Introduction
Atherosclerosis is a pathological process leading to
numerous fatal diseases, such as coronary heart disease, cerebral
infarction, and cerebral hemorrhage. The phenotypic change of
vascular smooth muscle cells (VSMCs) from a differentiated state to
a dedifferentiated state is critical to the progression of
atherosclerosis. Numerous studies have been carried out (1,2);
however, the molecular mechanisms underlying the phenotypic
transformation of VSMCs remain to be elucidated.
Galectin-3 (gal-3), which is a 29-35 kDa protein, is
a member of the β-galactoside-binding lectin family (3). Gal-3 is composed of a C-terminal
carbohydrate-recognition domain, a collagen-like internal R-domain,
and an N-terminal domain that promotes lectin oligomerization
(3,4). Gal-3 is present in the cytoplasm,
nucleus, extracellular space, and is also bound to the cell surface
(3). Gal-3 can be secreted into
the extracellular matrix and serum, where it binds to protein
laminin, fibronectin, and collagen IV. Although gal-3 is present in
almost every organ of the body, its contents vary greatly. A recent
study demonstrated that gal-3 may serve as a diagnostic and
prognostic marker for cardiovascular diseases (5). A previous study demonstrated that
gal-3 has an important pathophysiological role in cardiovascular
remodeling by promoting changes including cardiac hypertrophy,
fibrosis, arterial stiffness, as well as inflammation and oxidative
stress (6). Gal-3 may also serve
as a tool to predict future heart failure, and is associated with
heart failure morbidity and mortality (7). In addition, gal-3 has been shown to
induce angiogenesis and promote new blood vessel formation
(8).
Recent studies have reported that gal-3 has a role
in atherogenesis. An increase in the expression levels of gal-3 is
associated with the developmental process of atherogenesis
(9). In addition, gal-3 is
necessary for the inflammatory response induced by aldosterone in
VSMCs (6). Activation and
dedifferentiation of VSMCs are the principal pathological changes
that occur within the blood vessel wall during atherogenesis.
Activation of VSMCs also contributes to plaque instability
(1). However, the mechanism
underlying this pathological process remains to be elucidated.
Oxidized low-density lipoprotein (oxLDL) has been
widely reported to have a role in atherogenesis (10). In addition, oxLDL increases the
ability of macrophages to uptake lipid products (11), and promotes the migration and
proliferation of VSMCs (12).
Considering the importance of oxLDL in inducing atheroma, the
present study aimed to investigate whether gal-3 may function
together with oxLDL in atherogenesis. To demonstrate this
hypothesis, the role of gal-3 in the oxLDL-induced phenotypic and
functional changes of VSMCs was examined. The molecular mechanism
underlying this process was also investigated.
Materials and methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), and penicillin/streptomycin (10,000 U/ml each)
were purchased from Gibco Life Technologies (Carlsbad, CA, USA).
TRIzol® reagent for RNA isolation was purchased from
Invitrogen Life Technologies (Carlsbad, CA, USA). A Reverse
Transcriptase kit (cat. no. RR037A) and SYBR Premix Ex Taq
II (cat. no. RR820A) were purchased from Takara Biotechnology Co.,
Ltd. (Dalian, China). A Cell Counting kit-8 (CCK-8) assay was
purchased from Dojindo Molecular Technologies, Inc. (Kumamoto,
Japan). Oil Red O was purchased from Sigma-Aldrich (St. Louis, MO,
USA). OxLDL was purchased from Peking Union-Biology Co., Ltd.
(Beijing, China). The primary antibodies targeting smooth muscle
α-actin (SMA; cat. no. sc-53142), gal-3 (cat. no. sc-20157), and
GAPDH (cat. no. sc-48166) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The anti-β-catenin antibody
(cat. no. 8480) was purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). The monoclonal antibody targeting osteopontin
(OPN; cat. no. ab91655) was purchased from Abcam (Cambridge, UK).
The goat anti-rabbit secondary antibody (cat. no. A-21109) and the
goat anti-mouse secondary antibody (cat. no. A-21058) were
purchased from Invitrogen Life Technologies.
Cells culture
A primary culture of human umbilical smooth muscle
cells (HUSMCs) was established as previously described (13). Briefly, HUSMCs were collected by
explant outgrowth of a segment of human umbilical cord obtained
during a cesarean section procedure. Endothelial cells were removed
by scraping the luminal surface of the vessel with a cotton swab,
and the adventitia was mechanically stripped away. Primary cultures
were maintained in DMEM supplemented with 20% FBS and 1%
penicillin/streptomycin. The cells obtained between the 4th and
10th passage were used for further experimentation. The
experimental procedures of the present study complied with the
principles of the Declaration of Helsinki, and were approved by the
Ethics Committee of the Shanghai Ninth People's Hospital (Shanghai,
China). Written informed consent was obtained from all
patients.
Small interfering (si)RNA
transfection
Gal-3 expression was inhibited by transfection with
a siRNA specific to gal-3. Gal-3 siRNA was transiently transfected
into the cells using Lipofectamine® 2000 (Invitrogen
Life Technologies), according to the manufacturer's instructions.
Briefly, 5×105 HUSMCs per well were cultured in 6-well
plates to 75% confluence. The cells were then transfected with 100
pmol siRNA duplexes using 5 μl Lipofectamine®
2000 and DMEM. Following a 72 h incubation at 37°C the cells were
harvested for analysis. The human gal-3 siRNA sequence was
5′-CCUCGCAUGCUGAUAACAATT-3′, and the scrambled siRNA sequence was
5′-UUGUUAUCA GCAUGCGAGGTT-3′. siRNA were synthesized by Biotend
(Shanghai, China).
Cell proliferation assay
Cell proliferation was measured using the CCK-8
assay. Following cellular transfection with gal-3 siRNA or scramble
siRNA for 24 h, cells were plated into 96-well plates, at a density
of 5×103 cell/well. The HUSMCs were subsequently
incubated for 24 or 48 h in the absence or presence of 50
μg/ml oxLDL, and cell proliferation was measured using the
CCK-8 assay according to the manufacturer's instructions, at 0, 24
or 48 h.
Migration assay
The migration assay was performed as previously
described (14). Briefly,
following cellular transfection with gal-3 siRNA or scramble siRNA,
the HUSMCs were resuspended in 200 μl serum-free DMEM, and
5×104 HUSMCs were loaded into the upper chambers of a
transwell chamber (Corning Inc., Corning, NY, USA). The lower
chambers were filled with 400 μl DMEM in the presence or
absence of 10 μg/ml oxLDL. The cells were then incubated at
37°C for 24 h. The lower side of the filter was washed three times
with phosphate-buffered saline prior to being fixed with 4%
paraformaldehyde for 20 min. The nuclei were stained with
4′,6-diamino-2-phenylindole (DAPI; 1:1,000; Sigma-Aldrich) for 5
min at room temperature. The cells were counted using an Eclipse
TS100 microscope (Nikon Corporation, Tokyo, Japan) in three random
high-power fields (magnification, ×100) for each well.
Immunofluorescence staining and confocal
laser microscopy
The cells (5×105) were seeded onto
flame-sterilized coverslips and placed into 6-well tissue culture
plates. The cells were then transfected with gal-3 siRNA or
scramble siRNA for 24 h. The cells were subsequently fixed with 4%
paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100
(Sangon Biotech Co., Ltd., Shanghai, China) for 20 min, blocked
with 1% bovine serum albumin (Sigma-Aldrich) for 1 h, and incubated
with specific primary antibody overnight at 4°C. The cells were
incubated with Alexa Fluor 488-conjugated goat anti-rabbit
immunoglobulin G (Invitrogen Life Technologies) for 1 h at room
temperature. The nuclei were stained with DAPI (1:1,000) for 5 min
at room temperature. The protein expression levels of β-catenin
were then quantified using a FluoView™ FV1000 confocal laser
scanning microscope (Olympus Corporation, Tokyo, Japan).
Oil Red O staining assay
The Oil Red O staining assay was performed as
previously described (15).
Briefly, HUSMCs (5×105) were seeded on sterile
coverslips in 6-well plates. The cells were then transfected with
gal-3 siRNA or scramble siRNA for 24 h, prior to being treated with
50 μg/ml oxLDL for 48 h. The cells were washed three times
with PBS, fixed in 4% paraformaldehyde for 20 min and then stained
with 0.5% Oil Red O for 20 min in order to identify lipid droplets
in the cytoplasm using an Eclipse TS100 microscope at 40×
magnification.
Reverse transcription-quantitative
polymerase chain reac- tion (RT-qPCR) analysis
Total RNA was extracted using TRIzol®
reagent, according to the manufacturer's instructions. Total RNA
was reverse-transcribed into cDNA using a Reverse Transcriptase kit
(cat. no. RR037A), and RT-qPCR was performed using SYBR Premix
Ex Taq II (cat. no. RR820A), with gene-specific primers, on an
Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems
Life Technologies, Foster City, CA, USA), according to the
manufacturer's instructions. The primers targeting human gal-3,
β-catenin, calponin, SMA, and OPN were as follows: Gal-3, forward
5′-GGCCACTGATTGTGCCTTAT-3′, and reverse 5′-TGCAACCTTGAAGTGGTCAG-3′;
β-catenin, forward 5′-GCCGGCTATTGTAGAAGCTG-3′, and reverse
5′-GAGTCCCAAGGAGACCTTCC-3′; Calponin, forward
5′-ATGTGAGGAGGGAAGAGTGTG-3′, and reverse 5′-CGGTTGAAGTGAGCAGAGG-3′;
SMA, forward 5′-AGCGTGGCTACTCCTTCGTGAC-3′, and reverse
5′-GCTCGTTGCCGATGGTGATGAC-3′; OPN, forward
5′-TGAGTCTGGAAATAACTAATGTGTTTGA-3′, and reverse
5′-GAACATAGACATAACCCTGAAGCTTTT-3′; and GAPDH, forward
5′-TGATGACATCAAGAAGG TGGTGAAG-3′, and reverse 5′-TCCTTGGAGGCCA
TGTGGGCCAT-3′. The primers were synthesized by Sangon Biotech Co.,
Ltd. The PCR cycling conditions were as follows: Initial
denaturation at 95°C for 30 sec, followed by 40 cycles at 95°C for
5 sec, 60°C for 34 sec and 95°C for 15 sec, and finally 60°C for 1
min and 95°C for 15 sec. The relative mRNA expression levels were
calculated using the comparative cycle threshold (CT) method
(2−ΔΔCT) (16).
Western blot analysis
The cells were lysed using a lysis buffer containing
150 mM NaCl, 10 mM Tris (pH 7.5), 5 mM EDTA, 1% Triton X-100, 1 mM
phenylmethylsulfonyl fluoride, 10 mg/ml leupeptin, 10 mg/ml
pepstatin, and 10 mg/ml aprotinin for 30 min on ice. Protein
concentrations were measured using a Bicinchoninic Acid Protein
Assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA). The
lysates (20 μg) were separated by 10% SDS-PAGE, and
transferred onto nitrocellulose membranes (EMD Millipore,
Billerica, MA, USA). The membranes were then blocked with 5%
non-fat dry milk in Tris-buffered saline and Tween 20 (TBST; 100 mM
NaCl, 10 mM Tris-HCl, pH 7.4, and 0.1% Tween 20) for 1 h at room
temperature. The blots were incubated with the various primary
antibodies at a dilution of 1:1,000 in TBST, overnight at 4°C, and
then washed twice with TBST buffer at room temperature, prior to
incubation for 1 h with the appropriate peroxidase-conjugated
secondary antibodies (1:5,000). All signals were detected using
Odyssey® Clx Infrared Imaging system (LI-COR
Biosciences, Lincoln, NE, USA). Protein quantification was achieved
by measuring the band intensity using Quantity One®
4.6.2 software (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
All data are expressed as the mean ± standard
deviation. Statistical analyses were performed using SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA). The data were analyzed
using a one-way analysis of variance (ANOVA) and
Student-Newman-Keuls post-hoc test. All of the data did not pass
the normality test, therefore, a non-parametric ANOVA
(Kruskal-Wallis test) was used, as appropriate. P<0.05 was
considered to indicate a statistically significant difference. All
experiments were performed in triplicate.
Results
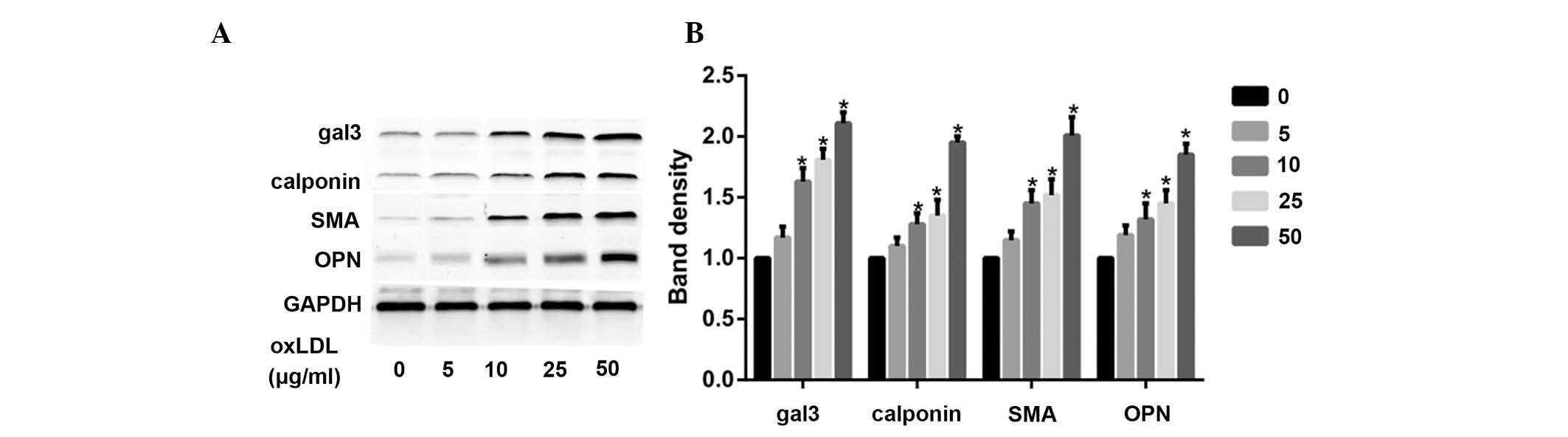
OxLDL induces the phenotypic
transformation of HUSMCs, and increases the expression levels of
gal-3
According to a previous study (17), minimally-oxidized LDL may induce
phenotypic transformation of coronary artery smooth muscle cells.
The present study demonstrated that oxLDL directly induced
significant changes in smooth muscle cell phenotype, following
cellular treatment with various oxLDL concentrations (0-50
μg/ml). Treatment with oxLDL induced a marked increase in
the expression levels of smooth muscle synthetic related protein
OPN, and contractile related proteins calponin and SMA. OxLDL also
increased the protein expression levels of gal-3 (Fig. 1A and B).
Silencing gal-3 reverses the
oxLDL-induced phenotypic trans- formation of HUSMCs
To investigate the role of gal-3 in the
oxLDL-induced phenotypic changes of HUSMCs, gal-3 knockdown was
performed. Following transfection with siRNA, the expression of
endogenous gal-3 was inhibited, and the phenotypic changes of
HUSMCs were assessed. Gal-3-specific siRNA reduced the mRNA and
protein expression levels of gal-3 by 95 and 45%, respectively. The
non-targeting siRNA had no effect on gal-3 expression. Neither
gal-3 nor the non-targeting siRNA induced a non-specific knockdown
of GAPDH. Furthermore, gal-3 knockdown significantly inhibited both
the oxLDL-induced mRNA and protein expression levels of OPN,
calponin, and SMA (Fig. 2A-C).
These results suggest that gal-3 may be involved in the
oxLDL-induced phenotypic transformation of HUSMCs.
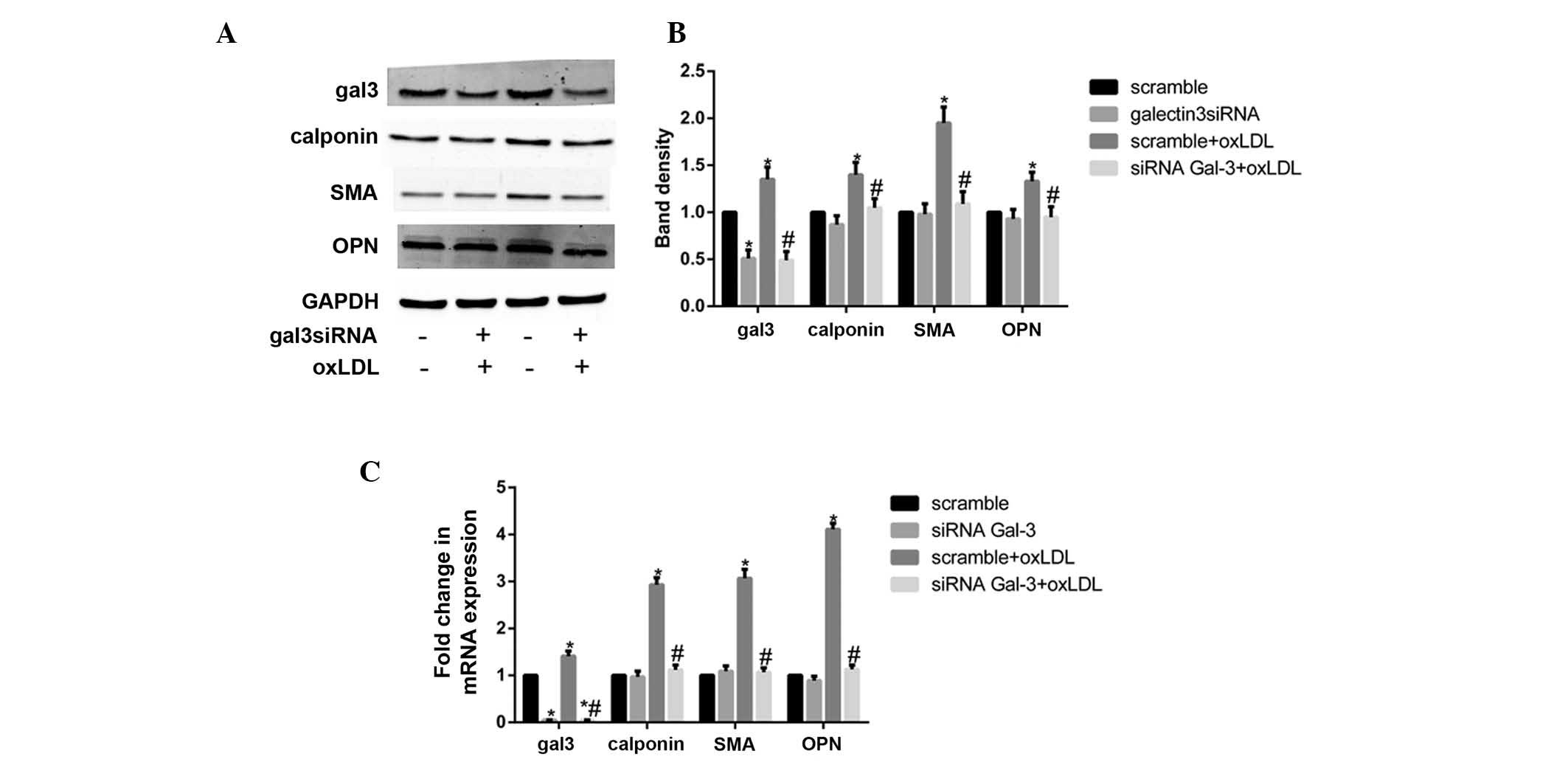 | Figure 2Silencing of galectin-3 (gal-3)
reversed the oxidized low-density lipoprotein (oxLDL)-induced
phenotypic transformation of human umbilical smooth muscle cells
(HUSMCs). HUSMCs were transfected with gal-3-specific small
interfering (si)RNA for 24 h, and then cultured with 50
μg/ml oxLDL for 48 h. The mRNA and protein expression levels
of gal-3, smooth muscle α-actin (SMA), calponin, and osteopontin
(OPN) were measured by reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting. (A and B)
Western blotting and (C) RT-qPCR results of gal-3, calponin, OPN
and SMA are shown. (B) The respective densitometric measurement
results are given. The protein expression levels of gal-3,
calponin, OPN and SMA were normalized to those of GAPDH. Band
density of HUSMCs transfected with scramble siRNA was defined as
control and set to 1. Data are presented as the mean ± standard
deviation. *P<0.05, vs. the control.
#P<0.05, vs. oxLDL. |
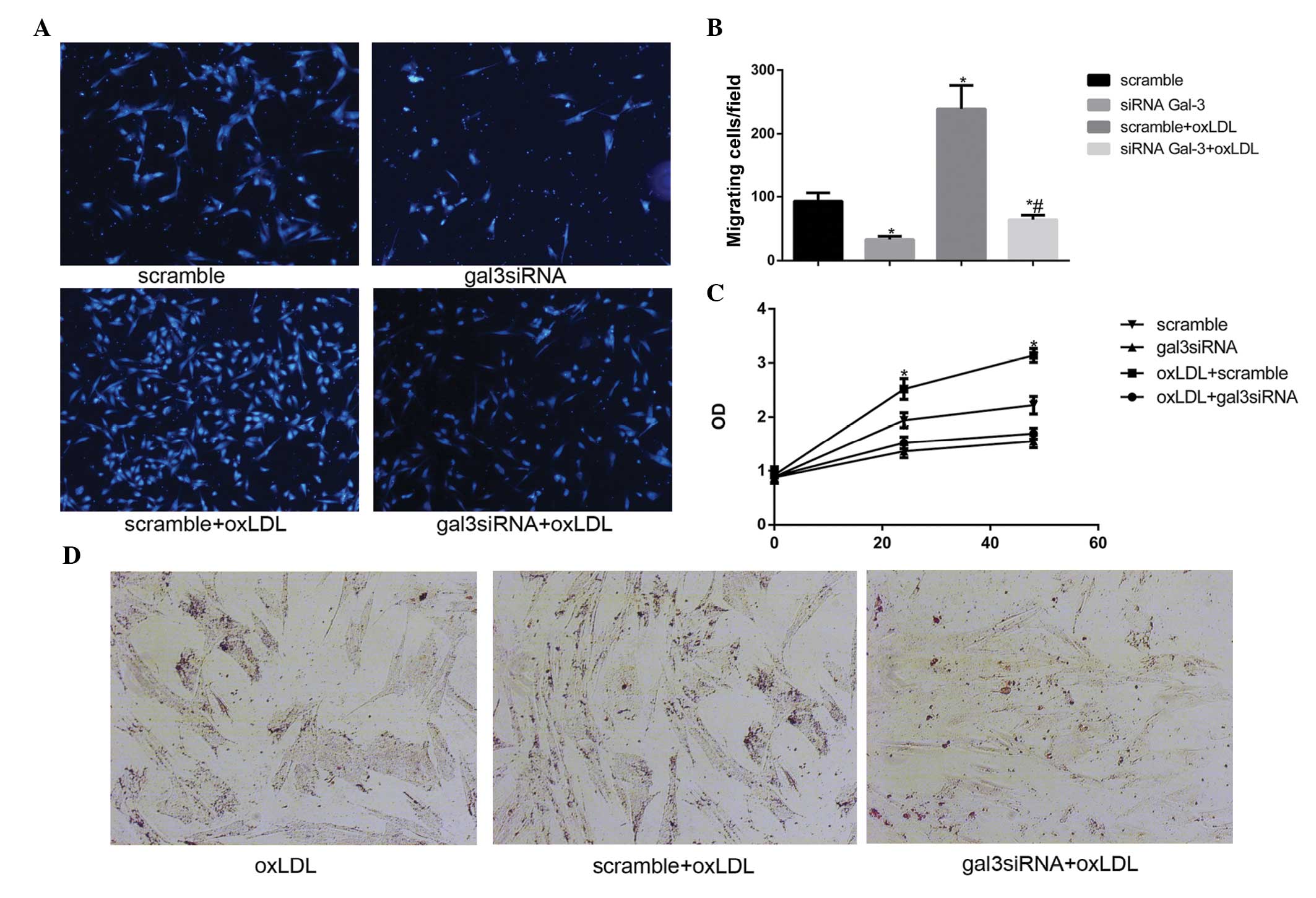
Silencing gal-3 reduces the oxLDL-induced
activation of HUSMCs
Previous studies (18–20)
have reported that oxLDL may promote the activation of VSMCs,
including migration, proliferation, and phagocytosis. The present
study investigated whether gal-3 was involved in the oxLDL-induced
activation of HUSMCs. Migration assays were performed in order to
determine the effects of gal-3 silencing on cell motility, and cell
numbers, which were determined using a microscope. Knockdown of
endogenous gal-3 markedly reduced oxLDL-mediated cell migration
in vitro (Fig. 3A and B). A
previous study reported that 50 μg/ml oxLDL induced the
proliferation of HUSMCs at 24 h (21). The present study demonstrated that
silencing gal-3 reduced cell proliferation, and 50 μg/ml
oxLDL induced proliferation at 24 h and 48 h (Fig. 3C). These results suggest that gal-3
may affect the proliferation of HUSMCs. The effects of gal-3
knockdown on cell phagocytosis were also investigated. The cells
were treated with oxLDL for 48 h, following which lipid
accumulation was stained with Oil Red O. A marked increase in Oil
Red O staining in the control cells incubated with oxLDL was
observed, as compared with the cells transfected with gal-3 siRNA
(Fig. 3D). These results suggest
that gal-3 has an important role in the oxLDL-induced activation of
HUSMC.
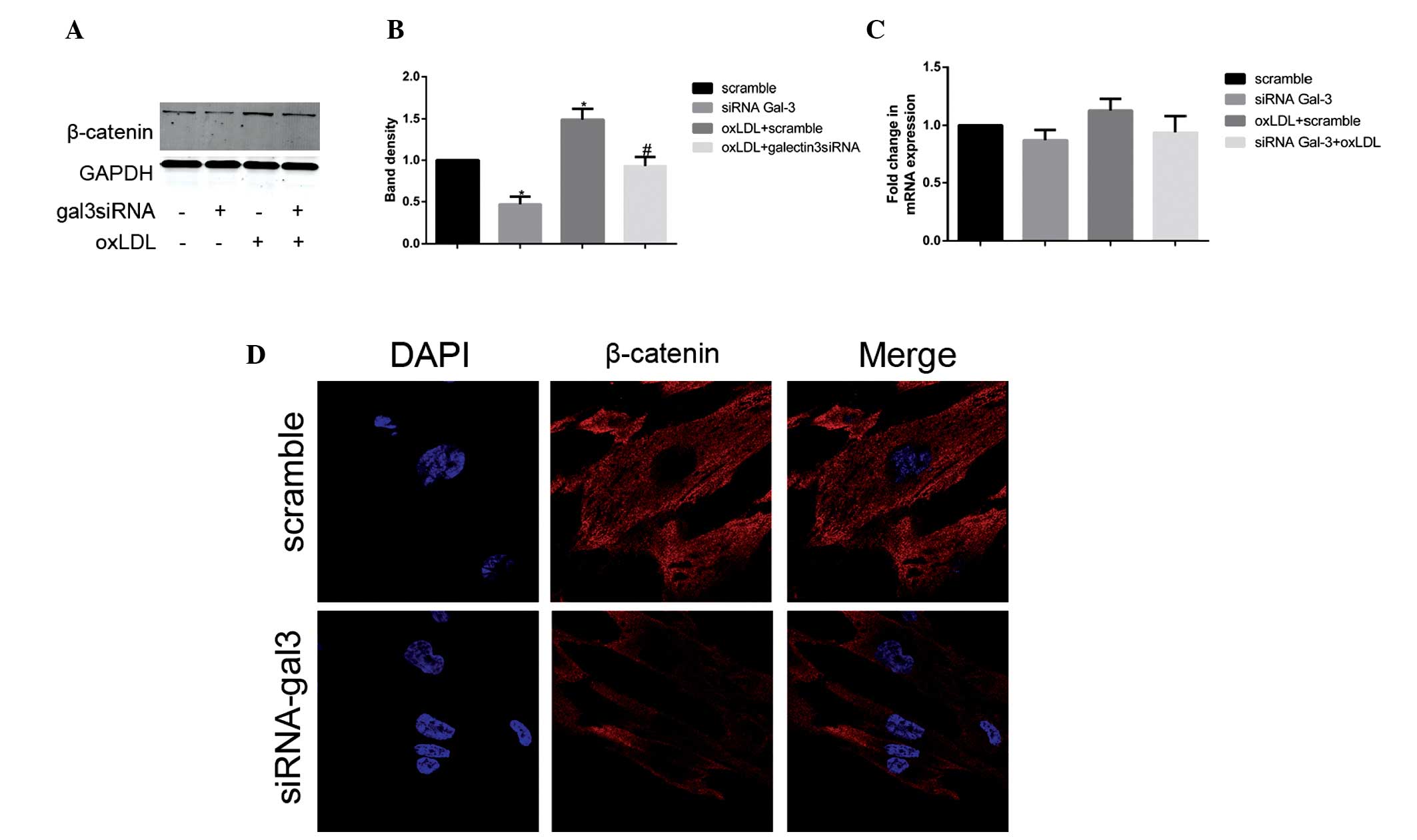
Gal-3 silencing inhibits the expression
of β-catenin
To further elucidate the molecular mechanism
underlying the gal-3-mediated oxLDL-induced activation and
transformation of HUSMCs, the present study investigated whether
Wnt/β-catenin signaling was involved in the process. The basis for
these experiments was provided by the well-documented role of gal-3
in Wnt/β-catenin signaling (22),
as well as the association between Wnt/β-catenin signaling and
phenotypic changes in murine VSMCs (23). Following treatment with oxLDL, the
protein expression levels of β-catenin increased; however, the mRNA
expression levels of β-catenin remained unchanged. The present
study subsequently investigated the expression levels of β-catenin
using RT-qPCR and western blotting, following gal-3 knockdown.
Silencing of gal-3 significantly reduced the oxLDL-induced protein
expression levels of β-catenin (Fig.
4A and B). However, no changes were observed in the mRNA
expression levels of β-catenin following treatment with gal-3 siRNA
(Fig. 4C). In the gal3
siRNA-transfected and control cells, the subcellular distribution
of β-catenin was examined by immunofluorescence staining and
confocal microscopy. Reduced expression levels of β-catenin were
observed not only in the cytoplasm but also in the nucleus
(Fig. 4D). These data suggest that
gal-3 has an important role in oxLDL-induced increases in β-catenin
expression.
Discussion
Increased expression levels of gal-3 have been
detected in atherosclerotic lesions (9), and the oxLDL-induced expression of
gal-3 has previously been described in macrophages (24). In addition, it has been suggested
that oxLDL-induced phenotypic transformation involves numerous
molecular mechanisms (25). The
present study provides, to the best of our knowledge, the first
report that gal-3 has a critical role in the oxLDL-induced
phenotypic transformation of HUSMCs. The canonical Wnt/β-catenin
signaling pathway was also reported to be associated with this
process.
Abnormal proliferation of SMCs in the subendothelial
space of arterial walls has an important role in the progression of
atherosclerotic lesions (26).
Numerous studies have demonstrated that oxLDL may promote cellular
proliferation and migration (4,21). A
recent study reported that gal-3 acts as an anti-adhesive factor,
and may promote SMC proliferation and migration, thus suggesting
that gal-3 may accelerate atherogenesis (9,27).
The present study demonstrated that silencing of gal-3 by siRNA
significantly reduced oxLDL-mediated cellular proliferation and
migration in vitro. The results of the present study
conclusively established that endogenous gal-3 may significantly
influence oxLDL-induced proliferation and migration. Foam cell
formation is an important contributor to atherosclerosis. Foam
cells were previously thought to originate primarily from
macrophages; however, recent studies have indicated that VSMCs are
also precursors of foam cells (28,29).
The phenotypic transformation of VSMCs is closely associated with
the phagocytosis of VSMCs, and this transformation has an important
role in regulating cellular lipid accumulation (28). It has been reported that gal-3 is
required for aldosterone-induced inflammation and fibrosis
(6). However, the association
between endogenous gal-3 and the phenotypic transformation of VSMCs
remains unknown. The results of the present study demonstrated that
silencing of gal-3 was able to suppress the phagocytosis of
HUSMCs.
Contractile and synthetic SMCs are located on the
opposite ends of the SMCs spectrum, with intermediate phenotypes
located in between (30). These
contractile and synthetic phenotypes are regarded as “idealized”
phenotypes, with heterogeneity in the phenotypes of smooth muscle
cells being reported in numerous studies (30–32).
In some instances, contractile differentiation may be upregulated
in the synthetic phenotype (30),
and contractile differentiation markers are expressed during matrix
synthesis (32). Similarly,
whereas higher growth rates and stronger migratory activity are
usually considered typical of the synthetic phenotype, OPN has also
been identified as a synthetic-associated protein (2). The results of the present study
suggested that oxLDL not only increased the characteristics of the
synthetic phenotype, but also promoted the activity of
contractile-associated proteins calponin and SMA. Gal-3 is widely
expressed in numerous types of cells, and its expression is closely
correlated with the proliferation and metastasis of these various
cell types (8,33). Until recently, the role of gal-3 in
HUSMCs was also poorly characterized. A previous study demonstrated
that VSMCs were able to express gal-3 under basal conditions
(6). The results of the present
study correlated with these results and also suggested that the
expression levels of gal-3 were moderately enhanced by oxLDL. The
present study also investigated the effects of gal-3 on the process
of oxLDL-induced phenotypic transformation of HUSMCs. The results
suggested that gal-3 knockdown significantly inhibited both the
oxLDL-induced mRNA and protein expression levels of OPN, calponin,
and SMA. Therefore, following gal-3-knockdown, oxLDL is more likely
to induce the contractile phenotype.
Previous studies have reported that β-catenin has a
crucial role in cell proliferation and migration (33,34).
Wnt/β-catenin signaling has also been shown to be associated with
OPN, SMA, and calponin expression in numerous types of cells
(23,30). It is well-documented that β-catenin
is regulated by gal-3 in colon and gastric cancer cells (22,35),
and gal-3 is considered an important factor that maintains the
stability of β-catenin (36). The
present study therefore aimed to investigate whether β-catenin was
affected by the silencing of gal-3 in HUSMCs. The results of the
present study demonstrated that knockdown of gal-3 decreased the
protein expression levels of β-catenin, but had no effect on its
mRNA expression levels. The present study also demonstrated that
oxLDL increased only the protein expression levels of β-catenin,
but not the mRNA expression levels. Following cellular transfection
with gal-3 siRNA, the oxLDL-induced increase in the protein
expression levels of β-catenin was also inhibited. These results
are concordant with those of previous studies (36,37).
Furthermore, binding of β-catenin to gal-3 has been shown to
increase glycogen synthase kinase 3β (GSK-3β) phosphorylation.
Inactivation of GSK-3β is able to inhibit the degradation of
β-catenin, and increase the cellular levels of β-catenin (38). These results suggest that
oxLDL-induced activation of the canonical Wnt/β-catenin is
dependent on the presence of gal-3 in HUSMCs. Canonical
Wnt/β-catenin signaling activation has been associated with
cellular β-catenin distribution (1,36).
The present study also aimed to detect β-catenin in both the
cytoplasm and the nuclei of HSMCs. The results demonstrated that
β-catenin was subcellularly distributed in HUSMCs, both in the
presence and absence of gal-3 siRNA transfection. In addition,
silencing of gal-3 induced a decrease in the expression levels of
β-catenin both in the cytoplasm and the nucleus.
In conclusion, the present study demonstrated that
gal-3 is an important factor in the oxLDL-induced phenotypic
transformation of VSMCs, and a mediator of oxLDL-induced activation
of Wnt/β-catenin signaling. However, the role of gal-3 in the
phenotypic transformation of HUSMCs remains to be determined in
vivo, and the mechanism by which the protein expression levels
of gal-3 and β-catenin affect the function and phenotype of HUSMC
merits further study. Recently, gal-3 has been identified as an
important factor in atherosclerosis and heart failure (5,39).
The findings of the present study may provide a novel therapeutic
strategy for the treatment of atherosclerosis.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81270376);
the Shanghai Hospital Development Center (grant no. SHDC12012312);
the Ministry of Education Science and Technology Development Center
(grant no. 20130073110016); and the Fund of the Ninth People's
Hospital (grant no. 2013A02).
References
|
1
|
Mill C and George SJ: Wnt signalling in
smooth muscle cells and its role in cardiovascular disorders.
Cardiovasc Res. 95:233–240. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rensen SS, Doevendans PA and van Eys GJ:
Regulation and characteristics of vascular smooth muscle cell
phenotypic diversity. Neth Heart J. 15:100–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dumic J, Dabelic S and Flögel M:
Galectin-3: An open-ended story. Biochim Biophys Acta.
1760:616–635. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Markowska AI, Liu FT and Panjwani N:
Galectin-3 is an important mediator of VEGF- and bFGF-mediated
angiogenic response. J Exp Med. 207:1981–1993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anand IS, Rector TS, Kuskowski M, Adourian
A, Muntendam P and Cohn JN: Baseline and serial measurements of
galectin-3 in patients with heart failure: Relationship to
prognosis and effect of treatment with valsartan in the Val-HeFT.
Eur J Heart Fail. 15:511–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calvier L, Miana M, Reboul P, Cachofeiro
V, Martinez-Martinez E, de Boer RA, Poirier F, Lacolley P, Zannad
F, Rossignol P and López-Andrés N: Galectin-3 mediates
aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc
Biol. 33:67–75. 2013. View Article : Google Scholar
|
|
7
|
Morrow DA and O'Donoghue ML: Galectin-3 in
cardiovascular disease: A possible window into early myocardial
fibrosis. J Am Coll Cardiol. 60:1257–1258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wesley UV, Vemuganti R, Ayvaci ER and
Dempsey RJ: Galectin-3 enhances angiogenic and migratory potential
of microglial cells via modulation of integrin linked kinase
signaling. Brain Res. 1496:1–9. 2013. View Article : Google Scholar
|
|
9
|
Arar C, Gaudin JC, Capron L and Legrand A:
Galectin-3 gene (LGALS3) expression in experimental atherosclerosis
and cultured smooth muscle cells. FEBS Lett. 430:307–311. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Massaeli H, Hurtado C, Austria JA and
Pierce GN: Oxidized low-density lipoprotein induces cytoskeletal
disorganization in smooth muscle cells. Am J Physiol.
277:H2017–H2025. 1999.PubMed/NCBI
|
|
11
|
Obradovic MM, Trpkovic A, Bajic V, Soskic
S, Jovanovic A, Stanimirovic J, Panic M and Isenovic ER:
Interrelatedness between C-reactive protein and oxidized
low-density lipoprotein. Clin Chem Lab Med. 53:29–34. 2015.
View Article : Google Scholar
|
|
12
|
Steinberg D: Oxidized low density
lipoprotein - an extreme example of lipoprotein heterogeneity. Isr
J Med Sci. 32:469–472. 1996.PubMed/NCBI
|
|
13
|
Auge N, Garcia V, Maupas-Schwalm F, Levade
T, Salvayre R and Negre-Salvayre A: Oxidized LDL-induced smooth
muscle cell proliferation involves the EGF receptor/PI-3 kinase/Akt
and the sphingolipid signaling pathways. Arterioscler Thromb Vasc
Biol. 22:1990–1995. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leavesley DI, Schwartz MA, Rosenfeld M and
Cheresh DA: Integrin beta 1- and beta 3-mediated endothelial cell
migration is triggered through distinct signaling mechanisms. J
Cell Biol. 121:163–170. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Argmann CA, Sawyez CG, Li S, Nong Z,
Hegele RA, Pickering JG and Huff MW: Human smooth muscle cell
subpopulations differentially accumulate cholesteryl ester when
exposed to native and oxidized lipoproteins. Arterioscler Thromb
Vasc Biol. 24:1290–1296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Karagiannis GS, Weile J, Bader GD and
Minta J: Integrative pathway dissection of molecular mechanisms of
moxLDL-induced vascular smooth muscle phenotype transformation. BMC
Cardiovasc Disord. 13:42013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Augé N, Maupas-Schwalm F, Elbaz M, Thiers
JC, Waysbort A, Itohara S, Krell HW, Salvayre R and Nègre-Salvayre
A: Role for matrix metalloproteinase-2 in oxidized low-density
lipoprotein-induced activation of the sphingomyelin/ceramide
pathway and smooth muscle cell proliferation. Circulation.
110:571–578. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guyton JR, Lenz ML, Mathews B, Hughes H,
Karsan D, Selinger E and Smith CV: Toxicity of oxidized low density
lipoproteins for vascular smooth muscle cells and partial
protection by antioxidants. Atherosclerosis. 118:237–249. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deng DX, Spin JM, Tsalenko A, Vailaya A,
Ben-Dor A, Yakhini Z, Tsao P, Bruhn L and Quertermous T: Molecular
signatures determining coronary artery and saphenous vein smooth
muscle cell phenotypes: Distinct responses to stimuli. Arterioscler
Thromb Vasc Biol. 26:1058–1065. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ding Z, Liu S, Yang B, Fan Y and Deng X:
Effect of oxidized low-density lipoprotein concentration
polarization on human smooth muscle cells' proliferation, cycle,
apoptosis and oxidized low-density lipoprotein uptake. J R Soc
Interface. 9:1233–1240. 2012. View Article : Google Scholar :
|
|
22
|
Shimura T, Takenaka Y, Tsutsumi S, Hogan
V, Kikuchi A and Raz A: Galectin-3, a novel binding partner of
beta-catenin. Cancer Res. 64:6363–6367. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Menini S, Iacobini C, Ricci C, Blasetti
Fantauzzi C, Salvi L, Pesce CM, Relucenti M, Familiari G, Taurino M
and Pugliese G: The galectin-3/RAGE dyad modulates vascular
osteogenesis in atherosclerosis. Cardiovasc Res. 100:472–480. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim K, Mayer EP and Nachtigal M:
Galectin-3 expression in macrophages is signaled by Ras/MAP kinase
pathway and up-regulated by modified lipoproteins. Biochim Biophys
Acta. 1641:13–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu J, Ren Y, Kang L and Zhang L: Oxidized
low-density lipoprotein increases the proliferation and migration
of human coronary artery smooth muscle cells through the
upregulation of osteopontin. Int J Mol Med. 33:1341–1347.
2014.PubMed/NCBI
|
|
26
|
Dzau VJ, Braun-Dullaeus RC and Sedding DG:
Vascular proliferation and atherosclerosis: New perspectives and
therapeutic strategies. Nat Med. 8:1249–1256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nachtigal M, Ghaffar A and Mayer EP:
Galectin-3 gene inactivation reduces atherosclerotic lesions and
adventitial inflammation in ApoE-deficient mice. Am J Pathol.
172:247–255. 2008. View Article : Google Scholar :
|
|
28
|
Xue JH, Yuan Z, Wu Y, Liu Y, Zhao Y, Zhang
WP, Tian YL, Liu WM, Liu Y and Kishimoto C: High glucose promotes
intracellular lipid accumulation in vascular smooth muscle cells by
impairing cholesterol influx and efflux balance. Cardiovasc Res.
86:141–150. 2010. View Article : Google Scholar
|
|
29
|
Rong JX, Shapiro M, Trogan E and Fisher
EA: Transdifferentiation of mouse aortic smooth muscle cells to a
macrophage-like state after cholesterol loading. Proc Natl Acad Sci
USA. 100:13531–13536. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carthy JM, Luo Z and McManus BM: WNT3A
induces a contractile and secretory phenotype in cultured vascular
smooth muscle cells that is associated with increased gap junction
communication. Lab Invest. 92:246–255. 2012. View Article : Google Scholar
|
|
31
|
Hao H, Gabbiani G and Bochaton-Piallat ML:
Arterial smooth muscle cell heterogeneity: Implications for
atherosclerosis and restenosis development. Arterioscler Thromb
Vasc Biol. 23:1510–1520. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rama A, Matsushita T, Charolidi N, Rothery
S, Dupont E and Severs NJ: Up-regulation of connexin43 correlates
with increased synthetic activity and enhanced contractile
differentiation in TGF-β-treated human aortic smooth muscle cells.
Eur J Cell Biol. 85:375–386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xie D, Yin D, Tong X, O'Kelly J, Mori A,
Miller C, Black K, Gui D, Said JW and Koeffler HP: Cyr61 is
overexpressed in gliomas and involved in integrin-linked
kinase-mediated Akt and beta-catenin-TCF/Lef signaling pathways.
Cancer Res. 64:1987–1996. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee KB, Ye S, Park MH, Park BH, Lee JS and
Kim SM: p63-Mediated activation of the β-catenin/c-Myc signaling
pathway stimulates esophageal squamous carcinoma cell invasion and
metastasis. Cancer Lett. 353:124–132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SJ, Choi IJ, Cheong TC, Lee SJ, Lotan
R, Park SH and Chun KH: Galectin-3 increases gastric cancer cell
motility by up-regulating fascin-1 expression. Gastroenterology.
138:1035–1045. 2010. View Article : Google Scholar
|
|
36
|
Kobayashi T, Shimura T, Yajima T, Kubo N,
Araki K, Tsutsumi S, Suzuki H, Kuwano H and Raz A: Transient gene
silencing of galectin-3 suppresses pancreatic cancer cell migration
and invasion through degradation of β-catenin. Int J Cancer.
129:2775–2786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang D, Chen ZG, Liu SH, Dong ZQ, Dalin
M, Bao SS, Hu YW and Wei FC: Galectin-3 gene silencing inhibits
migration and invasion of human tongue cancer cells in vitro via
downregu-lating β-catenin. Acta Pharmacol Sin. 34:176–184. 2013.
View Article : Google Scholar
|
|
38
|
Song S, Mazurek N, Liu C, Sun Y, Ding QQ,
Liu K, Hung MC and Bresalier RS: Galectin-3 mediates nuclear
beta-catenin accumulation and Wnt signaling in human colon cancer
cells by regulation of glycogen synthase kinase-3beta activity.
Cancer Res. 69:1343–1349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Weir RA, Petrie CJ, Murphy CA, Clements S,
Steedman T, Miller AM, McInnes IB, Squire IB, Ng LL, Dargie HJ and
McMurray JJ: Galectin-3 and cardiac function in survivors of acute
myocardial infarction. Circ Heart Fail. 6:492–498. 2013. View Article : Google Scholar : PubMed/NCBI
|