Introduction
The endoplasmic reticulum (ER) is one of the largest
cellular organelles and has diverse functions, such as regulating
the folding of membrane proteins and secretory proteins (1). Various stimuli, such as ischemia
(2), hypoxia (3), heat shock, genetic mutation (4), oxidative stress (5) and elevated protein synthesis could
result in ER dysfunction. Stresses that lead to the impairment of
ER function are collectively known as ER stress (6,7). ER
stress triggers an evolutionarily conserved response termed the
unfolded protein response (UPR), an adaptive mechanism that
initially promotes organelle recovery (8). In response to ER stress, there is
significant upregulation of various ER resident chaperones, such as
glucose-regulated protein 94-kDa (GRP94) and glucose-regulated
protein 78-kDa (GRP78) that inhibit protein synthesis and activate
protein degradation (9–10). When ER stress is excessive and/or
prolonged however, apoptotic cell death is triggered by
transcriptional induction of C/EBP homologous protein (CHOP) and/or
by the activation of c-JUN NH2-terminal kinase (JNK), and/or
caspase-12-dependent pathways (11).
Accumulating evidence demonstrates that ER
stress-induced apoptosis is the key contributor to cell loss in the
pathogenesis of a series of cardiovascular diseases, such as
ischemia/reperfusion heart diseases (12,13),
atherosclerosis (6,14,15),
acute coronary syndrome (16),
myocardial infarction (17,18)
and heart failure (19,20). Tunicamycin acts as a highly
specific ER stress inducer by inhibiting N-linked glycosylation of
protein. Since there is a lack of a systemic ER stress-induced
apoptotic model in cardiomyocytes, the present study constructed an
endoplasmic reticulum stress-induced apoptotic model using
tunicamycin in primary cultured rat neonatal cardiomyocytes. The
optimal treatment time and concentration of tunicamycin in
cardiomyocytes was investigated. Cell viability was detected using
an MTT assay and cell damage was observed using an LDH release
assay. Apoptosis was measured using a flow cytometry assay and
determining the activity of caspase-3. Finally, the expression of
ER stress markers, including GRP78 and CHOP was induced by
tunicamycin at different time points in cardiomyocytes.
Materials and methods
Reagents and antibodies
Tunicamycin, collagenase I,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
lactate dehydrogenase (LDH), dimethyl sulfoxide (DMSO),
5-bromo-2-deoxyuridine (BrdU) and trypsin were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Tunicamycin was dissolved in
DMSO. Dulbecco's modified Eagle's medium (DMEM) medium and fetal
bovine serum (FBS) were purchased from Gibco (Grand Island, NY,
USA). A bicinchoninic acid (BCA) protein assay kit was purchased
from Pierce (Rockford, IL, USA). Anti-GRP78 antibody was obtained
from Bioworld (St. Louis Park, MN, USA). Anti-CHOP and anti-β-actin
were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Goat anti-rabbit and goat anti-mouse antibodies conjugated to
IRDyeTM800 (Rockland Inc., IL, USA) were detected using
an Odyssey infrared imaging system (LI-COR Inc., Lincoln, NE,
USA).
Culture of primary neonatal rat
cardiomyocytes
Primary culture of neonatal rat cardiomyocytes was
prepared with the use of 53 neonatal Sprague-Dawley rats from the
Fourth Military Medical University (Xi'an, China), as described
previously (21). In brief,
neonatal rat ventricles were digested with collagenase I and
cardiomyocytes were purified by 1 h incubation at 37°C in a 5%
CO2 incubator. Cardiomyocytes were cultured in DMEM
medium (containing 0.1 mmol/l BrdU) with 10% FBS. Cardiomyocytes
amounted for 90–95% of total adherent cells and then were treated
with tunicamycin at multiple concentrations (25, 50, 100, 200 and
500 ng/ml) and time-points (24, 48, 72 and 96 h).. All procedures
involving animals were conducted in accordance with the Guide for
the Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH Publication no. 85–23, revised 1996), and
approved by the Fourth Military Medical University Committee on
Animal Care (Xi'an, China).
Cell viability determined by an MTT
assay
Cell viability was assessed by an MTT assay as
described previously (22).
Briefly, cardiomyocytes were seeded in 96-well plates at a density
of 5×104/well. After tunicamycin administration at the
concentrations and durations, described above, MTT solution (10
µl, 5 mg/ml in PBS) was added to each well and incubated at
37°C for 4 h. Then, the medium was replaced by 150 µl DMSO
per well. The plate was gently shaken for 5 min to completely
dissolve the precipitate. The absorbance was measured at 490 nm
using a microplate reader (Model 680; Bio-Rad, Hercules, CA, USA).
Cell viability was expressed as a percentage of the control.
LDH release assay
To determine cardiomyocyte injury, LDH release in
the medium was detected as described previously (21). The LDH release level was expressed
as the rate of LDH released in the medium to total cellular
LDH.
Apoptosis analysis by flow cytometry
assay
An Annexin V-fluorescein isothiocyanate (FITC)
Apoptosis Detection kit (Sigma-Aldrich) was used to detect
apoptosis as described previously (23). Following treatment, cardiomyocytes
were washed twice with cold PBS and resuspended in binding buffer.
FITC-Annexin V and propidium iodide were added according to the
manufacturer's instructions. The mixture was incubated for 10 min
in the dark at room temperature and then cellular fluorescence was
measured with a FACSscan flow cytometer (Becton Dickinson, Franklin
Lakes, NJ, USA). Annexin V labeled with a fluorophore could
identify cells in the early stage of apoptosis, and PI was
responsible for staining cells in the medium at late stages of
apoptosis. The apoptotic rate was calculated as the percentage of
Annexin V-positive and PI-negative cells divided by the total
number of cells in the gated region.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The cardiomyocytes were exposed to tunicamycin at
the corresponding concentrations and time-points, following which
the cells were removed through scraping with a cell scratch and the
addition of TRIzol to lyse the cells. cDNA synthesis was performed
using a QuantiTect Reverse Transcription kit with 1 µg total
RNA (Takara Bio, Inc., Shanghai, China). PCR was performed on a
Bio-Rad system using Power SYBR Green PCR Master mix (Applied
Biosystems, Foster City, CA, USA). cDNA was diluted at 1:5 for each
reaction and all qPCR performed using SYBR Green. The reaction
conditions were 10 min at 95℃, and then 40 cycles of 95°C for 15
sec and 60°C for 1 min. Rat β-actin was used to normalize sample
amplification. The following primer sequences were used: GRP78,
forward: 5′-CTACCGGGACGAGGTACTGG-3′ and reverse
5′-GGAAAAGGCGGTGAGGACTT-3′; CHOP, forward:
5′-CGGAGTGTACCCAGCACCATCA-3′ and reverse
5′-CCCTCTCCTTTGGTCTACCCTCA-3′; and β-actin. β-actin, forward
5′-AGAGGGAAATCGTGCGTGAC-3′ and reverse
5′-TTCTCCAGGGAGGAAGAGGAT-3′.
Western blot analysis for GRP78 and
CHOP
Cardiomyocytes were lysed using
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Haimen, China) in combination with a protease
inhibitor cocktail (Roche Diagnostics, Mannheim, Germany).
Electrophoresis and immunoblotting were conducted as described
previously (24). In brief,
following treatment with tunicamycin, cardiomyocytes were washed
three times with ice-cold PBS and isolated. Proteins were extracted
from cardiomyocytes and protein concentrations were determined
using a BCA protein assay kit. Protein samples were loaded on 12%
SDS-PAGE gels (Beyotime Institute of Biotechnology), and
transferred onto nitrocellulose membranes (Pierce Biotechnology,
Inc., Rockford, IL, USA). Following blocking with 5% non-fat milk
in TBS containing 0.1% Tween-20 (TBS-T) for 1 h at room
temperature, protein bands were reacted with rabbit anti-rat GRP78
antibody (dilution, 1:1,000; cat. no. BS1154; Bioworld), rabbit
anti-rat CHOP antibody (dilution, 1:500; cat. no. sc-575; Santa
Cruz Biotechnology, Inc.) or mouse anti-rat β-actin antibody
(dilution, 1:1,000; cat. no. sc-47778; Santa Cruz Biotechnology,
Inc) in TBS overnight at 4°C. Following washing three times with
TBST, the membranes were hybridized with goat anti-rabbit or goat
anti-mouse DyLIGHT800 antibodies for 1 h at room
temperature. Following three washes with TBST, protein bands were
visualized on infrared image system (Odyssey; LI-COR Biosciences,
Inc., Lincoln, NE, USA). For the densitometry analysis, optical
density was measured on the inverted digital images using Image J
1.46 software.
Statistical analysis
All experiments were performed at least five times.
Data are expressed as the mean ± standard error of the mean. The
results were compared by one-way analysis of variance followed by
Bonferroni's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
ER stress inducer tunicamycin results in
cardiomyocyte injury
An MTT assay and LDH release assay were used to
evaluate cell injury in tunicamycin-treated cardiomyocytes.
Tunicamycin, a pharmacological agent inhibiting N-linked protein
glycosylation, could be used to experimentally induce ER stress and
subsequent cell death. Concentrations of 25–500 ng/ml tunicamycin
were selected. Compared with the control group, 25 ng/ml
tunicamycin for 24–96 h had no effect on cell injury. However, cell
viability decreased, and LDH release increased after 48–96 h
exposure to tunicamycin at concentrations of 50, 100, 200 and 500
ng/ml (Fig. 1A and B). Treatment
with 100 ng/ml tunicamycin for 72 h resulted in a decrease of cell
viability to 57.4±3.2%. These results provide direct evidence that
tunicamycin led to significant cell injury in cardiomyocytes in a
time- and dose-dependent manner.
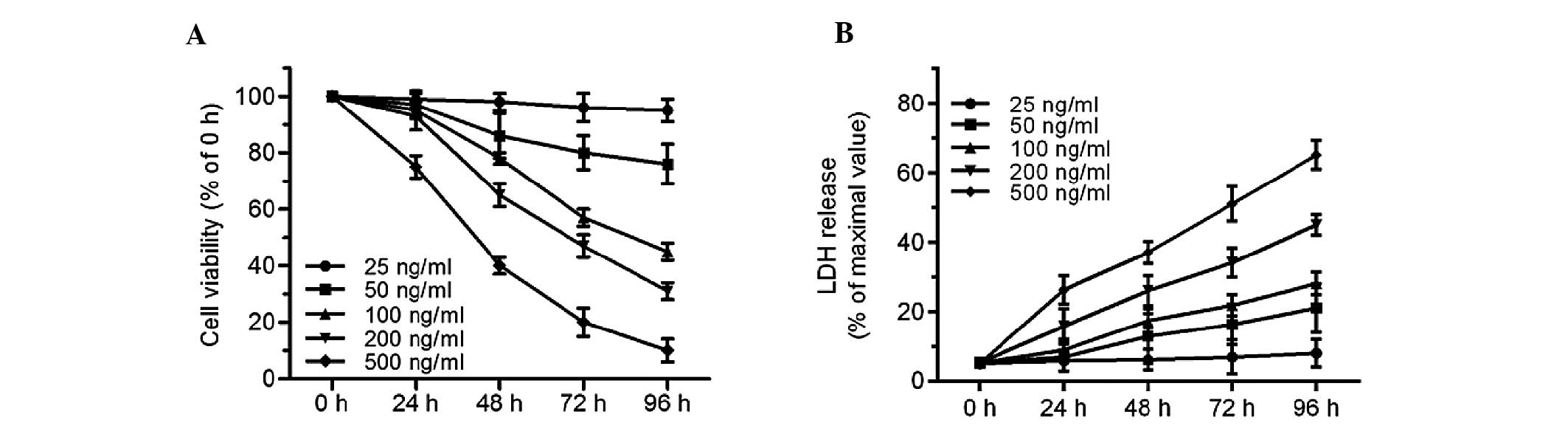 | Figure 1Tunicamycin induces cell injury in
primary cultured neonatal rat cardiomyocytes. Cardiomyocytes were
treated with tunicamycin (25, 50, 100, 200 and 500 ng/ml) for the
indicated times (0, 24, 48, 72, 96 h). (A) Cell viability was
measured by an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide assay and (B) cell injury was analyzed by the LDH release
assay. For cell viability, the viable cell number was expressed as
a percentage of the 0 h group. For the LDH release assay, all
values were compared with the 0 h group. n=5. LDH, lactose
dehydrogenase. |
Tunicamycin-induced cardiomyocyte injury
manifested as apparent apoptosis
To assess the feature of cell injury induced by
tunicamycin, a flow cytometry assay and analysis of caspase-3
activity were used to detect apoptosis. Compared with the control
group, treatment with 100 ng/ml tunicamycin for 24 h did not induce
marked apoptosis, as observed from flow cytometry assays (Fig. 2A) and activity of caspase-3
(Fig. 2B), whereas treatment with
100 ng/ml tunicamycin for 48–96 h significantly increased apoptosis
(Fig. 2A), and activity of
caspase-3 (Fig. 2B), particularly
for 72 h.
Tunicamycin induces upregulation of ER
chaperone GRP78 in cardiomyocytes
To confirm that tunicamycin induces ER stress,
endoplasmic reticulum chaperone GRP78 was assessed by RT-qPCR and
western blot analysis. As shown in Fig. 3A and B, there was a relatively low
level of GRP78 in normal cardiomyocytes. However, treatment with
tunicamycin (100 ng/ml) upregulated GRP78 at the mRNA and protein
levels. The mRNA and protein levels of GRP78 began to increase
following 6 h exposure to tunicamycin, it was then markedly
upregulated and reached the maximum at 24 h. Subsequently, its
expression significantly declined and returned to the basal level
at 72 h. These results indicate that exposure of cardiomyocytes to
tunicamycin induces upregulation of the ER resident molecular
chaperone GRP78 at the mRNA and protein levels, and induces ER
stress.
Tunicamycin induces CHOP expression in
cardiomyocytes
To elucidate that tunicamycin induces the expression
of CHOP in cardiomyocytes, CHOP levels were assessed by RT-qPCR and
western blot analysis. As shown in Fig. 4A and B, there was relatively low
expression of CHOP in normal cardiomyocytes. However, treatment
with tunicamycin (100 ng/ml) increased CHOP at the mRNA and protein
levels. CHOP levels began to increase at 6 h and reached a peak 24
h after exposure to 100 ng/ml tunicamycin. Subsequently, its
expression slowly declined. The results provide evidence that
tunicamycin triggers induction of CHOP in primary cultured neonatal
rat cardiomyocytes.
Discussion
In the present study, an endoplasmic reticulum
stress-induced apoptotic model was established in primary cultured
neonatal rat cardiomyocytes. Firstly, it was demonstrated that
tunicamycin resulted in cardiomyocyte injury in a time- and
dose-dependent manner. Secondly, apoptosis was the predominant mode
of cell death of cardiomyocytes induced by tunicamycin. Thirdly,
tunicamycin upregulated a number of ER stress markers, such as the
survival/rescue protein GRP78 and the apoptotic molecule CHOP.
Accumulating evidence demonstrates that ER
stress-induced apoptosis is the key contributor to cell loss in the
pathogenesis of a series of cardiovascular diseases (8,25,26).
However, the detailed mechanism of ER stress in cardiovascular
diseases remains unclear. Tunicamycin is a specific inhibitor of
N-linked glycosylation of protein, which is observed only in the
endoplasmic reticulum, showing that tunicamycin is a highly
specific ER stress inducer. Therefore, the present study aimed to
establish an ER stress-induced apoptotic model using tunicamycin in
primary cultured neonatal rat cardiomyocytes. Although tunicamycin
has been used to induce ER stress in other cell types, cells are
often regulated in a type- and stimulus-specific manner (27,28).
Therefore, in order to screen the optimal treatment time and
concentration of ER stress-induced cell injury by tunicamycin in
cardiomyocytes, cell viability was assessed by an MTT assay, and
cardiomyocyte injury was detected using an LDH release assay.
However, the pattern of cardiomyocyte injury-induced
by tunicamycin remains unclear. Flow cytometry and analysis of the
activity of caspase-3 were conducted to quantify the number of
apoptotic cells. The present data revealed that tunicamycin induced
cardiomycyte apoptosis. In addition, treatment of primary cultured
neonatal rat cardiomyocytes with 100 ng/ml tunicamycin for 72 h led
to the maximum rate of apoptosis.
Next, the effect of 100 ng/ml tunicamycin on ER
stress-related molecules in cardiomyocytes was investigated.
RT-qPCR and western blot analysis revealed that tunicamycin
increased GRP78 and CHOP levels, and induced ER stress in primary
cultured neonatal rat cardiomyocytes, which is consistent with the
results of a previous study (29).
GRP78 survival/rescue protein of cardiomyocytes treated with 100
ng/ml tunicamycin was rapidly upregulated, peaked at 24 h, and then
rapidly declined to near preinduction level at the mRNA and protein
levels. In addition, the level of apoptotic ER stress-related
molecule CHOP was low at the early phase, peaked 24 h following
treatment with 100 ng/ml tunicamycin and then decreased slowly.
These data indicated that GRP78 was rapidly upregulated in the
early stages of tunicamycin-induced ER stress in cardiomyocytes,
whereas CHOP gradually increased in the first 24 h. The levels of
GRP78 then rapidly decreased, whereas CHOP was slowly downregulated
by persistent tunicamycin-induced ER stress in cardiomyocytes.
However, how ER stress integrates its cytoprotective and
proapoptotic outputs to select between life or death cell fates
remains unknown.
In conclusion, the present study successfully
constructed an ERS-induced cardiomyocyte apoptotic model with
tunicamycin, and screened the optimal concentration and processing
time for detecting cell viability, apoptotic index and ERS
associated proteins GRP78 and CHOP. The results of the present
study provide important experimental data for preclinical and
clinical investigations of ERS-associated cardiovascular
diseases.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81070127 and 81270169). The
authors would like to thank Dr Xinliang Ma (Department of Emergency
Medicine, Thomas Jefferson University, Philadelphia, USA) and Dr
Jun Ren (Center for Cardiovascular Research and Alternative
Medicine, University of Wyoming College of Health Sciences,
Laramie, WY, USA) for commenting on the manuscript.
References
|
1
|
Millott R, Dudek E and Michalak M: The
endoplasmic reticulum in cardiovascular health and disease. Can J
Physiol Pharmacol. 90:1209–1217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peralta C and Brenner C: Endoplasmic
reticulum stress inhibition enhances liver tolerance to
ischemia/reperfusion. Curr Med Chem. 18:2016–2024. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ding W and Zhang X, Huang H, Ding N, Zhang
S, Hutchinson SZ and Zhang X: Adiponectin protects rat myocardium
against chronic intermittent hypoxia-induced injury via inhibition
of endoplasmic reticulum stress. PLoS One. 9:e945452014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gomes-Alves P, Couto F, Pesquita C, Coelho
AV and Penque D: Rescue of F508del-CFTR by RXR motif inactivation
triggers proteome modulation associated with the unfolded protein
response. Biochim Biophys Acta. 1804:856–865. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farrukh MR, Nissar UA, Afnan Q, et al:
Oxidative stress mediated Ca (2+) release manifests endoplasmic
reticulum stress leading to unfolded protein response in UV-B
irradiated human skin cells. J Dermatol Sci. 75:24–35. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang K and Kaufman RJ: From
endoplasmic-reticulum stress to the inflammatory response. Nature.
454:455–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fu HY, Okada K, Liao Y, et al: Ablation of
C/EBP homologous protein attenuates endoplasmic reticulum-mediated
apoptosis and cardiac dysfunction induced by pressure overload.
Circulation. 122:361–369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang L, Zhao D, Ren J and Yang J:
Endoplasmic reticulum stress and protein quality control in
diabetic cardiomyopathy. Biochim Biophys Acta. 1852:209–218. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Groenendyk J, Sreenivasaiah PK, Kim do H,
Agellon LB and Michalak M: Biology of endoplasmic reticulum stress
in the heart. Circ Res. 107:1185–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Avery J, Etzion S, DeBosch BJ, et al: TRB3
function in cardiac endoplasmic reticulum stress. Circ Res.
106:1516–1523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu D, Zhang M and Yin H: Signaling
pathways involved in endoplasmic reticulum stress-induced neuronal
apoptosis. Int J Neurosci. 123:155–162. 2013. View Article : Google Scholar
|
|
12
|
Thuerauf DJ, Marcinko M, Gude N, Rubio M,
Sussman MA and Glembotski CC: Activation of the unfolded protein
response in infarcted mouse heart and hypoxic cultured cardiac
myocytes. Circ Res. 99:275–282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Endo H, Murata K, Mukai M, Ishikawa O and
Inoue M: Activation of insulin-like growth factor signaling induces
apoptotic cell death under prolonged hypoxia by enhancing
endoplasmic reticulum stress response. Cancer Res. 67:8095–8103.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Erbay E, Babaev VR, Mayers JR, et al:
Reducing endoplasmic reticulum stress through a macrophage lipid
chaperone alleviates atherosclerosis. Nat Med. 15:1383–1391. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hotamisligil GS: Endoplasmic reticulum
stress and atherosclerosis. Nat Med. 16:396–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Myoishi M, Hao H, Minamino T, et al:
Increased endoplasmic reticulum stress in atherosclerotic plaques
associated with acute coronary syndrome. Circulation.
116:1226–1233. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Glembotski CC: Endoplasmic reticulum
stress in the heart. Circ Res. 101:975–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dickhout JG, Carlisle RE and Austin RC:
Interrelationship between cardiac hypertrophy, heart failure and
chronic kidney disease: endoplasmic reticulum stress as a mediator
of pathogenesis. Circ Res. 108:629–642. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Empel VP, Bertrand AT, Hofstra L,
Crijns HJ, Doevendans PA and De Windt LJ: Myocyte apoptosis in
heart failure. Cardiovasc Res. 67:21–29. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen M, Wang L, Yang G, et al: Baicalin
protects the cardiomyocytes from ER stress-induced apoptosis:
inhibition of CHOP through induction of endothelial nitric oxide
synthase. PLoS One. 9:e883892014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu B, Niu L, Shen MZ, et al: Decreased
astroglial monocarboxylate transporter 4 expression in temporal
lobe epilepsy. Mol Neurobiol. 50:327–338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fu HY, Minamino T, Tsukamoto O, et al:
Overexpression of endoplasmic reticulum-resident chaperone
attenuates cardiomyocyte death induced by proteasome inhibition.
Cardiovasc Res. 79:600–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu B, Wang L, Shen LL, et al:
RNAi-mediated inhibition of presenilin 2 inhibits glioma cell
growth and invasion and is involved in the regulation of Nrg1/ErbB
signaling. Neuro Oncol. 14:994–1006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou S, Yin X, Zheng Y, et al:
Metallothionein prevents intermittent hypoxia-induced cardiac
endoplasmic reticulum stress and cell death likely via activation
of Akt signaling pathway in mice. Toxicol Lett. 227:113–123. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang Y, Li C, Xiang X, et al: Ursolic acid
prevents endoplasmic reticulum stress-mediated apoptosis induced by
heat stress in mouse cardiac myocytes. J Mol Cell Cardiol.
67:103–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qi X, Vallentin A, Churchill E and
Mochly-Rosen D: DeltaPKC participates in the endoplasmic reticulum
stress-induced response in cultured cardiac myocytes and ischemic
heart. J Mol Cell Cardiol. 43:420–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Okada K, Minamino T, Tsukamoto Y, et al:
Prolonged endoplasmic reticulum stress in hypertrophic and failing
heart after aortic constriction: possible contribution of
endoplasmic reticulum stress to cardiac myocyte apoptosis.
Circulation. 110:705–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin JH, Li H, Yasumura D, et al: IRE1
signaling affects cell fate during the unfolded protein response.
Science. 318:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|

















