parameters and the formation of autophagosomes
were measured to evaluate the level of autophagy.
Materials and methods
Cell line
The INS-1 insulinoma pancreatic islet cell line was
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China) and was cultured at 37°C with 5%
CO2. The cells (1×106) were seeded into
6-well plates and treated with either 1 mM FFA (Gibco Life
Technologies, Carlsbad, CA, USA), 80 nM LRG (Novo Nordisk Company,
Aalborg Øst, Denmark) or 50 µM chloroquine Sigma-Aldrich,
St. Louis, MO, USA).
MTT assay
At a confluence of 75–80%, 0.25% trypsin was used to
digest the cells (37°C for 1 min). RPMI-1640 culture medium (Gibco
Life Technologies, Carlsbad, CA, USA), containing 10% fetal bovine
serum was added when the cells began to shrink and revert. The
cells were resuspended and the density was adjusted to
5×104/ml. The cells were cultured in a 96-well culture
plate (100 µl/well) overnight at 37°C to allow attachment.
The cells were then treated with either normal culture medium (CON
group), FFA (FFA group), LRG (LRG group) or LRG and FFA (FFA + LRG
group) for 36 h. Each group contained three or four repeats.
Following treatment, MTT (20 µl/well) was added, the culture
medium was carefully removed, and 150 µl dimethyl sulfoxide
was added to each well. The culture plate was then placed on a
shaker and agitated at a low speed (18 × g) for 10 min. The
absorbance density (490 nm) of each well was measured using an
enzyme-linked immunometric meter (Modulus™; YPH BIO technology,
Ltd., Beijing, China) at 490 nm.
Annexin V/propidium iodide (PI) double
staining flow cytometry
The cells (2×105) were stained with PI
and fluorescein isothiocyanate-annexin V (Sigma-Aldrich) for 15 min
and the percentages of apoptotic cells were analyzed by flow
cytometry using a BD FACSVerse™ flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA).
ECM
The cells in each group were treated for 36 h, as
described above, following which 0.25% trypsin was used for
collection. The cells were placed in an Eppendorf tube for fixation
(fixative provided by Weiya Electron Microscopy Center, Shanghai,
China), then were centrifuged at 5,000 × g for 15 min and
transferred to the Weiya Electron Microscopy Center for
observation, imaging and analysis.
Plasmid transfection
The LC3 expression plasmid containing GFP, provided
by Professor Jin Shengkan at the University of Medicine and
Dentistry of New Jersey (New Brunswick, NJ, USA) was transfected
into the INS-1 cells in the different treatment groups using
Lipofectamine™ 2000 Transfection reagent (Gibco Life Technologies).
A fluorescence microscope (CX41-32RFL; Olympus Corporation, Tokyo,
Japan) was used to detect the localization of GFP in the cultured
cells and determine the fluorescence, reflecting autophagy.
Immunoblotting
Whole cell lysates were prepared in
radio-immunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Haimen, China). The protein content was determined
using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) with 5% bovine serum albumin (Gibco Life
Technologies). Equal quantities of protein were separated using 8%
SDS-PAGE (Shanghai Reagent Manufactory, Shanghai, China) and
electro-transferred onto polyvinylidene fluoride membranes.
Following blocking with 5% non-fat milk, the membranes were
incubated with the following primary antibodies: anti-LC3B antibody
(2775S; Cell Signaling Technology, Inc., Danvers, TX, USA),
anti-CHOP antibody YT0912; ImmunoWay Biotechnology Company, Newark,
DE, USA), anti-GRP78 (G9043), anti-p62 (N1163), anti-ATG7 (A2856)
and anti-Beclin1 antibodies (SAB5300513) (Sigma-Aldrich) at 25°C
for 2 h. The membranes were then incubated with the following
secondary antibody: Goat anti-mouse immunoglobulin G (01-18-02;
Shanghai Reagent Manufactory, Shanghai, China), at 25°C for 2 h.
Subsequent to washing with Tris-buffered saline with 0.1% Tween-20
(Shanghai Reagent Manufactory), signals were detected using an
Enhanced Chemiluminescence kit (GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA).
In vivo experiments
Healthy 6-week-old specific pathogen-free male
ApoE−/− mice (n=36; body weight, 19.12±1.42 g) were
purchased from the Animal Center of Peking University Health
Science Center [Beijing, China; license number, SCXK (jing)
2011–2012]. The mice were maintained at 25°C. Animal care was
provided in accordance with the Guidelines for the Care and Use of
Laboratory Animals (38), and all
experiments were approved by the Ethics Committee of the Jinan
Military Area General Hospital of People's Liberation Army. The
baseline body weights of the mice were similar among the groups and
the animals were allowed free access to food and water. The mice
were randomly divided into four groups, each containing nine mice.
The CON group received normal maintenance food (mass proportion:
100% basic food; energy proportion: 10% fat, 19% protein and 71%
carbohydrate), while the HF group received high-fat food (mass
proportion: 68% basic food, 20% lard, 2% cholesterol, 10% yolk
powder and 0.1% cholate; energy proportion: 56% fat, 9% protein and
34% carbohydrate). The HF + LRG group received high-fat food and
LRG; and the HF + LRG + CQ group received high-fat food, LRG and
chloroquine. With the exception of mice in the CON group, all
animals received a 50 mg/kg STZ injection subsequent to being fed a
diet of high-fat food for 8 weeks, and blood was collected using a
tail-cut method 3 days later. A randomly detected fasting blood
glucose (FBG) ≥11.1 mmol/l was considered a successful model. The
mice in the CON and HF groups received an intraperitoneal injection
of normal saline (5 µl/g, twice/day) for 30 days, mice in
the HF + LRG group received intraperitoneal injections of LRG (1
mg/kg, twice/day) for 30 days, and mice in the HF + LRG + CQ group
received intraperitoneal injections of LRG (1 mg/kg, twice/day) and
chloroquine (50 mg/ml, once/3 days) for 30 days. The body weight,
food intake and water intake of each mouse was measured every 2
weeks. All mice were sacrificed 30 days following LRG/CQ
administration, prior to the collection of blood samples.
Collection of biological parameters
FBG was determined using the glucose oxidase method,
fasting insulin (FINS) and glycated hemoglobin (GHb) were
determined using enzyme-linked immunosorbent assay, plasma
triglyceride (TG), total cholesterol (TC), low density lipoprotein
cholesterin (LDL-C), high density lipoprotein cholesterin (HDL-C)
and FFA levels were determined using enzymatic methods.
Statistical analysis
SPSS software, version 16.0 (SPSS Inc., Chicago, IL,
USA) was used for statistical analyses. All data are expressed as
the mean ± standard deviation. Analysis of variance was used for
comparison among three or more different groups, Student's t-test
was used for comparison between two groups and non-parametric
analysis was used for data with unequal variances. P<0.05 was
considered to indicate a statistically significant difference.
Results
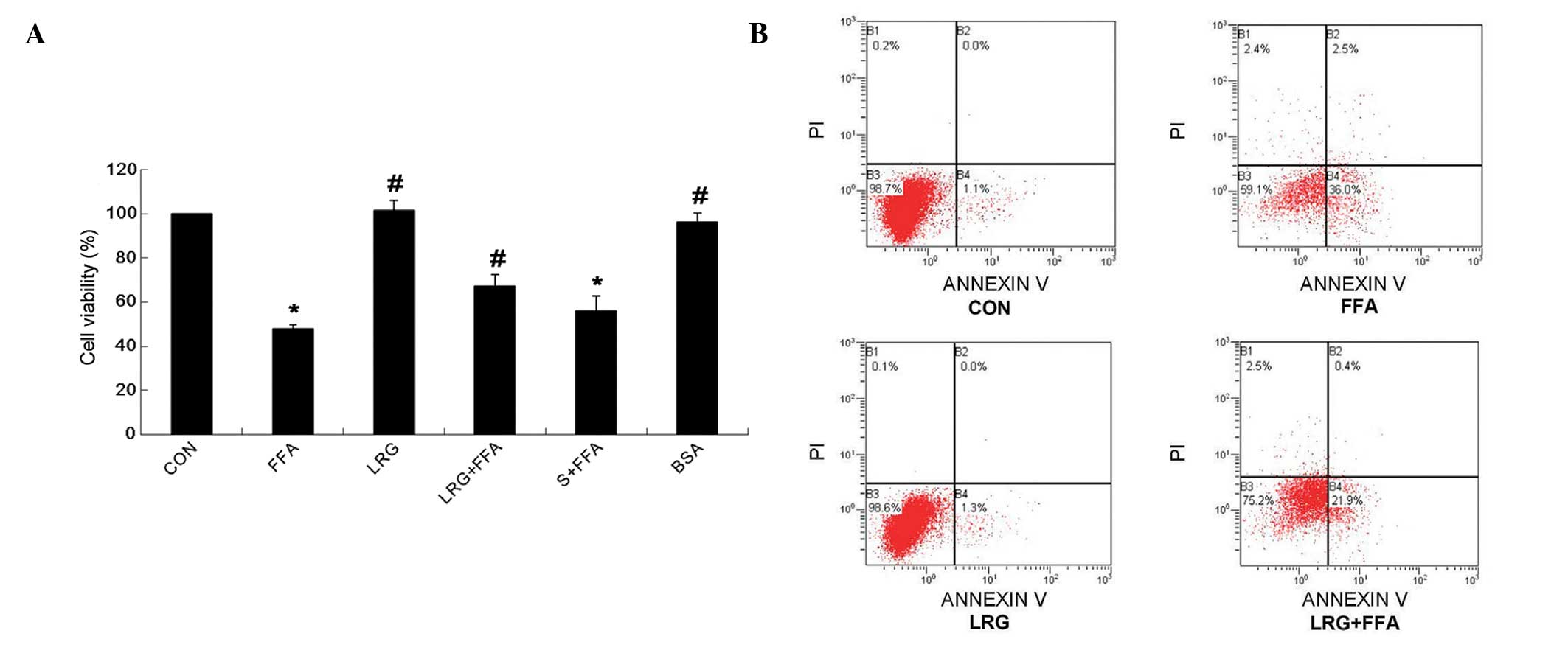
LRG reduces the apoptosis of INS-1 cells
induced by FFA
As shown in Fig. 1,
following treatment of the INS-1 cells with normal culture medium
(CON group), FFA (FFA group), LRG (LRG group) or LRG and FFA (FFA +
LRG group), the cell survival rate was significantly higher in the
FFA + LRG group, compared witht he FFA group (P<0.05; Fig. 1A). The annexin V/PI double labeling
flow cytometric analysis demonstrated that the apoptotic rate was
significantly lower in the FFA + LRG group, compared with the FFA
group (P<0.05). These observations suggested that LRG treatment
reduced the INS-1 cell apoptosis induced by FFA.
LRG protects INS-1 cells by activating
autophagy
Compared with the cells in the CON group, higher
levels of green fluorescence were observed in the INS-1 cells
treated with LRG (Fig. 2A). LC3B,
an indicator of autophagy, was increased in INS-1 cells treated
with 20 and 80 µmol/l LRG for 36 h (Fig. 2B and C). No changes were observed
in the expression of p62, another autophagy indicator, in the
LRG-treated INS-1 cells, however, the expression levels of ATG7 and
Beclin1 were significantly reduced (Fig. 2D). The ECM results demonstrated the
presence of LRG-induced autophagosomes in the ISN-1 cells. Multiple
vesicular bodies with double membranes were present in the LRG and
FFA + LRG groups (arrows in Fig.
2E). Excessive and aged organelles were identified inside the
bodies; and ER swelling and proliferation were also observed. No
autophagosomes were observed in the CON or FFA groups (Fig. 2E). Taken together, these
observations suggested that treating INS-1 cells with LRG
significantly activated autophagy in the cells.
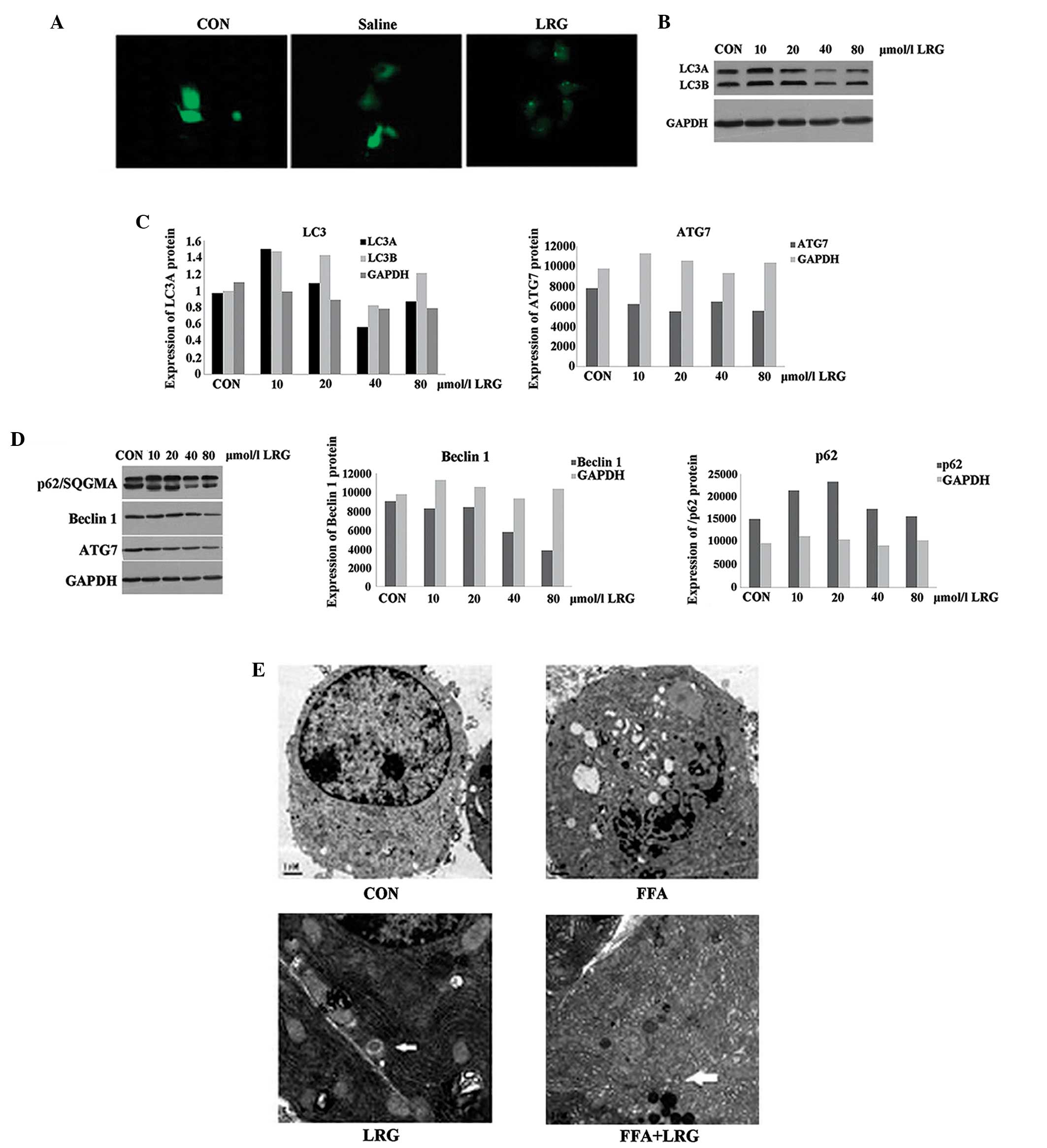 | Figure 2LRG protects INS-1 cells by
activating autophagy. (A) INS-1 cells were transfected with the
GFP-LC3 expression plasmid and then treated with normal saline or
LRG. Cells were observed under fluorescence microscopy
(magnification, ×200). (B) INS-1 cells were transfected with the
GFP-LC3 expression plasmid and then treated with different
concentrations of LRG: L1, 10; L2, 20; L3, 40 or or L4, 80
µmol/l for 36 h. The LC3 protein was detected using
immunoblotting. (C) Gray scanning of LC3A and LC3B normalized to
that of GAPDH. (D) INS-1 cells were transfected with the GFP-LC3
expression plasmid and were then treated with different
concentrations of LRG for 36 h. p62, The ATG7 and Beclin1 proteins
were detected using immunoblotting and gray scanning, normalized to
that of GAPDH; (E) ECM images of INS-1 cells in the CON, FFA, LRG
and FFA + LRG groups. Arrows indicate autophagosomes
(magnification, ×8,000). LRG, liraglutide; GFP, green fluorescent
protein; LC3, light chain 3; CON, control; FFA, free fatty
acids. |
CQ AND 3-MA autophagy inhibitors
significantly reduce the protective effects of LRG
The viability of the INS-1 cells in the CON, FFA,
LRG, FFA + LRG, CQ, CQ + FFA, CQ + LRG and CQ + FFA + LRG groups
were determined using MTT assays, and the results demonstrated that
the cell viability in the CQ + FFA + LRG group was significantly
lower than in the FFA + LRG group (P<0.05; Fig. 3A). Annexin V/PI double labeling
flow cytometry indicated that the apoptotic rate was significantly
higher in the CQ + FFA + LRG group than in the FFA + LRG group
(P<0.05; Fig. 3B). Another
autophagy inhibitor, 3-MA, was also used, instead of CQ, in the
present study to confirm these observations, and the results
indicated that the cell viability in the 3-MA + FFA + LRG group was
significantly lower than in the FFA + LRG group (P<0.05;
Fig. 3C). The results of the
annexin V/PI double labeling flow cytometry produced similar
results in the 3-MA and CQ groups (Fig. 3D). These observations demonstrated
that autophagy inhibitors were able to effectively reduce the
protective effects of LRG on the apoptosis of INS-1 cells induced
by FFA.
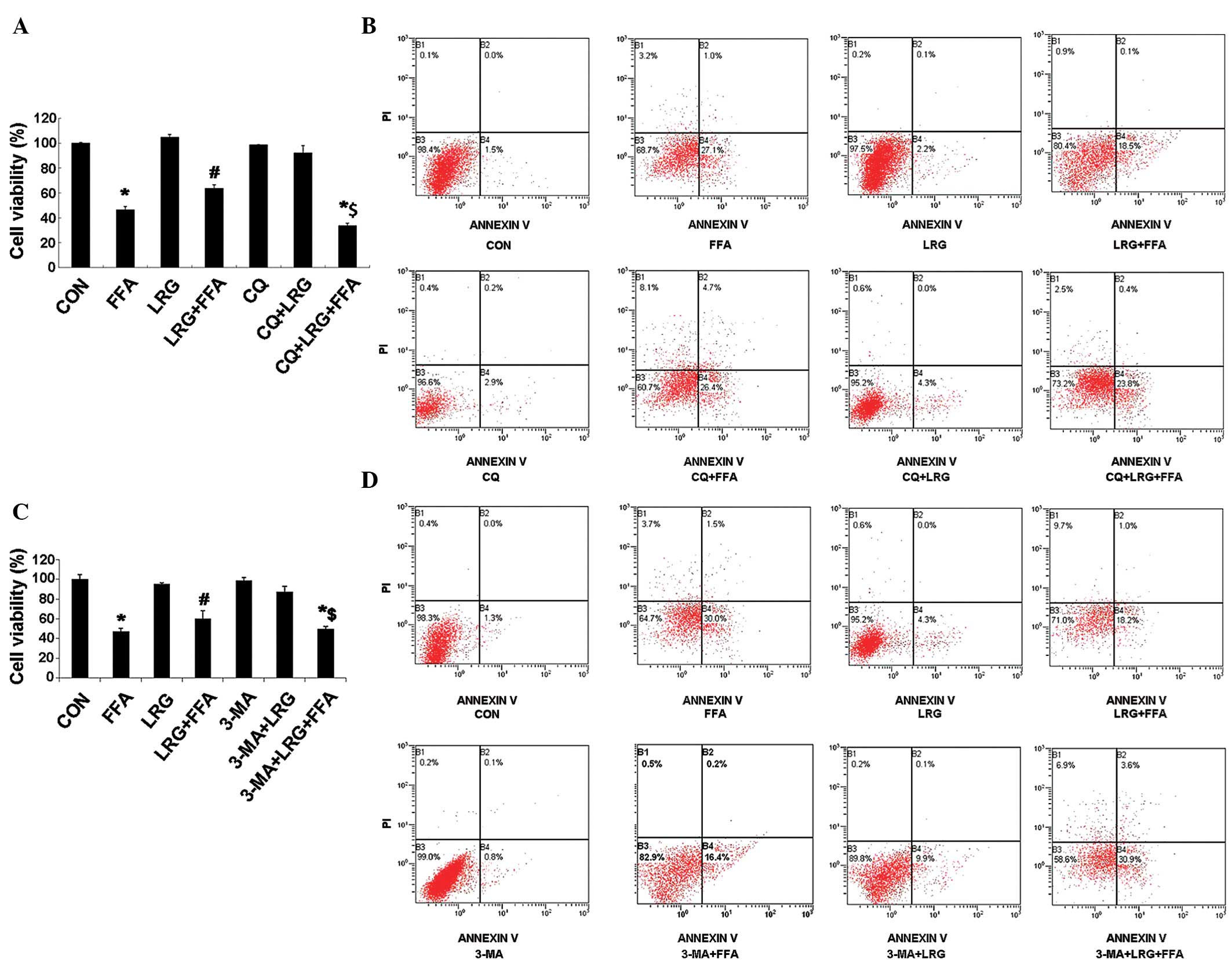 | Figure 3CQ and 3-MA autophagy inhibitors
significantly reduce the protective effects of LRG. (A) Viabilities
and (B) apoptotic rates of the INS-1 cells treated with normal
saline (CON group), FFA, LRG, FFA + LRG, CQ, CQ + LRG or CQ + FFA +
LRG for 24 h. (C) Viabilities and (D) apoptotic rates of the INS-1
cells treated with normal saline (CON group), FFA, LRG, FFA + LRG,
3-MA, 3-MA + LRG and 3-MA + FFA + LRG for 24 h. CQ, chloroquine;
3-MA, 3-methyladenine; LRG, liraglutide; CON, control; FFA, free
fatty acids; PI, propidium iodide. |
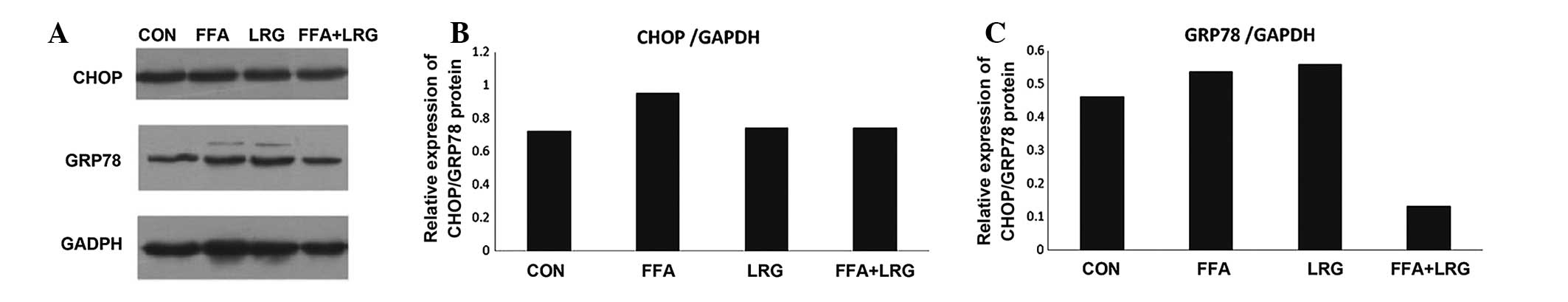
ER stress is involved in activating
autophagy of INS-1 cells, induced by LRG
As shown in Fig. 4,
the levels of GRP78 and CHOP, two ER stress-associated proteins,
were determined using immunoblotting. The results revealed that the
expression levels of GRP78 and CHOP were increased significantly in
the INS-1 cells treated with FFA, while treatment with LRG
significantly reduced the expression of GRP78 and CHOP, compared
with CON treatment. These observations suggested that LRG protected
pancreatic β-cells from FFA-induced apoptosis by activating
autophagy to overcome ER stress.
LRG reduces body weight and improves
glucolipid metabolism, which is reversed by the inhibition of
autophagy
The results of the in vivo experiments are
shown in Fig. 5. The body weights
of the mice in the HF + LRG group were significantly lower than in
the HF group (P<0.05); however, the difference between the HF +
LRG + CQ group and HF group was not statistically different
(P>0.05; Fig. 5A). Following an
8-week high-fat diet and intraperitoneal injection of STZ, the
levels of FBG, TC and LDL-C were significantly higher in the
ApoE−/− mice in the HF group, compared with those in the
CON group (P<0.05); no significant differences were observed in
body weight, TG, FFA, INS and GHb between these two groups
(P>0.05; Fig. 5B). Following
treatment of the mice in the different groups for 30 days, the
levels of FBG, TC, INS, TG, LDL-C and GHb were significantly lower
in the HF + LRG group, compared with those in the HF group
(P<0.05), however, no significant difference was observed in the
level of FFA. No significant differences in the parameters were
observed between the HF + LRG + CQ group and HF group
(P>0.05).
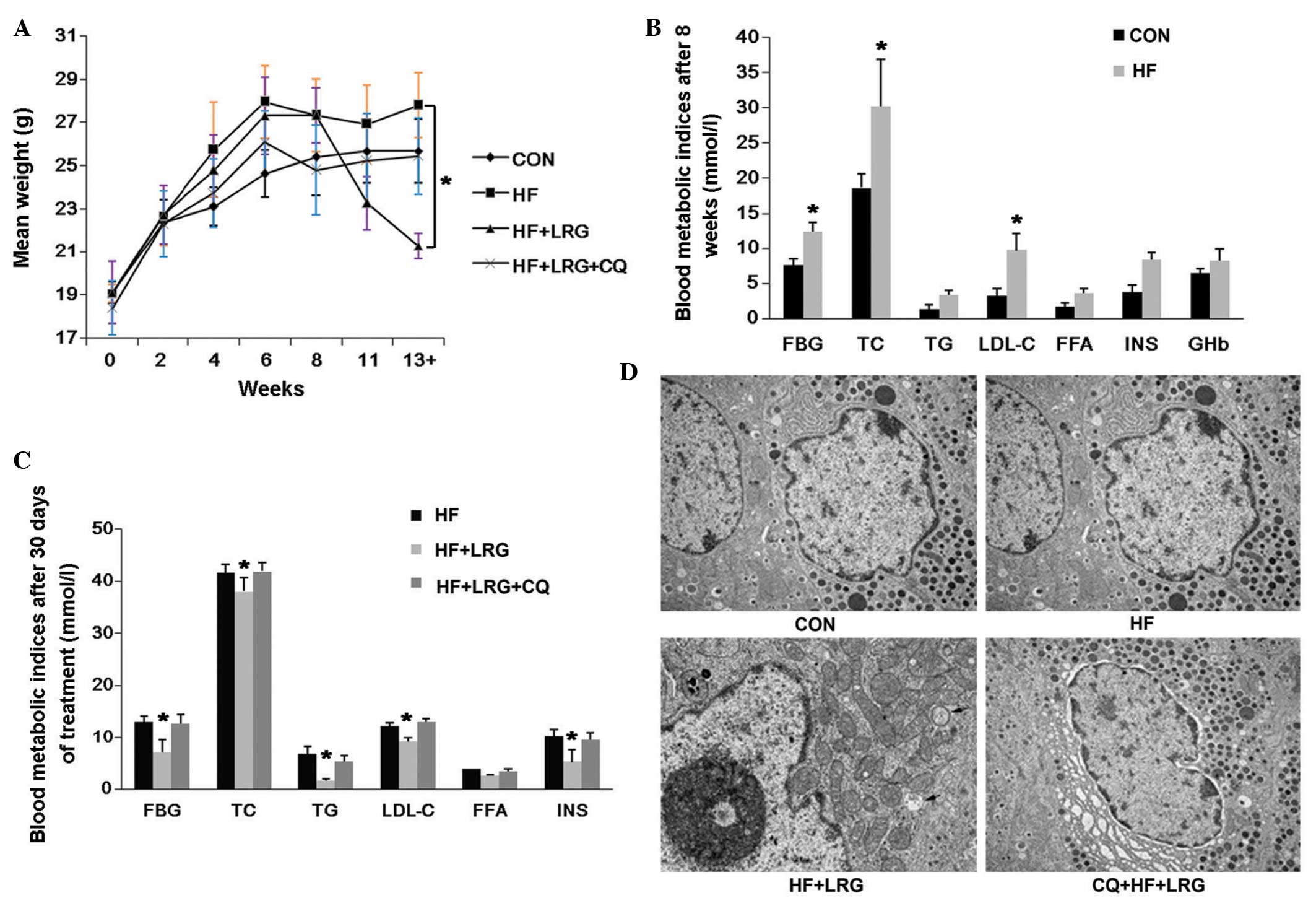 | Figure 5LRG reduces the body weight and
improves glucolipid metabolism of the mice, and these effects are
reversed by the inhibition of autophagy. (A) Body weights of mice
in the normal diet fed group (CON), mice fed with a high-fat diet
for 8 weeks and injected with streptozotocin (HF), mice fed a
high-fat and treated with intraperitoneal injection of LRG (1
mg/kg, twice/day) for 30 days (HF + LRG) and mice fed with a
high-fat and treated with intraperitoneal injection of LRG (1
mg/kg, twice/day) and CQ (50 mg/ml, once/3 days) for 30 days (HF +
LRG + CQ). (B) Blood metabolic indices of mice in the CON and HF
groups after 8 weeks of a normal or high-fat diet. (C) Blood
metabolic indices of mice in HF, HF + LRG and CQ + HF + LRG groups
after 30 days treatment with either normal saline, LRG or LRG + CQ.
(D) Electron microscopy images of pancreatic tissues of mice in the
CON, HF, HF + LRG and CQ + HF + LRG groups. Arrows indicates
autophagosomes (magnification, ×8,000). All data are expressed as
the mean ± standard deviation. LRG, liraglutide; CON, control; HF,
high-fat; CQ, chloroquine; FBG, fasting blood glucose; TC, total
cholesterol; TG, plasma triglyceride; LDL-C, low density
lipoprotein cholesterol; FFA, free fatty acids; INS, fasting
insulin; GHb, glycated hemoglobin. |
ECM examinations were also performed, and one image
was selected from each of the CON, HF, HF + LRG and HF + LRG + CQ
groups. No autophagosome-like micro-structures were identified in
the images of the CON group, however, several vesicular bodies with
double membranes were observed in the HF + LRG group, and excessive
and aged organelles were identified inside these bodies. These
observations demonstrated that LRG treatment induced the formation
of autophagosome in the pancreas of the ApoE−/− mice.
The results also indicated that, in mice in the HF + LRG + CQ
group, autophagy was inhibited and no autophagosomes were
identified. These findings suggested that LRG activated autophagy
in a high-fat high-glucose environment and inhibited the apoptosis
of the pancreatic β-cells, whereas the inhibition of autophagy
decreased the protective effects of LRG on the cells.
Discussion
Treating diabetes with GLP-1 has received increased
attention, and the long-acting GLP-1 analogue, LRG, exhibits the
pleiotropic effects of GLP-1, but is not as easily degraded
(1). Autophagy may not only
protect pancreatic cells from apoptosis induced by external
stimuli, but also maintains the number, structure and functionality
of pancreatic β-cells, and maintains internal homeostasis (11,12,39).
ApoE−/− mice have a high risk of developing lipid
metabolism disorders and have been reported to rapidly develop
hyperlipidemia and atherosclerosis. Previous studies have
demonstrated that estrogen has protective effects on lipid
metabolism, therefore, to evaluate the protective effects of LRG on
pancreatic β-cells, the present study used male ApoE−/−
mice, which were fed a high-fat diet to induce a high lipid model,
and STZ injections were administered to the mice to induce a mouse
model of diabetes (40).
The mechanisms by which LRG preserves pancreatic
β-cells have been investigated in other murine diabetes models. In
a previous study to determine the molecular mechanism by which LRG
preserves pancreatic β-cell mass, obese diabetic db/db mice were
treated with LRG for either 2 days or 2 weeks, while mice with
normal glycemic levels were treated with LRG for 2 weeks. LRG was
found to modify the expression levels of genes associated with cell
apoptosis, including Bcl2, caspase 8, caspase 3 and cadherin in
db/db mice. However, the mRNA levels of these genes were not
altered in mice with normal glycemic levels during short- or
long-term treatment. These observations suggested that LRG directly
suppressed β-cell apoptosis in mice under hyperglycemic conditions
(41). Another study evaluated the
protective effects of LRG in C57B1/6 J mice, in which the mice were
provided with drinking water treated with either corticosterone or
a vehicle for 5 weeks. The mice were then administered with
once-daily injections of either PBS or LRG and, in the C57B1/6 J
diabetes model, LRG promoted the function of the β-cells and slowed
the development of hyperglycemia (42).
The results of the cellular experiments in the
present study demonstrated that LRG protected the pancreatic
β-cells from FFA-induced apoptosis by activating autophagy, which
is associated with ER stress in cells. CQ and 3-MA, two autophagy
inhibitors, significantly reduced the protective effects of LRG.
The results of the animal experiments in the present study also
demonstrated that LRG reduced the body weight of the mice and
improved the glucolipid metabolism, following feeding of the mice
with a high-fat diet. However, the protective effects disappeared
when autophagy was inhibited. LRG also improved blood glucose and
regulated blood lipids in the diabetes model. These improvements
were significantly reduced following the inhibition of autophagy,
suggesting LRG protected the pancreatic β-cells by activating
autophagy.
In the present study, the expression of GRP78, an ER
stress switch protein, was increased in the FFA group, compared
with the CON group, which activated the ER stress pathway and
increased the expression of CHOP, in turn activating the downstream
pathway of apoptosis. The expression of GRP78 was significantly
reduced following the administration of LRG, and no excessive ER
stress was observed. The levels of CHOP and cellular apoptosis were
also significantly reduced. These observations were in accordance
with previous findings, which indicated that LRG protects
pancreatic β-cells from apoptosis by activating autophagy (43). However, the expression levels of
GRP78 and CHOP were not significantly different between the CON and
LRG groups, suggesting LRG did not activate the ER stress pathway
and inhibit the expression of apoptotic factors, unless the
apoptosis pathway had not already been activated. Previous studies
have also demonstrated that ER stress is involved and assists in
protecting the body against the adverse effects induced by damage;
however, in certain cases the damage was too severe for ER
homeostasis to be maintained, and subsequent apoptotic signal
activation results in ER-associated death (44–46).
A previous study demonstrated that ER stress also mediates another
pathway of cell death, namely autophagy (6), which is in accordance with the
observations of the present study.
ApoE−/− mice received a high-fat diet for
8 weeks followed by intraperitoneal injection of STZ to induce a
diabetes model, and LRG was administered for 30 days. The body
weight, FBG, TC, INS, TG and LDL-C of the mice in the HF + LRG
group were significantly lower, compared with those in the HF group
(P<0.05), while no significant difference was observed in FFA
between these two groups. High concentrations of FFA induced
insulin resistance and impaired the insulin producing function of
pancreatic β-cells. In the present study, LRG was unable to
effectively reduce the levels of FFA, suggesting that, although LRG
improved blood glucose, regulated blood lipids and reduce body
weight, it did not improve the sensitivity to insulin, which is a
limitation of LRG.
In conclusion, the in vitro and in
vivo experiments performed in the present study provided
results, which improve the current understanding of the mechanisms
involved in the protective effects of LRG on pancreatic β-cells,
and provided evidence for the prevention of β-cell apoptosis in
clinical practice.
Acknowledgments
This study was supported by the National Natural
Science Funds of China (grant nos. 31100849 and 81070619).
References
|
1
|
Salehi M, Aulinger BA and D'Alessio DA:
Targeting beta-cell mass in type 2 diabetes: promise and
limitations of new drugs based on incretins. Endocr Rev.
29:367–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ritzel RA: Therapeutic approaches based on
beta-cell mass preservation and/or regeneration. Front Biosci
(Landmark Ed). 14:1835–1850. 2009. View
Article : Google Scholar
|
|
3
|
Berlie H, Hurren KM and Pinelli NR:
Glucagon-like peptide-1 receptor agonists as add-on therapy to
basal insulin in patients with type 2 diabetes: a systematic
review. Diabetes Metab Syndr Obes. 5:165–174. 2012.PubMed/NCBI
|
|
4
|
Grieve DJ, Cassidy RS and Green BD:
Emerging cardiovascular actions of the incretin hormone
glucagon-like peptide-1: potential therapeutic benefits beyond
glycaemic control? Br J Pharmacol. 157:1340–1351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laybutt DR, Preston AM, Akerfeldt MC, et
al: Endoplasmic reticulum stress contributes to beta cell apoptosis
in type 2 diabetes. Diabetologia. 50:752–763. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ogata M, Hino S, Saito A, et al: Autophagy
is activated for cell survival after endoplasmic reticulum stress.
Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi SE, Lee SM, Lee YJ, et al: Protective
role of autophagy in palmitate-induced INS-1 beta-cell death.
Endocrinology. 150:126–134. 2009. View Article : Google Scholar
|
|
8
|
Marchetti P and Masini M: Autophagy and
the pancreatic beta-cell in human type 2 diabetes. Autophagy.
5:1055–1056. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hartley T, Brumell J and Volchuk A:
Emerging roles for the ubiquitin-proteasome system and autophagy in
pancreatic beta-cells. Am J Physiol Endocrinol Metab. 296:E1–E10.
2009. View Article : Google Scholar
|
|
10
|
Masini M, et al: Autophagy in human type 2
diabetes pancreatic beta cells. Diabetologia. 52:1083–1086. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ichimura Y, Kumanomidou T, Sou YS, et al:
Structural basis for sorting mechanism of p62 in selective
autophagy. J Biol Chem. 283:22847–22857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marsh BJ, Soden C, Alarcon C, et al:
Regulated autophagy controls hormone content in secretory-deficient
pancreatic endocrine beta-cells. Mol Endocrinol. 21:2255–2269.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fujitani Y, Kawamori R and Watada H: The
role of autophagy in pancreatic beta-cell and diabetes. Autophagy.
5:280–282. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh R, Kaushik S, Wany Y, et al:
Authophagy regulates lipid metabolism. Nature. 458:1131–1135. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eskelinen EL: Maturation of autophagic
vacuoles in Mammalian cells. Autophagy. 1:1–10. 2005. View Article : Google Scholar
|
|
18
|
Heckmann BL, Yang X, Zhang X and Liu J:
The autophagic inhibitor 3-methyladenine potently stimulates
PKA-dependent lipolysis in adipocytes. Br J Pharmacol. 168:163–171.
2013. View Article : Google Scholar :
|
|
19
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mariño G and López-Otín C: Autophagy:
Molecular mechanisms, physiological functions and relevance in
human pathology. Cell Mol Life Sci. 61:1439–1454. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scarlatti F, Maffei R, Beau I, Codogno P
and Ghidoni R: Role of non-canonical Beclin 1-independent autophagy
in cell death induced by resveratrol in human breast cancer cells.
Cell Death Differ. 15:1318–1329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Edinger AL and Thompson CB: Defective
autophagy leads to cancer. Cancer Cell. 4:422–424. 2003. View Article : Google Scholar
|
|
24
|
Liang XH, Jackson S, Seaman M, et al:
Induction of autophagy and inhibition of tumorigenesis by beclin 1.
Nature. 402:672–676. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee DY, Lee J and Sugden B: The unfolded
protein response and autophagy: Herpesviruses rule! J Virol.
83:1168–1172. 2009. View Article : Google Scholar :
|
|
26
|
Li J, Ni M, Lee B, Barron E, Hinton DR and
Lee AS: The unfolded protein response regulator GRP78/BiP is
required for endoplasmic reticulum integrity and stress-induced
autophagy in mammalian cells. Cell Death Differ. 15:1460–1471.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Geng J, Baba M, Nair U and Klionsky DJ:
Quantitative analysis of autophagy-related protein stoichometry by
fluorescence microscopy. J Cell Biol. 182:129–140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pickford F, Masliah E, Britschgi M, et al:
The autophagy-related protein beclin 1 shows reduced expression in
early Alzheimer disease and regulates amyloid beta accumulation in
mice. J Clin Invest. 118:2190–2199. 2008.PubMed/NCBI
|
|
29
|
Arsov I, Li X, Matthews G, et al:
BAC-mediated transgenic expression of fluorescent autophagic
protein Beclin 1 reveals a role for Beclin 1 in lymphocyte
development. Cell Death Differ. 15:1385–1395. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gaytan M, Morales C, Sanchez-Criado JE and
Gaytan F: Immunolocalization of beclin 1, a bcl-2-binding,
autophagy-related protein, in the human ovary: possible relation to
life span of corpus luteum. Cell Tissue Res. 331:509–517. 2008.
View Article : Google Scholar
|
|
31
|
Dawson TR, Lazarus MD, Hetzer MW and Wente
SR: ER membrane-bending proteins are necessary for de novo nuclear
pore formation. J Cell Biol. 184:659–675. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deniaud A, Sharaf el dein O, Maillier E,
et al: Endoplasmic reticulum stress induces calcium-dependent
permeability transition, mitochondrial outer membrane
permeabilization and apoptosis. Oncogene. 27:285–299. 2008.
View Article : Google Scholar
|
|
33
|
Yi M, Chi MH, Khang CH, et al: The ER
chaperone LHS1 is involved in asexual development and rice
infection by the blast fungus Magnaporthe oryzae. Plant Cell.
21:681–695. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Biol. 21:1249–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SJ, Winter K, Nian C, Tsuneoka M, Koda
Y and McIntosh CH: Glucose-dependent insulinotropic polypeptide
(GIP) stimulation of pancreatic beta-cell survival is dependent
upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB)
signaling, inactivation of the forkhead transcription factor Foxo1
and down-regulation of bax expression. J Biol Chem.
280:22297–22307. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hattori Y, Jojima T, Tomizawa A, et al: A
glucagon-like peptide-1 (GLP-1) analogue, liraglutide, upregulates
nitric oxide production and exerts anti-inflammatory action in
endothelial cells. Diabetologia. 53:2256–2263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gaspari T, Liu H, Welungoda I, et al: A
GLP-1 receptor agonist liraglutide inhibits endothelial cell
dysfunction and vascular adhesion molecule expression in an ApoE−/−
mouse model. Diab Vasc Dis Res. 8:117–124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Curran KA and Miller J: Guidelines for
Animal-Assisted Interventions in Health Care Facilities. Am J
Infect Control. 37:257–258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bernales S, Schuck S and Walter P:
ER-phagy: selective autophagy of the endoplasmic reticulum.
Autophagy. 3:285–287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Takács G, Papp S, Lukáts B, et al:
Homeostatic alterations after IL-1beta microinjection into the
nucleus accumbens of the rat. Appetite. 54:354–362. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shimoda M, Kanda Y, Hamamoto S, et al: The
human glucagon-like peptide-1 analogue liraglutide preserves
pancreatic beta cells via regulation of cell kinetics and
suppression of oxidative and endoplasmic reticulum stress in a
mouse model of diabetes. Diabetologia. 54:1098–1108. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fransson L, Dos Santos C, Wolbert P,
Sjoholm A, Rafacho A and Ortsater H: Liraglutide counteracts
obesity and glucose intolerance in a mouse model of
glucocorticoid-induced metabolic syndrome. Diabetol Metab Syndr.
6:32014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen ZF, Li YB, Han JY, et al: Liraglutide
prevents high glucose level induced insulinoma cells apoptosis by
targeting autophagy. Chin Med J (Engl). 126:937–941. 2013.
|
|
44
|
Hetz C: The unfolded protein response:
controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.PubMed/NCBI
|
|
45
|
Zhang SH, Reddick RL, Piedrahita JA and
Maeda N: Spontaneous hypercholesterolemia and arterial lesions in
mice lacking apolipoprotein E. Science. 258:468–471. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li L, Kim J and Boussiotis VA:
IL-1β-mediated signals preferentially drive conversion of
regulatory T cells but not conventional T cells into
IL-17-producing cells. J Immunol. 185:4148–4153. 2010. View Article : Google Scholar : PubMed/NCBI
|