Introduction
Naked plasmid delivery systems have been recognized
as an appealing alternative to viral vectors, due to their ability
to avoid the immunogenicity and toxicity problems commonly
associated with viral systems (1,2).
Since 1989, >1,800 clinical trials have been approved worldwide,
with ~20% of these trials assessing the use of plasmid DNA as a
vector system (3–5). DNA vectors generally encode
antibiotic resistance genes in the plasmid backbone (6). Recently, the widespread use of
antibiotics has resulted in the emergence of so-called super
bacteria, which are resistant to multiple drugs (7). As a result of enhanced antibiotic
management, the use of antibiotics has been banned in the
production process of therapeutic plasmids in the European Union,
except at the seeding stage. In addition, regulatory authorities in
Europe and the US have voiced their concerns regarding the use of
antibiotics or antibiotic selection markers in plasmids destined
for clinical use (8,9). Therefore, the development of various
antibiotic marker-free selection approaches for the production of
therapeutic plasmids has become an attractive solution (6).
Numerous strategies have been assessed regarding the
production of antibiotic marker-free plasmids, including the use of
auxotrophic complementation, toxin-antitoxin-based systems,
RNA-based selection markers, and overexpression of growth essential
genes (10). The present study
aimed to develop a diaminopimelic acid (DAP) auxotrophy-based
plasmid system, in order to produce plasmids for therapeutic use.
In prokaryotes, the asd gene encodes aspartate-semialdehyde
dehydrogenase (ASD; EC 1.2.1.11) (11). ASD is involved in cata-lyzing the
intermediate steps leading to the synthesis of DAP, which in turn
is an important component of the peptidoglycans that are used in
cell wall synthesis by gram-negative bacteria. Bacteria that are
deficient in ASD are unable to synthesize a functional cell wall,
and therefore would be unable to grow, due to cell lysis (12,13).
Previous studies have reported the creation of auxotrophic S.
typhimurium and E. coli strains with defects in the
asd or QAPRTase genes for use in antibiotic marker-free
plasmids (14,15); however, their plasmid
production-associated performance has not been fully characterized.
Therefore, the present study aimed to engineer a widely used
recombinant auxotrophic DH10B E. coli strain. The DH10B
strain has previously been engineered for the propagation of large
insert DNA library clones (16),
and is commonly used throughout the research community due to its
useful properties, including its high transformation efficiency,
its ability to take up and stably maintain large plasmids, its lack
of methylation-dependent restriction systems (MDRS), and its
lacZ-based α-complementation colony screening properties
(17).
In the present study, the cell wall auxotrophic
mutant DH10BΔasd was generated to host a eukaryotic
asd expression vector, thereby creating a balanced-lethal
system similar to those reported previously (18,19).
Numerous parameters associated with the stability of the host, and
the yield of the plasmid, were evaluated to determine the potential
of the system in large-scale plasmid production.
Materials and methods
Media, chemicals and reagents
Ampicillin (Amp), streptomycin (Stm), spectinomycin
(Spm), and kanamycin (Km)-resistant (R) transformants
were selected on a Luria Bertani (LB) agar medium (Kemin Bio Tech.,
Shanghai, China), containing the antibiotics at 100, 100, 50, and
25 µg/ml, respectively. All antibiotics were purchased from
Sigma-Aldrich (St. Louis, MO, USA). DAP (Sigma-Aldrich) was used at
100 µg/ml, and L-arabinose (Sigma-Aldrich) was used at 0.4%.
The oligonucleotides were purchased from Beijing Sunbiotech Co.,
Ltd. (Beijing, China). The restriction enzymes were obtained from
New England Biolabs Inc. (Ipswich, MA, USA), and the Taq Plus DNA
polymerase was purchased from Takara Biotechnology Co., Ltd
(Dalian, China).
Bacteria and plasmids
The DH10B (F-mcrA
Δ(mrr-hsdRMS-mcrBC)
Φ80lacZΔM15ΔlacX74 recA1endA1
araD139 Δ(ara leu) 7,697 galU galK
rpsL nupG λ-) E. coli strain was purchased
from Invitrogen Life Technologies (Carlsbad, CA, USA), and was
maintained in LB medium. The pKD46 and pKan-kil plasmids were
provided from Dr. Shanhu Li (China Academy of Military Medical
Science), and were previously described (16), and the pDV-cmv-gene plasmid
was constructed in our laboratory (unpublished data; China
Institute for Food and Drug control).
Disruption of asd
A Red recombinase-based homologous recombination
technique was used to knockout the chromosomal asd in the
DH10B cells, as reported previously (13,20).
The details of the asd knockout are presented in Fig. 1. Briefly, transformants carrying a
pKD46 Red helper plasmid were induced in 5 ml super optimal broth
(Hope Bio Tech., Qingdao, China) cultures with Amp and L-arabinose
at 30°C. The plasmid was subsequently transformed with linear DNA
fragments carrying kan-kil and asd-homologous
extensions using a Gene Pulser Xcell Electroporation system with
0.1 cm chambers according to the manufacturer's instructions
(Bio-Rad Laboratories Inc., Hercules, CA, USA). The fragment was
obtained by polymerase chain reaction (PCR), using Taq Plus DNA
polymerases with the following primer sequences: Forward, 5′-ATT
TAT ACA GCA CAC ATC TTT GCA GGA AAA AAA CGC TTA TTA GTG AAT GCT TTT
GCT TG-3′; and reverse, 5′-CGC ACT AAC AGG GGC GGC ATC GCG CCC CAG
ATT TAA TGA ACA ACC TCC TTA GTA CAT GC-3′, and pkan-kil as a
template. The electroporated cells
(1×109−2×109 CFU/ml) were plated out to
select for the KmR and DAP-dependent transformants, and
subsequently colony-purified at 37°C, prior to being tested for Amp
sensitivity to isolate the cells that had lost the helper plasmid.
The helper plasmid cells were then made competent again and
received another round of electroporation with the linear fragments
carrying the homologous extensions without the kan-kil
region. The fragment was obtained by PCR using Taq Plus DNA
polymerases with the following primer (Sun Bio Tech, Beijing,
China) sequences: Forward, 5′-CGG CAC ATT TAT ACA GCA CAC ATC TTT
GCA GGA AAA AAA CGC TTT CAT TAA ATC TGG GG-3′; and reverse, 5′-CTG
CGC TTA CTC CTG TAT TAC GCA CTA ACA GGG GCG GCA TCG CGC CCC AGA TTT
AAT GA-3′. The PCR mixture was comprised of 12.5 µl Q5
High-Fidelity 2X Master mix (Takara Biotechnology Co., Ltd), 10
µM forward primer (1.25 µl), 10 µM reverse
primer (1.25 µl), 1 µl template DNA and made up to 25
µl with nuclease-free water. The transformants were
subsequently induced at 37°C for kil expression and
homologous recombination, in order to remove the kan-kil
fragment that was previously integrated into the E. coli
genome (Fig. 1).
PCR verification
PCR was used to confirm that all mutants had the
correct structure. A freshly isolated colony was suspended in 50
µl water, from which 3 µl aliquots were used in
separate 25 µl PCR reaction mixtures following a 2 min
pre-incubation 'hot start' at 95°C. The primers (Sun Bio Tech) used
were as follows: Forward, 5′-ATT TAT ACA GCA CAC ATC TTT GCA GGA
AAAAAA CGC TTA TTA GTG AAT GCT TTT GCT TG-3′; and reverse, 5′-CGC
ACT AAC AGG GGC GGC ATC GCG CCC CAG ATT TAA TGA ACA ACC TCC TTA GTA
CAT GC-3′. Control colonies were tested side-by-side, and the
amplified products were analyzed by agarose gel
electrophoresis.
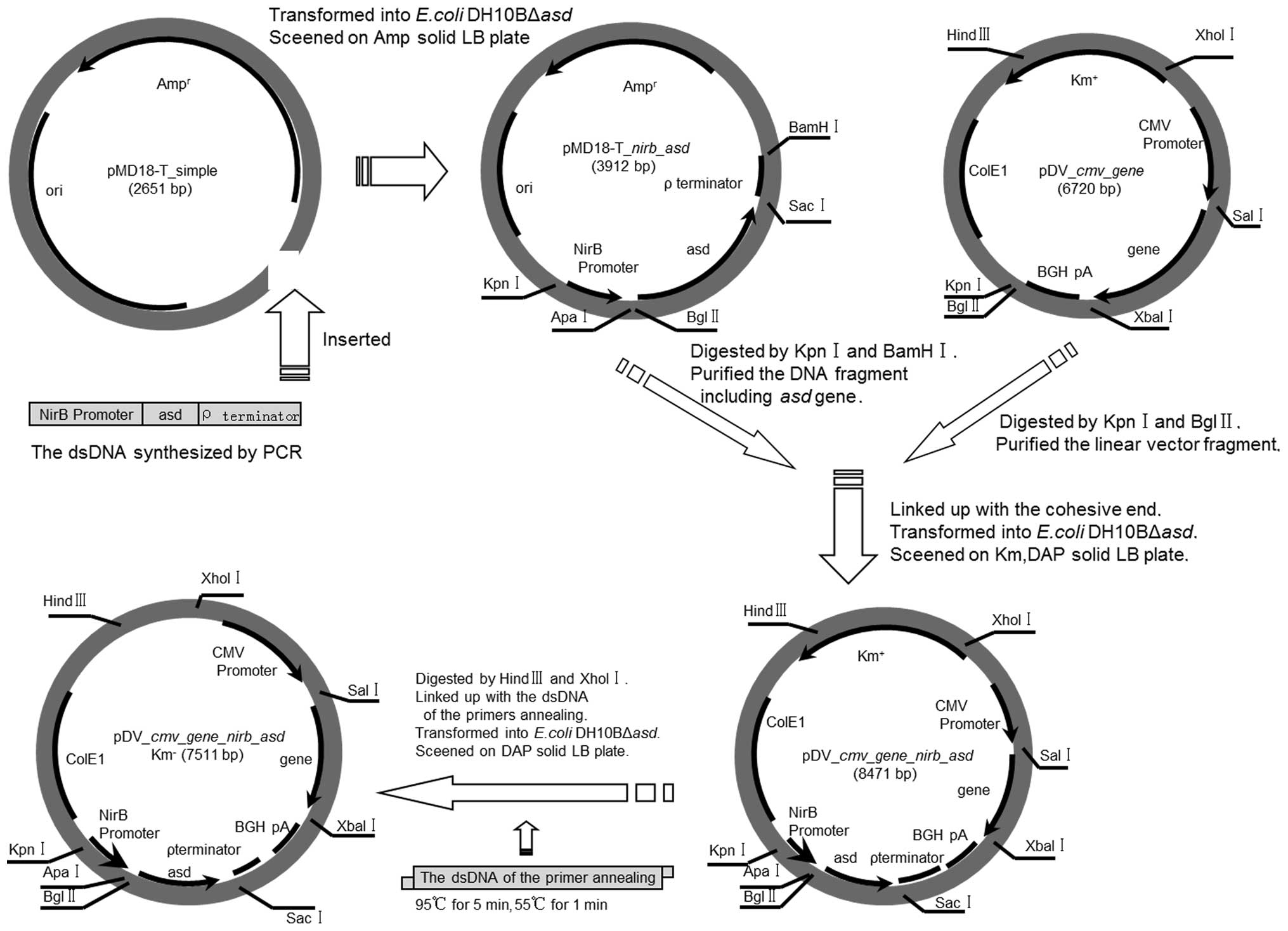
Construction of the antibiotic
resistant-marker-free plasmid
Detailed steps for the construction of the plasmid
are presented in Fig. 2. Briefly,
the DNA sequence of the entire expression cassette, including the
nirB15 promoter (21), the
Shine-Dalgarno sequence, the asd coding region
(Salmonella enterica subsp. enterica serovar typhi
str. Ty2, GenBank: AAO71451.1), and the terminator was chemically
synthesized to incorporate the appropriate restriction sites
(Genewiz, Inc., South Plainfield, NJ, USA). The sequence was
ligated into the pMD 18-T-simple plasmid (Sun Bio Tech) and
confirmed by sequencing at the junction regions, prior to double
digestion with KpnI and BamHI, in order to release
the expression cassette. The released fragment was ligated to the
similarly digested pDV-cmv-gene plasmid in order to generate
pDV-cmv-gene-nirb-asd Km+. The Km
resistance gene (Km+) in the resultant plasmid
was then removed by digestion with HindIII and XhoI,
and the purified plasmid was ligated by annealing a dsDNA fragment
carrying complementary restriction sites to the linearized plasmid
prior to ligation. The sequences of the dsDNA are as follows:
Sense, 5′-TCG AGT AGA TAA CTG ACT AGC ATA ACC CCT TGG GGC CTC TAA
ACG GGT CTT GAG GGG TTT TTT A-3′; and antisense, 5′-AGC TTA AAA AAC
CCC TCA AGA CCC GTT TAG AGG CCC CAA GGG GTT ATG CTA GTC AGT TAT CTA
C-3′. The ligation products were subsequently transformed into the
DH10BΔasd cells and spread onto LB agar plates to recover
DAP-independent colonies, prior to being further colony tested on
LB agar plates for Km-sensitivity, in order to identify any
potential antibiotic resistant-marker-free vectors.
Cell growth test
DH10BΔasd cells were streaked onto agar
plates containing LB, DAP+LB,
Stm+DAP+LB, and
Spm+DAP+LB, and their growth was subsequently
recorded after overnight culture at 37°C.
Cell lysis test
To test the autolysis of DH10BΔasd cells the
cells were grown on DAP+LB agar plates, prior to being
washed three times with sterile distilled water, and were then
resuspended in sterile distilled water. Three types of media
containing: Water only, LB, and LB + Amp, were inoculated with 100
µl aliquots prior to culture at 37°C. Subsequently, 10
µl aliquots were taken from each culture at various times,
and plated onto LB agar medium, prior to overnight incubation at
37°C. For each assessment three colony plates were counted to
determine the number of colonies.
Plasmid structure and sequence
analysis
To verify the structure of the expected plasmid,
purified pDV-cmv-gene-nirb-asd Km−
DNA was digested in three double enzyme reactions: SalI +
BglII, HindIII + BglII, and HindIII +
SalI. The digested products were subsequently analyzed by
agarose gel electrophoresis. The selected clones were fully
sequenced on both strands at the Beijing Genomics Institute
(Beijing, China).
Plasmid yield and supercoiled
plasmid
The DH10BΔasd cells containing
pDV-cmv-gene-nirb-asd Km+ and
pDV-cmv-gene-nirb-asd Km− were
cultured overnight at 37°C with agitation at 150 rpm, in liquid LB
medium with or without Km (100 µg/ml). The cells were then
harvested by centrifugation and diluted with 0.9% NaCl solution
with the same OD600 values for both cultures (OD=2.0).
Plasmid DNA was prepared from 2 ml aliquots of the cells
(1×109−2×109 CFU/ml) using the Qiagen
Miniprep kit (Qiagen, Inc., Valencia, CA, USA) and quantified using
a UV spectrometer (DU800; Beckman Coulter, Inc., Fullerton, CA,
USA) at a wavelength of 260 nm. The plasmids were subsequently
separated on 1% agarose gel, and the resulting images were scanned
and analyzed to determine the amount of DNA in the supercoiled
conformation using MultiGauge (Version 3.1, Fujifilm, Tokyo,
Japan).
Plasmid retention
After 10, 20 and 30 passages, cells from the rapidly
growing cultures were taken and inoculated in LB medium with
inoculant-medium ratios of 1:1,000, 1:100 and 1:10. The cells were
subsequently cultured for 5 h at 37°C with 150 rpm agitation. LB
agar and DAP+ LB agar plates were then spread with 100
µl aliquots from each of the cultures prior to further
overnight culture at 37°C. The number of colonies on both plates
were counted and used to calculate the percentage of the bacterial
cells that had retained the plasmid and could grow normally in LB
medium.
Doubling time
Aliquots of 10 µl of cells were removed
following 0, 1.5, 3 and 4.5 h cultures in LB broth with 200 rpm
agitation at 37°C. The cells were subsequently diluted 100 times,
spread onto LB agar plates, and cultured at 37°C. Following
overnight growth, the colonies were counted and used to calculate
the cell doubling time using the Gompert function (F =
K·ab·t) (22).
Storages
DH10BΔ(asd
pDV-cmv-gene-nirb-asd km−) cells
were stored as glycerol stock at 25, 4, −20 and −70°C for 12 months
and their survival rate was subsequently tested by plating 100
µl of the thawed cells onto LB agar plates, prior to
counting the number of colonies formed after overnight culture.
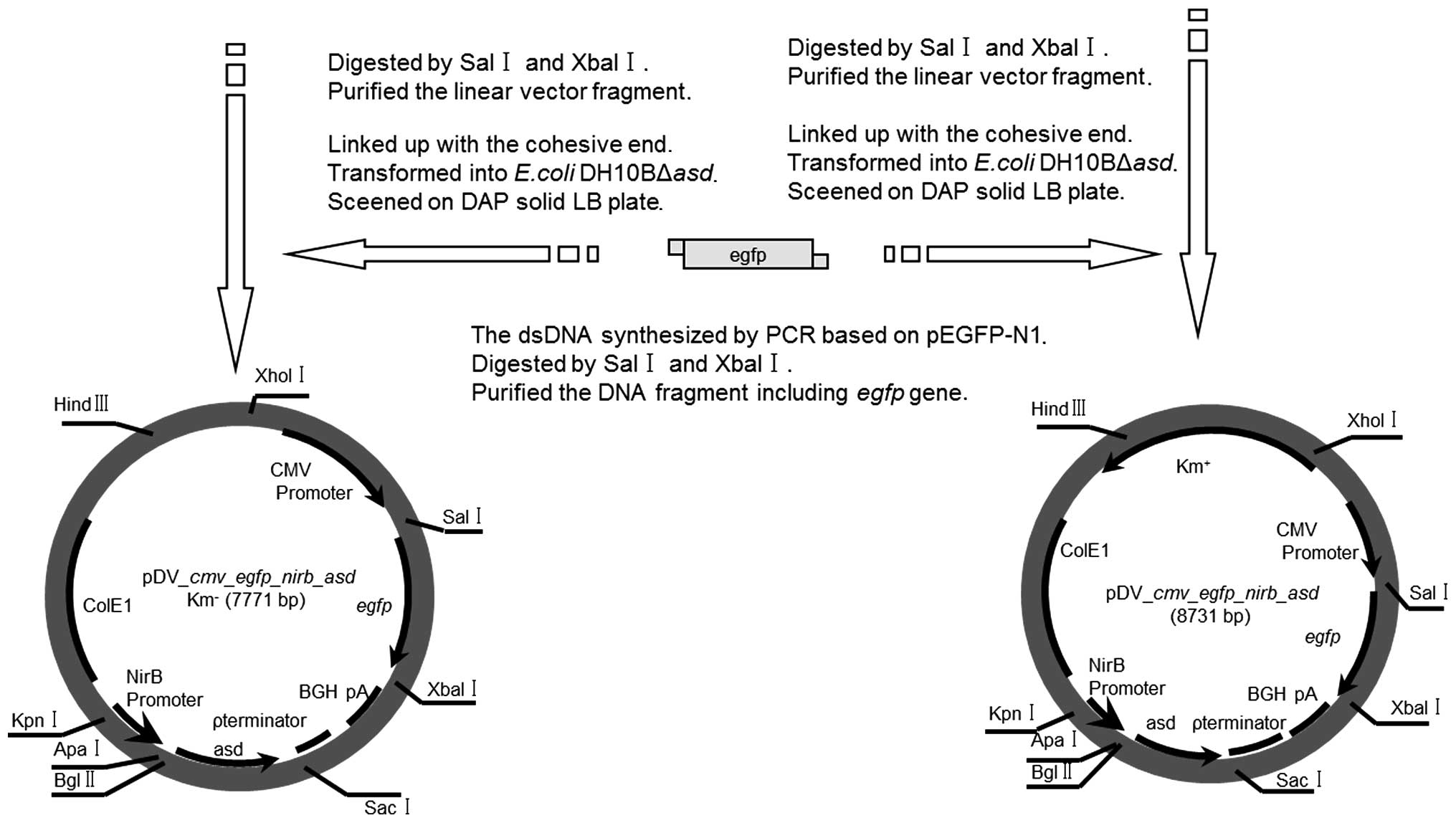
Construction of egfp expression
antibiotic resistant-marker-free plasmid and lipofection of HeLa
cells
The egfp gene was ligated into the
pDV-cmv-gene-nirb-asd Km− and
pDV-cmv-gene-nirb-asdKm-plasmids (Fig. 3), and the resulting transformants
containing the egfp gene were selected by PCR, and sequenced
using egfp gene-specific primers (Sun Bio Tech). Based on
the concentration and molecular mass, the copies of the two types
of plasmid were adjusted to the same order of magnitude. The
lipofection of the HeLa cells with the two plasmids was carried out
using Lipofectamine® reagent (Invitrogen Life
Technologies), according to the manufacturer's instructions.
ddH2O was used as a control. The egfp gene was
also ligated into the pDV-cmv-gene-nirb-asd
and pDV-cmv-gene plasmids, which were then transformed into
the DH10BAΔasd E. coli strain. DH10BΔasd cells were
streaked onto agar plates containing LB medium, and their growth
was recorded after overnight culture at 37°C.
Statistical analysis
The data were analyzed using SPSS version 19.0 (IBM
SPSS, Armonk, NY, USA), and P<0.05 was considered to indicate a
statistically significant difference. Curve fitting was conducted
using OriginPro v7.0 (OriginPro, Northampton, MA, USA).
Results
Screening for asd disruption mutants
Following several preliminary gene disruption
experiments, namely the use of the pKD46 plasmid to provide Red
recombinase and FRT-flanked Km resistance gene fragments carrying
the homologous extensions to asd, hundreds of colonies were
obtained that could grow on LB medium supplemented with Km and DAP.
Thus indicating that the Km gene was successfully inserted into the
DH10B genome. The Km-resistant cells were subjected to a second
round of recombination by electroporating a linear DNA fragment
containing asd-homologous extensions without the
kan-kil region, in order to remove the kan-kil
fragment that had previously been inserted into the bacterial
genome. Following several experiments used to comparatively screen
the transformants on LB agar plates containing Km+
DAP+, DAP+, and LB only, a few colonies were
obtained that could grow on DAP+ LB, but not on
Km+ DAP+ LB, or LB. These results indicate
that the cells in these colonies had lost resistance to Km as a
result of the secondary recombination, and were dependent on the
exogenous supply of DAP. This is due to the expected disruption of
asd in the strain. The colonies were therefore selected as
candidates for asd-deficient lines for further confirmation
at the DNA level.
PCR verification
asd locus-specific PCRs were performed using
genomic DNA extracted from several candidate template colonies. As
shown in Fig. 4, a band
corresponding to the expected 77 bp product was observed in the
asd deletion candidate colony, whereas a 1,184 bp PCR
product was amplified with DNA from the wildtype DH10B cells,
indicating that asd-specific disruption had occurred in the
cell line. Therefore, the cell line was named DH10BΔasd.
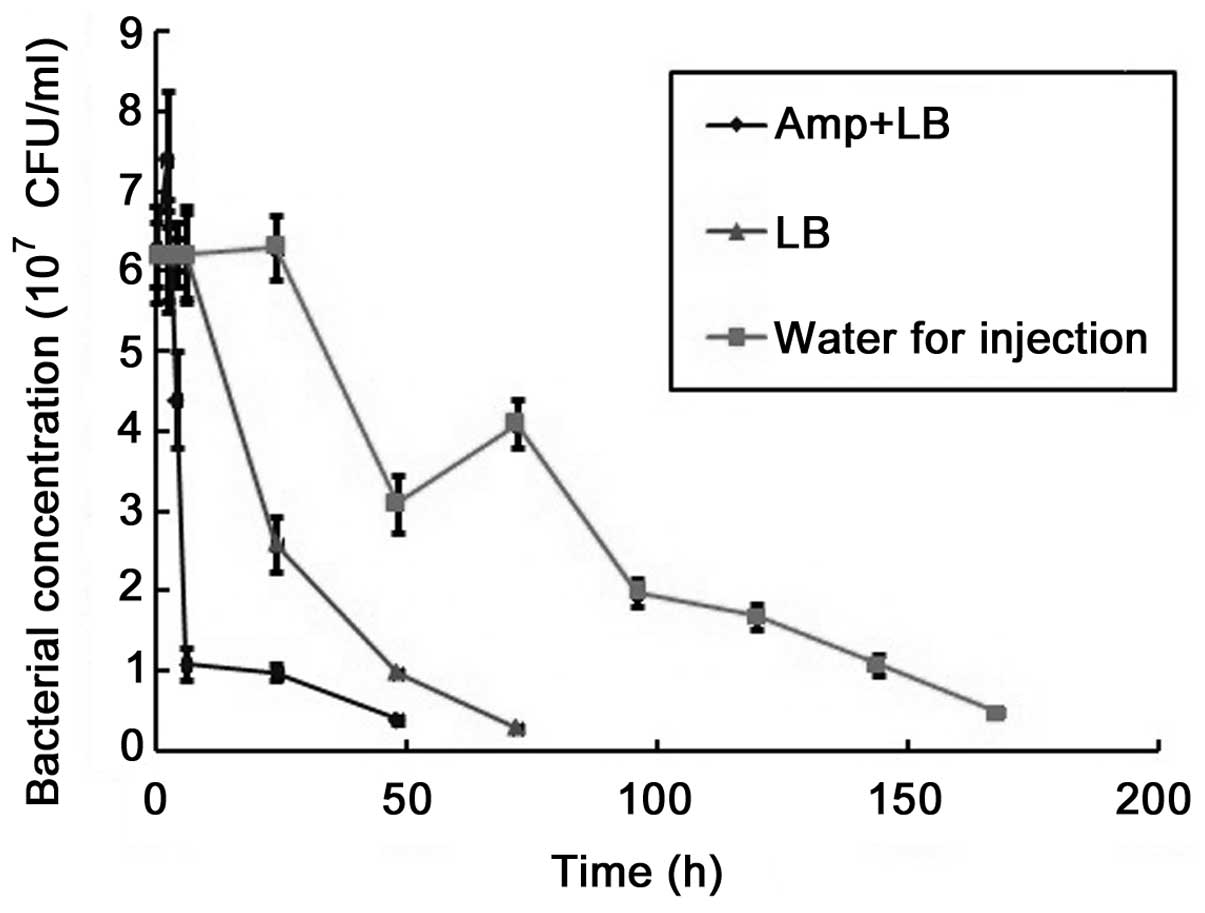
Cell lysis test
Deletion or disruption of functional asd in
E. coli cells is expected to result in the failure of the
bacterium to synthesize a functional cell wall, due to the
disruption of DAP synthesis, thus resulting in cell death. To
acquire a better understanding regarding how cell death occurred in
the asd-disrupted cells, cell survival duration was
determined in various growth conditions by exposing them to water
and LB media, with or without Amp, for various durations. As
demonstrated in Fig. 5, the cells
gradually lost their viability following exposure to these media,
but at different rates. The half-life of the cells was ~2–3 h and
≤1 day in the LB medium with and without Amp, respectively. In
distilled water, the half-life of the cells was 3–4 days.
Construction of marker-free expression
vectors
The pDV cmv gene was used as a starter
plasmid for construction of an antibiotic resistance-marker-free
vector. Following several steps of DNA manipulation, as described
in the Materials and Methods, the DH10BΔasd cell colonies
that contained the target plasmid
pDV-cmv-gene-nirb-asd Km− were
recovered, since they could grow in the LB medium without requiring
extra supplementation of DAP. The nirB-asd cassette was
expected to provide ASD activity in the DH10BΔasd cells. To
verify the structure of the plasmid, DNA was prepared from a number
of colonies and was restricted using restriction enzymes in three
reactions: SalI + BglII, HindIII +
BglII, and HindIII + SalI. The digested
products were analyzed and plasmids with the expected bands and
sizes were identified (Fig. 6).
Sequencing of the plasmids further confirmed that the entire
sequences were correct (data not shown).
Plasmid yield and conformation
To investigate whether the elimination of antibiotic
pressure during the plasmid amplification process would have an
impact on the plasmid yields in this symbiotic system, the plasmid
yield of the resultant constructs were compared prior to and
following the removal of Km+ in the backbone, in LB
medium with or without Km in the DH10BΔasd cells. The
results demonstrated that the plasmid yields were similar in the
two culture conditions (0.527, vs. 0.552 µg/ml of culture in
LB with or without Km, respectively), and did not statistically
significantly differ from each other (P>0.05). The examination
of the conformation of the plasmids extracted from the cultures
under the two conditions demonstrated a similar percentage (~60%)
of supercoiled plasmid DNA (data not shown).
Plasmid retention
Differing from the typical plasmid amplification
protocols, where antibiotics are added to the culture media in
order to selectively proliferate the bacterial cells that contain
plasmids bearing antibiotic resistant genes, maintenance of the
plasmids in this symbiotic system relies solely on DAP production
from the host plasmid containing an asd expression cassette
located in the asd-disrupted cells. It is therefore
important to investigate how well the plasmid can be retained under
the symbiotic, but non-selective condition. The cells containing
pDV-cmv-gene-nirb-asd Km− were
grown to various passages in LB medium, and were then plated out
onto LB agar plates with or without DAP after diluting the culture
up to 1,000 times. The colonies that appeared in LB medium were
considered to contain the plasmid, whereas those that appeared on
the LB + DAP medium were considered to be either with or without
plasmid. The results shown in Table
I indicate that the plasmid retention rates (ratio of colony
counts on medium without and with DAP) were statistically the same
(100, vs. 99%) between the 10th and the 30th passages when they
were inoculated at a 1:1,000 dilution. However, the plasmid
retention rates were reduced to ~80% when inoculated at a 1:10
dilution.
 | Table IPlasmid retention ratios of the
asd deletion mutant strain after various generations of
passages, and after culture at various dilutions. |
Table I
Plasmid retention ratios of the
asd deletion mutant strain after various generations of
passages, and after culture at various dilutions.
| Passage | Dilution at
inoculation
|
|---|
| 1:1,000 | 1:100 | 1:10 |
|---|
| 10 | 1.00±0.01 | 0.98±0.02 | 0.82±0.04 |
| 20 | 0.99±0.02 | 0.97±0.03 | 0.83±0.03 |
| 30 | 0.99±0.01 | 0.98±0.03 | 0.80±0.05 |
Bacterial storability
As a cell line deficient in cell wall composition,
it is important to analyze bacterial storability at various
temperatures for its further use in a plasmid production system.
The results summarized in Table
II demonstrated that the cells rapidly lost their viability
when stored at higher temperatures (25° and 4°C), but maintained
stable colony forming ability when stored at −70°C. The half-life
of the cells was 2 and 15 days when the cells were stored at 25°C
and 4°C, respectively, and increased to 2 months when stored at
−20°C, and to 10 years when stored at −70°C.
| Table IIColony forming ability of the
DH10BΔasd (pDV-cmv-gene-nirb-asd
km−) cells at various storage temperatures, and after
various durations. |
Table II
Colony forming ability of the
DH10BΔasd (pDV-cmv-gene-nirb-asd
km−) cells at various storage temperatures, and after
various durations.
| Storage
(months) | Colony forming
units/ml culture after storage at certain temperatures
|
|---|
| −70°C | −20°C | 4°C | 25°C |
|---|
| 0 |
6.4×108±0.5×108 |
6.4×108±0.5×108 |
6.4×108±0.5×108 |
6.4×108±0.5×108 |
| 1 |
6.5×108±0.5×108 |
5.1×108±0.4×108 |
1.6×108±0.2×108 |
2.1×104±0.2×104 |
| 3 |
6.4×108±0.4×108 |
2.5×108±0.4×108 |
1.1×107±0.4×107 | 5.0±2.0 |
| 6 |
6.2×108±0.3×108 |
7.9×107±0.3×108 |
1.6×105±0.2×105 | – |
| 12 |
6.0×108±0.5×108 |
1.4×107±0.2×107 | 50.0±4.0 | – |
Doubling time
To examine the growth speed that would impact
overall plasmid productivity, the present study determined the
number of cells in the culture by counting their colony forming
units at various time points. The doubling time computed based on
the compertz function, was 29.9 min when the cells were grown in LB
medium.
Expression of egfp gene in HeLa
cells
The expression of enhanced green fluorescence
protein (EGFP) is shown in Fig. 7.
The cells transfected with the
pDV-cmv-egfp-nirb-asd Km−
plasmid exhibited green fluorescence under blue light, indicating a
good expression of EGFP protein in the cells. As assessed with the
naked eye, the efficiency of transfection with the two types of
plasmid appeared similar. The DH10BΔasd cells with the
pDV-cmv-egfp-nirb-asd gene grew well on
LB medium, whereas the cells transformed with the
pDV-cmv-egfp gene did not, which is indicative of a
normally functioning asd gene following the insertion of
egfp.
Discussion
Due to increasing concerns regarding horizontal
antibiotic resistance gene transfer, it has become crucial to
develop plasmid vectors that are free of antibiotic resistance
genes for potential use in human gene therapy (6,7,10).
The present study aimed to develop an antibiotic
resistant-marker-free vector that can be produced in large
quantities, in a similar manner to regular plasmids that carry
antibiotic selection makers. This can be achieved so that the
system has both a specific host that can retain and amplify the
plasmid, and a plasmid that can provide all of the essential
elements for survival of the host cells. Such systems have
previously been explored in S. typhimurium and E.
coli (6,10,18,20).
In the present study, using two rounds of Red recombinase-induced
homologous recombination, the asd gene was initially knocked
out in the E. coli cells, in order to render them dependent
on exogenous DAP supply. A plasmid was subsequently constructed
that contained an asd expression cassette to provide ASD
once transformed into the cells. Therefore, a symbiotic plasmid
production system that met all of the targets was generated.
Initially, an asd-disrupted strain was
engineered from commonly used DH10B E. coli cells. As a
laboratory strain for prokaryotic plasmid amplification, the strain
has a ribosomal mutation conferring resistance to Stm, which is
useful for selective bacterial maintenance and many other routine
features, including large scale plasmid production (6,16,17).
It also avoids any potential issues associated with JM83 plasmid
instability, the derivative of which was used to construct the
asd-deficient strain in the early study (14). In order to disrupt endogenous
asd expression in the DH10B cells, a modified prophage
recombination system was used to introduce a Km-resistance gene
into the DH10B cells, as previously reported (13,20).
The successful functioning of this step was demonstrated by the
large number of Km resistant colonies that were recovered following
the electroporation of linear DNA fragments containing the
kan-kil genes. The kan-kil fragment contains a DNA
sequence consisting of an aminoglycoside phosphotransferase
expression cassette for Km resistance (23), and a λ phage lysis gene cassette
under the control of a heat-inducible promoter (24). The former was used to select
homologous recombination events targeting the asd gene, and
the latter was used to remove the introduced Km-resistance gene by
killing the non-recombinants in the second round of homologous
recombination. However, removal of the inserted Km resistance gene
appeared to be poorly effective, and most of the colonies recovered
at the beginning of the study were false positives. These results
were likely due to the low efficiency of kil gene lysis, due
to the low copy or mostly single copy number of the gene within the
cells. To overcome this, several rounds of induction at 37̊C
followed by the plating of the transformants onto LB medium
supplemented with DAP was subsequently performed. Following several
attempts, a small number of colonies were eventually obtained and
their presence confirmed by asd locus-specific PCR and
positive Km sensitivity tests. Exposure of the cells to water and
LB medium with and without Amp led to cell death, indicating that
the loss of asd had compromised cell survival. These results
were most likely due to the inability of the cells to synthesize a
functional cell wall. However, as opposed to antibiotic-induced
cell death, the loss of viability in these cells appeared to be
slow, particularly when cultured in water. These observations were
likely due to the residual DAP in the cells, and the low metabolic
activity of the cells in water, which prevented them from actively
synthesizing a new cell wall. Nevertheless, further investigation
is required in order to elucidate the effects of asd
disruption on cell wall synthesis.
The nirB promoter was chosen to provide ASD
activity and to drive asd expression, as it has previously
been demonstrated to exhibit leaking expression in aerobic
conditions (25), thus allowing
many of the screenings and tests to be carried out under normal
aerobic conditions. The starter plasmid pDV-cmv gene
contained a relax ColE1 replicon (26,27),
and would therefore provide between 300 and 500 copies of the
nirB promoter to direct the expression of the cassette in
the E. coli cells. This resulted in the leaked production of
ASD at a sufficient enough quantity to allow DAP to be synthesized
for cell wall integrity. Therefore, under aerobic conditions, DH10B
Δasd cells transformed with the marker-free plasmid
pDV-cmv-gene-nirb-asd Km− were
expected to grow normally, even in the absence of DAP. The
experiments demonstrated that colonies appeared on the LB medium in
the absence of DAP whilst containing the correct form of
pDV-cmv-gene-nirb-asd Km− (Fig. 6), thus indicating successful
symbiosis in the vector/host combination. Yield and conformation of
the plasmid DNA was also found to be similar when produced in the
symbiotic condition under antibiotic selection, indicating that
symbiosis is robust enough to replace antibiotic selection for
plasmid production. Furthermore, plasmids containing the asd
gene were constructed with EGFP expression, to test whether the
plasmids would function regularly in other cells. In the present
study HeLa cells were chosen for this experiment. The results
indicated that the plasmid was expressed normally in the HeLa
cells, and that the insertion of the egfp gene into the
plasmid did not influence asd gene function.
One particularly important consideration for an
antibiotic resistant-marker-free plasmid production system is how
well the plasmid can be maintained in the host cell in the absence
of negative selection from antibiotics, whilst relying solely on
symbiosis. The results of the present study demonstrated that the
plasmid can be retained in the asd-disrupt cells without
noticeable loss for ≤30 generations of cell divisions when
inoculated at a 1:1,000 (v-v, inoculants: Medium) ratio. However,
the plasmid loss became evident when the inoculation ratio was 1:10
(Table I). This may be due to
cross-cell feeding of DAP from the cells with the plasmid to the
cells that had lost the plasmid, thereby increasing the chance of
survival of the cells that had lost the plasmid. Such cross-feeding
would be dependent on the number of cells within the plasmid (such
as the cells in the inoculants), leading to an increase in plasmid
loss in an inoculant quantity-dependent manner as seen in the
present study and as reported previously (27).
Another feature that would have a marked impact on
the practical use of this system is the survivability and
storability of the asd-deficient cells, particularly in the
case of long term storage. While these cells quickly lost their
viability at room or refrigerator temperature (25 and 4°C), little
change was observed in viability after 12 months storage at −70°C
(Table 2), indicating that they
may be stored for long term use. This also implies that the
disruption of asd would not compromise the viability of the
cells, as long as the lost asd function is complemented by
the plasmid. The pDV-cmv-gene-nirb-asd
Km− plasmid is expected to be of a value similar to that
of the eukaryotic expression vector pVXA1, which can be used in
preventive research such as the production of melanoma and
Alzheimer's DNA vaccines, as well as in the treatment of hemagioma
and artery terminal ischemia using animal expression models.
In conclusion, the present study developed a
symbiotic vector/host system for the use of marker-free plasmids in
gene therapy. The vector may be further manipulated in order to
carry additional gene(s) of interest for various purposes, and may
be produced in large quantities in the symbiotic host cells under
antibiotic-free conditions. This method would considerably reduce
the risk of antibiotic resistant gene transfer during the
therapeutic process, and would markedly improve therapeutic
safety.
Acknowledgments
The present study was financially supported by the
National Health and Family Planning Commission of the People's
Republic of China (grant no. 2012ZX09304010), and the National
High-tech R&D Program, Ministry of Science and Technology of
the People's Republic of China (grant no. 2012AA020805).
References
|
1
|
Tolmachov OE: Building mosaics of
therapeutic plasmid gene vectors. Current gene therapy. 11:466–478.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nahaei M, Valizadeh H, Baradaran B, et al:
Preparation and characterization of chitosan/beta-cyclodextrin
nanoparticles containing plasmid DNA encoding interleukin-12. Drug
research (Stuttg). 63:7–12. 2013. View Article : Google Scholar
|
|
3
|
Przybylowski M, Bartido S, Borquez-Ojeda
O, Sadelain M and Riviere I: Production of clinical-grade plasmid
DNA for human Phase I clinical trials and large animal clinical
studies. Vaccine. 25:5013–5024. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Norell H, Poschke I, Charo J, et al:
Vaccination with a plasmid DNA encoding HER-2/neu together with low
doses of GM-CSF and IL-2 in patients with metastatic breast
carcinoma: a pilot clinical trial. J Transl Med. 8:532010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smith LR, Wloch MK, Ye M, et al: Phase 1
clinical trials of the safety and immunogenicity of adjuvanted
plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin.
Vaccine. 28:2565–2572. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oliveira PH and Mairhofer J: Marker-free
plasmids for biotechnological applications - implications and
perspectives. Trends Biotechnol. 31:539–547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pruden A, Pei R, Storteboom H and Carlson
KH: Antibiotic resistance genes as emerging contaminants: Studies
in northern Colorado. Environ Sci Technol. 40:7445–7450. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
7.0 EP: Gene transfer medical
products for human use. 648, 2011.
|
|
9
|
U.S. Food and Drug Administration:
Guidance for human somatic cell therapy and gene therapy. 1998.
|
|
10
|
Vandermeulen G, Marie C, Scherman D and
Préat V: New generation of plasmid backbones devoid of antibiotic
resistance marker for gene therapy trials. Mol Ther. 19:1942–1949.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Haziza C, Stragier P and Patte JC:
Nucleotide sequence of the asd gene of Escherichia coli: Absence of
a typical attenuation signal. EMBO J. 1:379–384. 1982.PubMed/NCBI
|
|
12
|
Narayanan K and Warburton PE: DNA
modification and functional delivery into human cells using
Escherichia coli DH10B. Nucleic Acids Res. 31:e512003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Datsenko KA and Wanner BL: One-step
inactivation of chromosomal genes in Escherichia coli K-12 using
PCR products. Proc Natl Acad Sci USA. 97:6640–6645. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Curtiss R III, Galan JE, Nakayama K and
Kelly SM: Stabilization of recombinant avirulent vaccine strains in
vivo. Res Microbiol. 141:797–805. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dong WR, Xiang LX and Shao JZ: Novel
antibiotic-free plasmid selection system based on complementation
of host auxotrophy in the NAD de novo synthesis pathway. Appl
Environ Microbiol. 76:2295–2303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Durfee T, Nelson R, Baldwin S, et al: The
complete genome sequence of Escherichia coli DH10B: Insights into
the biology of a laboratory workhorse. J Bacteriol. 190:2597–2606.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grant SG, Jessee J, Bloom FR and Hanahan
D: Differential plasmid rescue from transgenic mouse DNAs into
Escherichia coli methylation-restriction mutants. Proc Natl Acad
Sci USA. 87:4645–4649. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Galán JE, Nakayama K and Curtiss R III:
Cloning and characterization of the asd gene of Salmonella
typhimurium: Use in stable maintenance of recombinant plasmids in
Salmonella vaccine strains. Gene. 94:29–35. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Z, Wang C, Xue Y, et al:
Immunogenicity and protective efficacy of pertactin recombinants
against Bordetella bronchiseptica challenge. Wei Sheng Wu Xue Bao.
50:1239–1245. 2010.In Chinese. PubMed/NCBI
|
|
20
|
Arias-Lopez C, Lazaro-Trueba I, Kerr P, et
al: p53 modulates homologous recombination by transcriptional
regulation of the RAD51 gene. EMBO Rep. 7:219–224. 2006. View Article : Google Scholar
|
|
21
|
Makoff AJ and Oxer MD: High level
heterologous expression in E. coli using mutant forms of the lac
promoter. Nucleic Acids Res. 19:2417–2421. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Teeter KC, Thibodeau LM, Gompert Z,
Buerkle CA, Nachman MW and Tucker PK: The variable genomic
architecture of isolation between hybridizing species of house
mice. Evolution. 64:472–485. 2010. View Article : Google Scholar
|
|
23
|
Kobayashi F, Yamaguchi M and Mitsuhashi S:
Drug resistance to aminoglycosidic antibiotics in Pseudomonas
aeruginosa and its lability. Jpn J Microbiol. 16:425–431. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yagisawa M, Yamamoto H, Naganawa H, Kondo
S and Takeuchi T: A new enzyme in Escherichia coli carrying
R-factor phosphorylating 3′-hydroxyl of butirosin A, kanamycin,
neamine and ribostamycin. J Antibiot (Tokyo). 25:748–750. 1972.
View Article : Google Scholar
|
|
25
|
Wang H and Gunsalus RP: The nrfA and nirB
nitrite reductase operons in Escherichia coli are expressed
differently in response to nitrate than to nitrite. J Bacteriol.
182:5813–5822. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bowers LM, Lapoint K, Anthony L,
Pluciennik A and Filutowicz M: Bacterial expression system with
tightly regulated gene expression and plasmid copy number. Gene.
340:11–18. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu YC and Liu ST: A sequence that affects
the copy number and stability of pSW200 and ColE1. J Bacteriol.
192:3654–3660. 2010. View Article : Google Scholar : PubMed/NCBI
|