Introduction
Hepatocyte growth factor/scatter factor (HGF/SF),
which is mainly secreted by Kuppfer cells, endothelial cells,
fibroblasts and hepatic stellate cells in the liver, has been shown
to have a role in embryonic organ development, adult organ
regeneration and wound healing (1). HGF regulates cell growth, cell
motility and morphogenesis by binding to its unique receptor c-Met
and by activating a tyrosine kinase-signaling cascade (2–4).
Owing to its functions, including stimulating mitogenesis, cell
motility and matrix invasion, HGF has a central role in
angiogenesis, tumorigenesis and tissue regeneration.
Hepatocellular carcinoma (HCC) is one of most common
malignant tumors; the incidence rate and mortality of HCC is the
fifth- and third-highest in the world, respectively (5). Although several advanced treatments
are now available, including surgical resection, liver
transplantation and ablation therapies (6), and in spite of the development of
molecular-target drugs such as sorafenib (7), the five-year overall survival rate of
patients with advanced HCC remains poor (8). The main reason for the poor survival
rate of advanced HCC is the high rate of recurrence and metastasis
after local treatment (9).
The tumor microenvironment is composed of various
stromal cells, including myofibroblasts, vascular cells and immune
cells, and it has an important role in not only supporting the
growth and survival of tumor cells but also in triggering tumor
recurrence and metastasis (10).
Within the tumor microenvironment, several of the growth factors
secreted by stromal or tumor cells, including HGF, insulin-like
growth factor 1 (IGF1), epidermal growth factor (EGF), vascular
endothelial growth factor (VEGF) and platelet-derived growth factor
(PDGF), may induce a similar signaling cascade downstream of
receptor tyrosine kinase (RTK) and trigger synergistic tumor
recurrence and metastasis (11).
The important role of the HGF/c-Met signaling pathway in
carcinogenesis and development has been well established. For
example, this pathway activates mitogen-activated protein kinase
(MAPK) and phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of
the rapamycin (mTOR) pathways as well as enables cross-talk with
epidermal growth factor and transforming growth factor (TGF)-β
ligands. Overactivation of these pathways promotes proliferation,
survival, migration, invasiveness and angiogenesis of certain
tumors (11,12). In general, the HGF expression
levels in the liver cancer microenvironment after surgery show
marked increases, implicating HGF as one of the main causes of HCC
recurrence and metastasis (13).
In addition to genetic alteration, abnormality of
epigenetic mechanisms is also frequent in carcinogenesis and cancer
progression, including dysregulation of histone modification and
DNA methylation (14). DNA
methylation is one of the most important epigenetic mechanisms that
modulate gene expression in a plethora of physiological and
pathological processes, including carcinogenesis (15). Accurate DNA methylation patterns
are established by the de novo DNA methyltransferases
(DNMTs) DNMT3A and DNMT3B, and then subsequently maintained by
DNMT1 (16). DNMTs are
overexpressed in various cancer types, including colorectal
(17), prostate (18), hepatocellular (19), breast (20), gastric (21) and lung cancers (22), and their overexpression is
significantly correlated with poor histological differentiation and
prognosis (17–22). At present, it is generally accepted
that tumor environmental cues together with cell-intrinsic
alterations contribute to the epigenetic changes in carcinogenesis
as well as recurrence and metastasis of cancer, as these changes
induce adaptations of cancer cells for successful invasion of the
stroma, entry and survival in the lymphatic or blood vessels,
followed by spread and colonization of distant or different organs
(23,24). It is thus clear that the tumor
micro-environment, particularly the growth factors, are able to
affect the functions of epigenetic molecules. However, the
potential tumor suppressor genes (TSGs) regulated by HGF via an
epigenetic mechanism have remained to be identified. The present
study investigated the target TSGs of HGF by altering DNA
methylation.
Materials and methods
Ethics statement
The present study was approved by the Xiamen
University Medical Ethics Committee (Xiamen, China), and written
informed consent was obtained from all participants or their
representatives if direct consent could not be obtained.
Cell culture and reagents
The HCC cell line HepG2 was obtained from the Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China) and cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco-BRL, Invitrogen Life Technologies, Carlsbad, CA, USA). The
immortalized normal human liver cell line HL-7702 was obtained from
the China Center for Type Culture Collection (Wuhan, China) and
cultured in RPMI-1640 medium (Gibco-BRL). Media were supplemented
with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA) and 1%
penicillin-streptomycin (Gibco-BRL). The cells were incubated at
37°C in an incubator with 5% CO2. Recombinant human HGF
was obtained from Millipore (Billerica, MA, USA).
5′Aza-2′-deoxycytidine (5′Aza) was obtained from Sigma-Aldrich (St
Louis, MO, USA).
Tissue samples and clinical
characteristics of patients
Tumor samples were obtained from the Chronic Liver
Disease Biological Tissue Bank, Department of Hepatobiliary
Surgery, Zhongshan Hospital of Xiamen University (Xiamen, China),
which were obtained during surgery between 2008 and 2011. Paraffin
blocks of tumor tissue from 89 patients were prepared for
immunohistochemical (IHC) assays. The patients were aged between 30
and 70 and 72 were male and 17 were female. The patients were
assessed at two-month intervals after surgery in outpatient clinics
or by routine telephonic inquiry. The end of the follow-up period
was December 2011 for all patients. The overall survival was
calculated from the day of surgery to the day of succumbing to the
disease or final follow-up.
RNA isolation and reverse transcription
quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted with the TRIzol reagent
(Invitrogen Life Technologies). cDNA was synthesized using the
PrimeScript RT Reagent kit with gDNA Eraser (Takara Bio, Inc.,
Otsu, Japan). qPCR was performed using the StepOne™ Real-Time PCR
system (Applied Biosystems, Foster City, CA, USA), SYBR®
Green (Takara Biotechnology Co., Ltd., Dalian, China) and SYBR
Select Master mix (Takara Bio, Inc., Otsu, Japan) with the
following cycling condiditons: 95º°C for 30 sec and 60°C for 1 min.
The primers were designed and synthesized by BGI company (Shenzhen,
China). β-actin mRNA was used as an endogenous control. The
relative expression of RNA was calculated using the comparative Ct
method (2−ΔΔCT).
Microarray analysis
Expression profiling analysis was performed on
HL-7702 cells and HGF-treated HL-7702 cells. To simulate
hepatocarcinogenesis and to explore the long-term effect of HGF,
HL-7702 cells were cultured in RPMI-1640 medium supplemented with
1% FBS and 50 ng/ml HGF for 4 weeks. The medium was replaced every
day with fresh medium containing the same concentration of HGF.
Total RNA from each sample was quantified by using the NanoDrop
ND-1000 spectrometer (Thermo Fisher Scientific), and the RNA
integrity was assessed by standard denaturing agarose gel
electrophoresis (Biowest SAS, Nuaillé, France). The Human GeneArray
1.0 ST platform (Agilent Technologies, Santa Clara, CA, USA) was
used for the microarray analysis. The sample preparation and
microarray hybridization were performed based on the manufacturer's
instructions. The labeled cRNAs were hybridized onto the Whole
Human Genome Oligo Microarray (4×44K; Agilent Technologies). The
slides were washed and the arrays were scanned by the Agilent
Scanner G2505B (Agilent Technologies). Agilent Feature Extraction
software (version 10.7.3.1; Agilent Technologies) was used to
analyze the acquired array images. Raw signal intensities were
normalized using the quantile method in the GeneSpring GX software
(version 11.5.1; Agilent Technologies), and low-intensity genes
were filtered. P<0.05 was considered to indicate significant
changes in gene expression and when >2.0-fold changes in the
ratio of means were observed.
Methylation arrays
Genome DNA was extracted and bisulfite-converted
using the Epitect Fast DNA Bisulfite kit (Qiagen, Hilden, Germany).
DNA methylation microarray analysis was performed at Kangchen
Biotech Inc. (Shanghai, China).
Statistical analysis
All statistical analyses were performed using the
SPSS software (version 19.0) for Windows (International Business
Machines, Armonk, NY, USA). The survival curves were calculated by
the Kaplan-Meier method, and comparison was performed by a log rank
test. Values for parametric variables are expressed as the mean ±
standard error. P<0.05 was considered to indicate a
statistically significant difference between values.
Results
Gene expression profiling reveals
multiple functions of HGF
To explore the underlying molecular mechanisms of
HGF-induced carcinogenesis, the transcriptome following HGF
treatment was examined using a microarray analysis. Comparison of
the transcriptomes of untreated HL-7702 and HGF-treated HL-7702
cells identified 6,874 genes with a >2-fold change (P<0.05),
of which 2,902 genes were upregulated and 3,972 genes were
downregulated (Fig. 1A).
To obtain further insight into the biological
function of the differentially expressed genes, 6,874 genes were
subjected to Gene Ontology (GO) analysis by using the GeneSpring GX
software. Unexpectedly, the genes were markedly enriched in
multiple metabolic processes, including cellular, macromolecular
and mRNA metabolic processes, as well as translation (Fig. 1B). The genes associated with
metabolism were found to be significantly affected by HGF. Next, a
pathway analysis was performed based on differentially expressed
genes by using the latest Kyoto Encyclopedia of Genes and Genomes
database (http://www.genome.jp/kegg/).
Consistent with those of the GO analysis, the results of the
pathway analysis revealed that metabolic pathways involving
ribosomes, oxidative phosphorylation, amino sugars and nucleotide
sugar metabolism had high enrichment scores, with a significance in
the order in which they are stated (Fig. 1C). The genes were also markedly
enriched in the P53 pathway, which is closely associated with HCC,
suggesting that HGF influences the recurrence and development of
HCC, mainly through the P53 pathway (Fig. 1C). A list of cancer-associated
pathways showing significantly up- or downregulated genes is shown
in Fig. 1D, including P53, cell
adhesion molecules as well as the Hedgehog- and MAPK signaling
pathways. However, HGF was also shown to influence several
non-tumorous pathways, including those involved in Parkinson's and
Huntington's disease, implying that HGF may have a role in nervous
system diseases (Fig. 1C). The
findings of the present study suggest that HGF may have multiple
roles in these signaling pathways in HCC or in diseases of the
nervous system.
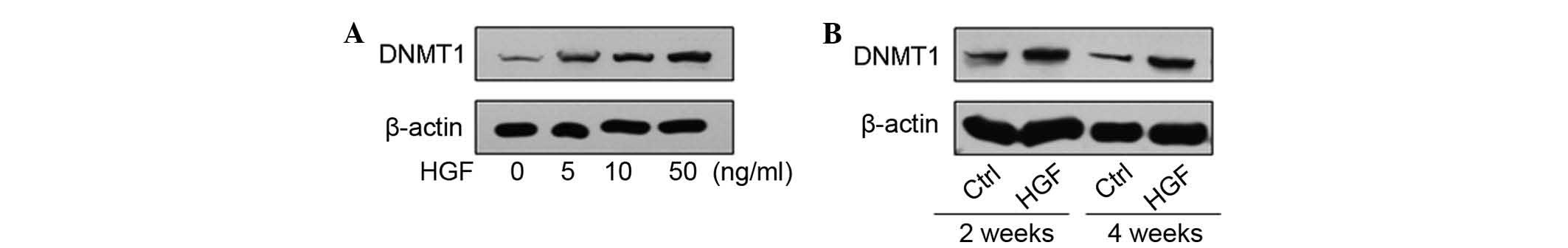
HGF induces DNMT1 overexpression in HCC
patients, which correlates with the malignant potential and poor
prognosis of HCC
The results revealed that several differentially
expressed genes induced by HGF treatment had a role in the DNA
methylation pathway (Fig. 1D).
DNMT1 is closely associated with various cancers; it is a key
regulator in DNA hypermethylation (17). The DNMT1 protein levels was
obviously increased following HGF treatment in the present study
(Fig. 2). To unravel the clinical
significance of abnormal DNMT1 expression in HCC, 89 primary HCC
samples were examined by IHC staining. The results revealed that
DNMT1 was more easily detectable in the nuclei of tumorous tissues
than in paired non-tumorous tissues in 74 out of 89 (83.1%) HCC
cases. Among these, 59 (66.2%) cases were strongly positive (++)
and 15 (16.8%) were moderately positive (+) (Fig. 3A). Furthermore, a correlation
analysis between DNMT1 expression and clinical features was
performed for the 89 HCC cases. Elevated levels of serum
alpha-fetoprotein (AFP) indicate a high risk of liver cancer
(21). As shown in Fig. 3B, the serum AFP levels were
significantly elevated in the DNMT1-overexpressing groups, with
median serum AFP levels of 9.87, 88.89 and 825.5 ng/ml in the
DNMT1-negative, -moderately positive and -strongly positive groups,
respectively (P=0.02). The proportion of low and intermediate
differentiation gradually increased with DNMT1 overexpression
(Fig. 3C; P=0.008). Furthermore,
Kaplan-Meier analysis revealed that the three-year recurrence-free
survival rate was significantly lower for HCC patients in the
strongly positive DNMT1 group as compared with that in the
DNMT1-negative HCC patients (Fig.
3D; P=0.002). The three-year overall survival rate of patients
with DNMT1-positive HCC was also significantly lower than that of
patients with DNMT1-negative HCC (Fig.
3D; P=0.034). These cumulative findings suggested the clinical
significance of DNMT1 as a biomarker for HCC diagnosis as well as
prognosis.
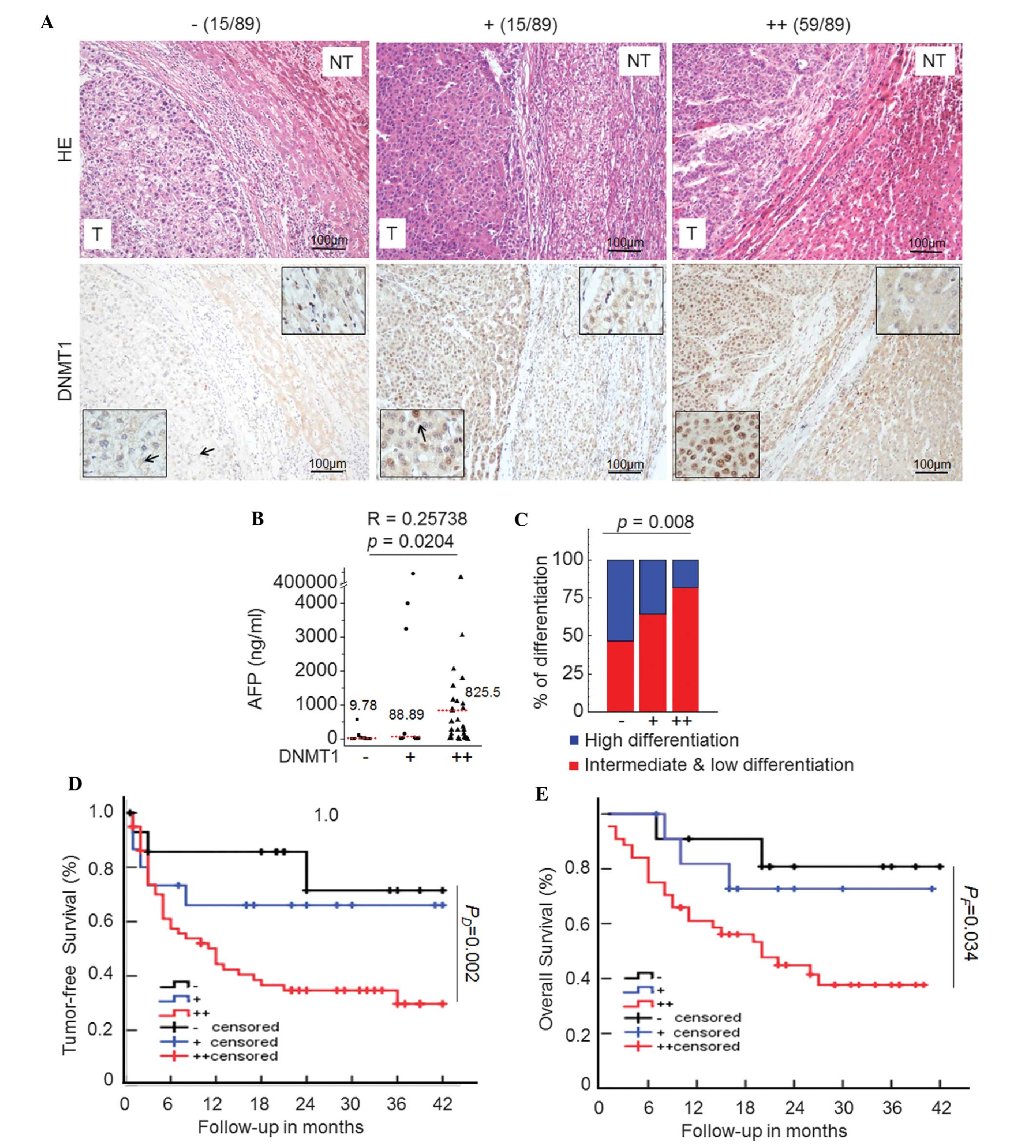 | Figure 3Upregulated DNMT1 expression
correlates with malignant indicators of HCC. (A)
Immunohistochemical staining of DNMT1 (brown) in paraffin sections
of cancer tissues and adjacent tissues from HCC patients (scale
bar, 100 µm). According to the DNMT1 protein expression
levels, 89 cases were divided into three groups (negative,
moderately positive and strongly positive). (B) Scatter plots of
serum AFP levels in the three study groups. In each panel, the
dotted line indicates the median (r=0.26, P=0.02; Spearman
correlation). (C) Distribution of histopathological differentiation
and classification in the three study groups. The differentiation
status is indicated as follows: Blue, high differentiation; red,
intermediate and low differentiation. Differences in
differentiation among the groups were significant (P=0.008). The
Kaplan-Meier curves for (D) tumor-free survival and (E) overall
post-operative survival indicate that HCC patients with marked
DNMT1 overexpression had a worse prognosis as compared with those
in the negative group. Arrows indicate representative cells. DNMT,
de novo DNA methyltransferase; HCC, hepatocellular
carcinoma; AFP, alpha-fetoprotein; H&E, hematoxylin and eosin;
T, tumorous; NT, non-tumorous. |
HGF induces changes in DNA
methylation
Extensive low-level methylation of genomic DNA is a
characteristic of a malignant tumors (25). Silencing of TSGs due to promoter
hypermethylation is closely associated with tumorigenesis (26). To explore the effect of HGF on
genomic methylation, a genome-wide DNA microarray analysis was
performed. The level of DNA methylation in a number of loci was
significantly altered in the HGF group as compared to that in the
control group (Fig. 4). After HGF
treatment, a total of 2,878 genes were hypermethylated and 2,302
genes were hypomethylated. Parts of known TSG promoters, including
PCGF2, PTEN, and PANX2, were markedly
hypermethylated following HGF treatment (Fig. 4). These results suggested that HGF
may alter the expression of TSGs via an epigenetic mechanism
involving DNA hypermethylation.
Integrated analysis of gene expression
and DNA methylation reveals potential TSGs in HCC
One of the most important contributors to
tumorigenesis is the downregulation of TSGs by hypermethylation of
CpG islands (27). To reveal the
epigenetic mechanism of HGF-mediated repression of TSG
transcription via DNA hypermethylation, DNA methylation and gene
expression profiles were subjected to integrated analysis. The
results showed that the expression of a total of 89 genes was
decreased by promoter hypermethylation after HGF treatment
(Fig. 5A). 35 of these
differentially expressed genes, were associated with cell
proliferation, apoptosis, cell cycle and metastasis based on
previous studies (Fig. 5B)
(28–30). In addition, GO analysis revealed
that the 89 differentially expressed genes were markedly enriched
in the adhesion process (Fig. 5C),
which is consistent with the findings reported by a previous study
asserting that HGF promotes tumor metastasis (31). Furthermore, pathway analysis showed
that the P53 pathway, hedgehog pathway and cell adhesion (Fig. 5C), which influenced HCC occurrence
and metastasis, also had high enrichment scores. The 35 potential
TSGs, which were associated with cell proliferation, apoptosis,
cell cycle, and metastasis, were selected for the confirmation of
microarray profiles in the HL-7702 and HepG2 cell lines. Among
these 35 genes, the expression of 17 genes was downregulated
following HGF treatment for 4 days, whereas these genes were
upregulated following treatment with the DNA demethylation agent
5′Aza (Fig. 5D). The expression
levels of PTEN, PNMT, MYOCD, LHX9 and
PANX2 were obviously changed after HGF or 5′Aza treatment.
These results identified potential TSGs regulated by HGF via DNA
hypermethylation in HCC.
Discussion
Metabolism is a fundamental aspect of every
essential cell function. Metabolic reprogramming induced by growth
factors including vascular endothelial growth factor-B has also
been reported recently (32). The
initiation and development of cancer is frequently associated with
the upregulation of catabolic pathways (33). One such important pathway is
aerobic glycolysis that preferentially metabolizes glucose to
lactate, even in the presence of excess oxygen, and provides
substances which are essential for cancer cell survival (33). In the present study, gene
expression profiling demonstrated that HGF mainly participates in
cellular metabolic processes, including macromolecular, mRNA- and
translation-associated metabolic processes. HGF is a growth factor
that exerts multiple effects; it promotes cell proliferation,
migration and angiogenesis in cancer (34). Previous studies have mainly focused
on dysregulation of certain tumor-associated signaling pathways
influenced by HGF (35).
Therefore, little is known about the effects of HGF-induced
metabolic reprogramming on cancer development and recurrence. The
present study provided novel clues for the exploration of the
underlying mechanisms of HGF-induced tumorigenesis from a metabolic
perspective.
Emerging evidence suggests an association between
DNA hypermethylation and hepatocarcinogenesis (36). For example, DNMT1 overexpression
was observed in 43% of HCC cases whose three-year overall survival
rate was <40% (19). However,
the mechanistic and prognostic significance of DNA hypermethylation
in human HCC has remained to be elucidated. The findings of the
present study demonstrated the clinical significance of aberrant
DNA methylation in HCC diagnosis and prognosis. In 82.8% of HCC
samples, DNMT1 protein was overexpressed. The results also revealed
a significant positive association between DNMT1 overexpression and
poor HCC prognosis. The Kaplan-Meier curves for post-operative
overall survival indicated a worse prognosis for HCC patients with
DNMT1 overexpression compared with those negative for DNMT1. The
three-year overall survival rate was only 40% in the DNMT1-positive
group, which was significantly lower (P=0.002) than that in the
DNMT1-negative group (~80%). Serum AFP is currently the most widely
used sero-logical tumor marker for HCC diagnosis. In the present
study, median AFP levels were 9.78, 88.89 and 825.5 ng/ml in the
DNMT1-negative, -moderately positive and -strongly positive group,
respectively, and these differences were significant (P=0.0204).
The normal range for AFP is 10–20 ng/ml, and a level >400 ng/ml
is usually considered diagnostic for HCC. In the present study, the
percentage of patients with AFP levels >400 ng/ml was 54.5% in
the strongly positive group, which was significantly higher than
that in the negative group (16.7%), suggesting that DNMT1 is
clinically significant as an additional indicator in routine HCC
diagnosis.
Evidence of epigenetic dysregulation in cancer,
including aberrant histone modification and DNA methylation, has
been rapidly expanding (37). The
epigenetic reprogramming by the tumor microenvironment has a
critical role in tumor growth and survival. Growth factors bind to
their corresponding receptors and subsequently activate downstream
signaling cascades. More importantly, these signaling pathway
activations give rise to comprehensive epigenetic reprogramming.
For example, TGF-β is an important factor that regulates
proliferation, apoptosis, extracellular matrix composition and
epithelial mesenchymal transition (38) through upregulation of SNAIL and
TWIST family proteins and through recruitment of epigenetic
molecules, including G9a, DNMTs (39), BMI1 (40) and EZH2 (41). However, the exact underlying
mechanisms of HGF on tumor initiation and development through
epigenetic reprogramming remain elusive. The present study
demonstrated that HGF can alter the global chromosomal DNA
methylation status. The methylation of promoters of certain known
TSGs, including PCGF2, PTEN and PANX2, were
enriched after HGF treatment. Methylation of the cytosine residues
in the DNA of TSGs has been recognized as a silencing mechanism of
fundamental importance in tumorigenesis and metastasis (42). DNA methylation and gene expression
profiles were subjected to integration analysis to finally identify
the following potential TSGs silenced by HGF via DNA methylation
changes: PTEN, PNMT, MYOCD, LHX9 and
PANX2. MYOCD encodes a nuclear protein functioning as
a transcriptional co-activator of serum response factor (SRF) and
modulates the expression of cardiac and smooth muscle-specific
SRF-target genes (43). However,
to the best of our knowledge, there is no evidence of any role of
MYOCD in any type of cancer; this aspect therefore requires further
study. LHX9, which encodes LIM-homeodomain 9 transcription
factor, was found to be involved in cell differentiation of several
neural cell types (44). It has
been reported that LHX9 expression was reduced by promoter
hypermethylation in malignant childhood gliomas. Restoration of
LHX9 expression inhibited glioma cell migration and
invasion, suggesting the implication of LHX9 on the migratory
phenotype of cancer (45). Panx2,
which is a member of the gap-junction protein family, showed an
overall reduction in gliomas and can thus help predict
post-diagnosis survival of patients with glial tumors. In addition,
restoration of Panx2 reduces oncogenicity in vivo and in
vitro (46). MYOCD,
LHX9 and PANX2 may thus be novel TSGs that are
regulated by the HGF-DNA methylation pathway in HCC.
In conclusion, the results of the present study
suggested a significant disruption of TSGs by hypermethylation.
Several of these genes have not been previously implicated in HCC.
To the best of our knowledge, the present study was the first to
associate aberrant DNA methylation with gene expression induced by
HGF in order to identify potential TSGs in HCC. The findings of the
present study are valuable in the identification of key TSGs.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81172286 and
81372618), the '973' Program (grant no. 2013CB910803) and the
National Key Sci-Tech Special Project of China (grant no.
2012ZX10002-011-005c).
References
|
1
|
Nakamura T: Structure and function of
hepatocyte growth factor. Prog Growth Factor Res. 3:67–85. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matsumoto K and Nakamura T: Hepatocyte
growth factor: Molecular structure, roles in liver regeneration and
other biological functions. Crit Rev Oncog. 3:27–54. 1992.
|
|
3
|
Pavan S, Musiani D, Torchiaro E, Migliardi
G, Gai M, Di Cunto F, Erriquez J, Olivero M and Di Renzo MF: HSP27
is required for invasion and metastasis triggered by hepatocyte
growth factor. Int J Cancer. 134:1289–1299. 2014. View Article : Google Scholar
|
|
4
|
Weidner KM, Hartmann G, Sachs M and
Birchmeier W: Properties and functions of scatter factor/hepatocyte
growth factor and its receptor c-Met. Am J Respir Cell Mol Biol.
8:229–237. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Caldwell S and Park SH: The epidemiology
of hepatocellular cancer: from the perspectives of public health
problem to tumor biology. J Gastroenterol. 44(Suppl 19): 96–101.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Belghiti J and Fuks D: Liver resection and
transplantation in hepatocellular carcinoma. Liver Cancer. 1:71–82.
2012. View Article : Google Scholar
|
|
7
|
Palmer DH: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:2498author reply
2498–2499. 2008.PubMed/NCBI
|
|
8
|
Kishi Y, Hasegawa K, Sugawara Y and Kokudo
N: Hepatocellular carcinoma: Current management and future
development-improved outcomes with surgical resection. Int J
Hepatol. 2011:7281032011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ueno M, Uchiyama K, Ozawa S, Hayami S,
Shigekawa Y, Tani M and Yamaue H: Adjuvant chemolipiodolization
reduces early recurrence derived from intrahepatic metastasis of
hepatocellular carcinoma after hepatectomy. Ann Surg Oncol.
18:3624–3631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park CC, Bissell MJ and Barcellos-Hoff MH:
The influence of the microenvironment on the malignant phenotype.
Mol Med Today. 6:324–329. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu CT, Wu JR and Wu WS: The role of
endosomal signaling triggered by metastatic growth factors in tumor
progression. Cell Signal. 25:1539–1545. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scagliotti GV, Novello S and von Pawel J:
The emerging role of MET/HGF inhibitors in oncology. Cancer Treat
Rev. 39:793–801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Osada S, Kanematsu M, Imai H and Goshima
S: Clinical significance of serum HGF and c-Met expression in tumor
tissue for evaluation of properties and treatment of hepatocellular
carcinoma. Hepatogastroenterology. 55:544–549. 2008.PubMed/NCBI
|
|
14
|
Reik W: Stability and flexibility of
epigenetic gene regulation in mammalian development. Nature.
447:425–432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Johnson AA, Akman K, Calimport SR, Wuttke
D, Stolzing A and de Magalhães JP: The role of DNA methylation in
aging, rejuvenation and age-related disease. Rejuvenation Res.
15:483–494. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ting AH, Jair KW, Suzuki H, Yen RW, Baylin
SB and Schuebel KE: CpG island hypermethylation is maintained in
human colorectal cancer cells after RNAi-mediated depletion of
DNMT1. Nat Genet. 36:582–584. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Patra SK, Patra A, Zhao H and Dahiya R:
DNA methyltransferase and demethylase in human prostate cancer. Mol
Carcinog. 33:163–171. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saito Y, Kanai Y, Nakagawa T, Sakamoto M,
Saito H, Ishii H and Hirohashi S: Increased protein expression of
DNA meth-yltransferase (DNMT) 1 is significantly correlated with
the malignant potential and poor prognosis of human hepatocellular
carcinomas. Int J Cancer. 105:527–532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Girault I, Tozlu S, Lidereau R and Bièche
I: Expression analysis of DNA methyltransferases 1, 3A and 3B in
sporadic breast carcinomas. Clin Cancer Res. 9:4415–4422.
2003.PubMed/NCBI
|
|
21
|
Etoh T, Kanai Y, Ushijima S, Nakagawa T,
Nakanishi Y, Sasako M, Kitano S and Hirohashi S: Increased DNA
methyltransferase 1 (DNMT1) protein expression correlates
significantly with poorer tumor differentiation and frequent DNA
hypermethylation of multiple CpG islands in gastric cancers. Am J
Pathol. 164:689–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin RK, Hsu HS, Chang JW, Chen CY, Chen JT
and Wang YC: Alteration of DNA methyltransferases contributes to
5′CpG methylation and poor prognosis in lung cancer. Lung Cancer.
55:205–213. 2007. View Article : Google Scholar
|
|
23
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Taddei ML, Giannoni E, Comito G and
Chiarugi P: Microenvironment and tumor cell plasticity: An easy way
out. Cancer Lett. 341:80–96. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gaudet F, Hodgson JG, Eden A,
Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H and Jaenisch R:
Induction of tumors in mice by genomic hypomethylation. Science.
300:489–492. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Narimatsu T, Tamori A, Koh N, Kubo S,
Hirohashi K, Yano Y, Arakawa T, Otani S and Nishiguchi S: p16
promoter hyper-methylation in human hepatocellular carcinoma with
or without hepatitis virus infection. Intervirology. 47:26–31.
2004. View Article : Google Scholar
|
|
27
|
Zhang Z, Chen Y, Tang J and Xie X:
Frequent loss expression of dad2 and promotor hypermethylation in
human cancers: A meta-analysis and systematic review. Pak J Med
Sci. 30:432–437. 2014.PubMed/NCBI
|
|
28
|
Yu G, Yao W, Gumireddy K, et al:
Pseudogene PTENP1 functions as a competing endogenous RNA to
suppress clear-cell renal cell carcinoma progression. Mol Cancer
Ther. 13:3086–3097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Christoforou N, Chellappan M, Adler AF,
Kirkton RD, Wu T, Addis RC, Bursac N and Leong KW: Transcription
factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing
effect of GATA4, TBX5, and MEF2C during direct cellular
reprogramming. PLoS One. 8:e635772013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu C, Shao P, Bao M, et al: miR-154
inhibits prostate cancer cell proliferation by targeting CCND2.
Urol Oncol. 32:e9–e16. 2014. View Article : Google Scholar
|
|
31
|
Ogunwobi OO, Puszyk W, Dong HJ and Liu C:
Epigenetic upregulation of HGF and c-Met drives metastasis in
hepatocellular carcinoma. PLoS One. 8:e637652013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kivelä R, Bry M, Robciuc MR, Räsänen M,
Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A,
Mäyränpää MI, et al: VEGF-B-induced vascular growth leads to
metabolic reprogramming and ischemia resistance in the heart. EMBO
Mol Med. 6:307–321. 2014.PubMed/NCBI
|
|
33
|
Koppenol WH, Bounds PL and Dang CV: Otto
Warburg's contributions to current concepts of cancer metabolism.
Nat Rev Cancer. 11:325–337. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Suárez-Causado A, Caballero-Díaz D,
Bertrán E, et al: HGF/c-Met signaling promotes liver progenitor
cell migration and invasion by an epithelial-mesenchymal
transition-independent, phosphatidyl inositol-3 kinase-dependent
pathway in an in vitro model. Biochim Biophys Acta. S0167-4889(15):
00163–9. 2015.
|
|
35
|
Lordick F: Targeting the HGF/MET pathway
in gastric cancer. Lancet Oncol. 15:914–916. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tischoff I and Tannapfe A: DNA methylation
in hepatocellular carcinoma. World J Gastroenterol. 14:1741–1748.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Egger G, Liang G, Aparicio A and Jones PA:
Epigenetics in human disease and prospects for epigenetic therapy.
Nature. 429:457–463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Massagué J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer and heritable disorders. Cell.
103:295–309. 2000. View Article : Google Scholar
|
|
39
|
Dong C, Wu Y, Yao J, Wang Y, Yu Y,
Rychahou PG, Evers BM and Zhou BP: G9a interacts with Snail and is
critical for Snail-mediated E-cadherin repression in human breast
cancer. J Clin Invest. 122:1469–1486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY,
Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, et al: Bmi1 is
essential in Twist1-induced epithelial-mesenchymal transition. Nat
Cell Biol. 12:982–992. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tong ZT, Cai MY, Wang XG, Kong LL, Mai SJ,
Liu YH, Zhang HB, Liao YJ, Zheng F, Zhu W, et al: EZH2 supports
nasopharyngeal carcinoma cell aggressiveness by forming a
co-repressor complex with HDAC1/HDAC2 and Snail to inhibit
E-cadherin. Oncogene. 31:583–594. 2012.
|
|
42
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
43
|
Wang D, Chang PS, Wang Z, Sutherland L,
Richardson JA, Small E, Krieg PA and Olson EN: Activation of
cardiac gene expression by myocardin, a transcriptional cofactor
for serum response factor. Cell. 105:851–862. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Failli V, Rogard M, Mattei MG, Vernier P
and Rétaux S: Lhx9 and Lhx9alpha LIM-homeodomain factors: Genomic
structure, expression patterns, chromosomal localization and
phylogenetic analysis. Genomics. 64:307–317. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vladimirova V, Mikeska T, Waha A,
Soerensen N, Xu J, Reynolds PC and Pietsch T: Aberrant methylation
and reduced expression of LHX9 in malignant gliomas of childhood.
Neoplasia. 11:700–711. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lai CP, Bechberger JF and Naus CC:
Pannexin2 as a novel growth regulator in C6 glioma cells. Oncogene.
28:4402–4408. 2009. View Article : Google Scholar : PubMed/NCBI
|