Introduction
In recent years, the incidence of diabetes mellitus
and osteoporosis has increased, as a result of longer life
expectancy and a lifestyle characterized by low levels of physical
activity and increased high-energy food intake (1). Osteoporosis, which is the most common
type of bone disease, is associated with low bone mineral density
(BMD) and systemic impairment of bone mass, strength and
microarchitecture, which increases the propensity of fragility
fractures (2). Osteoporosis is
clinically defined as having a BMD ≥2.5 standard deviations below
the young female adult mean (T-score, −2.5) (3). The etiology of osteoporosis is
attributed to various endocrine, metabolic, and mechanical factors,
and can occur at any age; however, it is predominantly diagnosed in
elderly and diabetic patients (4).
Diabetes mellitus is a risk factor for osteoporotic fractures.
Diabetes mellitus affects bone metabolism, and may lead to
osteopenia and osteoporosis (5,6). The
causes of diabetes-induced osteoporosis are varied, and include
insulin deficiency, disordered calcium-phosphate metabolism and
excessive secretion of parathyroid hormone (7–9).
However, the underlying molecular link between diabetes and
osteoporosis remains to be elucidated.
In patients with diabetes mellitus, during
long-lasting hyperglycemia, glucose forms covalent adducts with
plasma proteins through a non-enzymatic process, the products of
which are termed advanced glycation end products (AGEs) (10). AGEs may be critical mediators in
the pathogenesis and development of osteoporosis and other
age-associated chronic degenerative diseases (11). The present study aimed to determine
whether AGEs serve as a causal link between diabetes and
osteoporosis.
Zinc, which is an essential nutritional factor in
the growth of humans and animals, has been considered a novel
supplementation factor in the prevention and treatment of
osteoporosis (12). A previous
study demonstrated that zinc was able to inhibit high
glucose-induced apoptosis by suppressing oxidative stress in renal
tubular epithelial cells (13).
Furthermore, zinc supplementation has been observed to exert
positive effects in diabetes, including a modest, but significant,
reduction in fasting glucose, and a trend towards decreased
glycated hemo-globin (14). The
present study aimed to use the MC3T3-E1 mouse osteoblastic cell
line to investigate the role of zinc in AGE-induced cell apoptosis,
as well as investigate novel ways to treat diabetes-induced
osteoporosis.
Materials and methods
Reagents
α-minimal essential medium (α-MEM),
penicillin/streptomycin (5,000 U/ml penicillin; 5,000 U/ml
streptomycin) and fetal bovine serum (FBS) were obtained from Gibco
Life Technologies (Grand Island, NY, USA). ZnSO4, MTT,
PD98059, LY294002, Triton X-100, bovine serum albumin (BSA) and
dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Mouse monoclonal IgG anti-caspase 3 (sc-65496),
anti-caspase 9 (sc-56073), anti-B-cell lymphoma 2-associated X
protein (Bax) (sc-20067), anti-cytochrome c (cyto-C)
(sc-13561), anti-β-actin (sc-47778), anti-extracellular
signal-regulated kinases (ERK) (sc-514302), anti-phosphorylated
(p)-ERK (sc-377400) and anti-AKT (sc-377457) antibodies, and the
rabbit polyclonal IgG anti-p-AKT antibody (sc-135650), were
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Enhanced Chemiluminescence (ECL) kit was purchased from Pierce
Biotechnology, Inc. (Rockford, IL, USA). All reagents used were
trace element analysis grade. All water used was glass
distilled.
Cell culture
MC3T3-E1 mouse preosteoblasts (cat. no. CRL-2593;
American Type Culture Collection, Manassas, VA, USA) were cultured
in α-MEM, supplemented with 10% heat-inactivated FBS and
antibiotics (100 units penicillin/streptomycin) at 37°C in a
humidified incubator containing 5% CO2. For experiments,
the cells were cultured for 24 h in 3 ml α-MEM, supplemented with
10% FBS, in order to obtain monolayers. The cells were subsequently
rinsed with phosphate-buffered saline (PBS), the medium was then
replaced and the cells were cultured further. In order to
investigate the effects of zinc on the signaling pathways of
PI3K/AKT and MAPK/ERK, a specific PI3K/AKT inhibitor, LY294002 (10
µM LY294002, 1×105 cells) and a specific MAPK/ERK
inhibitor, PD98059 (20 µM PD98059, 1×105 cells)
were used.
Groups
The groups were as follows: Control group, MC3T3-E1
cells (1×105) cultured with α-MEM, supplemented with 10%
heat-inactivated FBS for 48 h; BSA group, MC3T3-E1 cells
(1×105) treated with BSA for 48 h; AGEs group, MC3T3-E1
cells (1×105) treated with 500 µg/ml AGEs
(Anyan-bio Technology, Shanghai, China) for 48 h; AGEs + zinc
group, MC3T3-E1 cells (1×105) treated with 500
µg/ml AGEs in the presence of 50 µM ZnSO4
for 48 h.
Cell viability
Cell viability was measured using a quantitative
colorimetric MTT assay, with 1×105 cells/ml in 96-well
plates. Briefly, 10 µl MTT (final concentration, 5 mg/ml)
was added to the medium and the cells were incubated at 37°C for 4
h. The medium was then aspirated from each well and 100 µl
DMSO was added to dissolve the formazan crystals. The optical
density of each well was read at 492 nm using a microplate reader
(Multiskan MK3; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Detection of intracellular reactive
oxygen species (ROS) levels
The concentrations of ROS were determined based on
the oxidation of 2,7-dichlorodihydrofluorescein (DCFH; Nanjing
Jiancheng Bioengineering Research Institute, Nanjing, China).
Briefly, MC3T3-E1 cells (4×106) were incubated with 10
µmol/l DCFH-DA probe at 37°C for 35 min, following which the
cells were washed three times with PBS to remove residual probe.
DCFH-DA is deacetylated intracel-lularly by nonspecific esterase
and is further oxidized by ROS to the fluorescent compound
2,7-dichlorofluorescein (DCF). DCF fluorescence was detected using
flow cytometry with a FACSCalibur (BD Biosciences, Franklin Lakes,
NJ, USA), and analyzed using Quantity One software, version 4.62
(Bio-Rad Laboratories, Inc.).
Analysis of apoptosis
To quantify the number of apoptotic cells, flow
cytometry (ACCURI™ C6; BD Biosciences) was performed using a
Fluorescein Isothiocyanate (FITC) Annexin V Apoptosis Detection kit
(Nanjing KeyGen Biotech Co., Ltd., Nanjing, China), according to
the manufacturer's instructions. The cells were resuspended in 300
µl 1X binding buffer and transferred into a sterile flow
cytometry glass tube. Annexin V/FITC (10 µl) was added to
the cells and incubated in the dark for 30 min at room temperature.
The cells (1×105) were then treated with 5 µl
propidium iodide (Sigma-Aldrich), and incubated at room temperature
for 10 min in the dark. Following incubation, the cells were
analyzed using flow cytometry. The percentage of cellular apoptosis
was determined using the FITC Annexin V Apoptosis Detection
kit.
Western blotting
The MC3T3-E1 cells were washed with PBS, scraped,
collected and lysed using ice-cold lysis buffer (Beyotime Institute
of Biotechnology, Haimen, China), and centrifuged at 750 × g for 8
min at 0°C to yield the whole cell extract. The resuspended cells
were then homogenized with ten strokes of a Teflon homogenizer
(PT1200E; Kinematica, Luzern, Switzerland), and the homogenates
were centrifuged twice at 750 × g for 10 min at 4°C. The
supernatants were further centrifuged at 10,000 × g for 15 min at
4°C, to obtain the mitochondrial pellets. Cytosolic fractions were
obtained following further centrifugation at 100,000 × g for 1 h at
4°C. The samples were then denatured in boiling water for 5 min,
the protein concentration of cell lysates was quantified using the
Bicinchoninic Acid kit (Beyotime Institute of Biotechnology) and
equal amounts of protein (10 µg) were separated by 10%
SDS-PAGE (Sigma-Aldrich) and transferred to nitrocellulose
membranes (Sigma-Aldrich). The protein concentration of cell
lysates was quantified using the Bicinchoninic Acid kit (Beyotime
Institute of Biotechnology) and equal amounts of protein(10
µg) were separated by SDS-PAGE. The membranes were incubated
overnight at 4°C with anti-Bax, anti-cyto-C, anti-caspase 3,
anti-caspase 9, anti-ERK, anti-p-ERK, anti-AKT, anti-p-AKT, and
anti-β-actin antibodies. The antibodies were used at a dilution of
1:400 in Tris-buffered saline Tween (Sigma-Aldrich), containing 50
mM Tris-HCl, 150 mM NaCl, and 0.05% (w/v) Tween 20, (pH 7.4)
containing 5% (w/v) BSA. The membranes were then incubated for 2 h
with horseradish peroxidase-conjugated anti-rabbit or anti-mouse
secondary IgG antibodies (1:1,000; Santa Cruz Biotechnology, Inc.)
at room temperature. The immunoreactive bands were visualized using
an ECL kit.
Statistical analysis
Statistical analyses were performed using SPSS
version 18 (SPSS Inc., Chicago, IL, USA). Data are expressed as the
mean ± standard deviation of at least three independent
experiments. The variance was homogenous permitting the use of
standard analysis of variance (ANOVA). Following determination of
statistical significance using ANOVA, individual comparisons were
made using Tukey's multiple comparison test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Zinc protects MC3T3-E1 cells from
AGE-induced apoptosis
AGEs induce cell apoptosis; however, whether AGEs
induce the apoptosis of mouse osteoblasts remains to be elucidated.
To determine whether AGEs induce osteoblast apoptosis, the present
study treated MC3T3-E1 cells with various doses of AGEs for 24, 48
and 72 h. Cell viability assays demonstrated that the AGEs
inhibited cell growth in a dose- and time-dependent manner
(Fig. 1A). The half maximal
inhibitory concentration values of AGEs in the MC3T3-E1 cells at
24, 48 and 72 h were 936, 725 and 590 µg/ml, respectively.
For subsequent experiments in the present study, 500 µg/ml
AGEs were used (the concentration that apoptosis is clear). In
order to determine the role of AGEs in promoting apoptosis and the
anti-apoptotic effects of zinc, the MC3T3-E1 cells were treated
with either BSA, or with 500 µg/ml AGEs in the presence or
absence of 30 or 50 µM ZnSO4 for 48 h, and cell
viability was detected using an MTT assay. When treated with BSA
alone, the MC3T3-E1 cells proliferated at the same rate as the
control cells, whereas treatment with 500 µg/ml AGEs
significantly suppressed the viability of the MC3T3-E1 cells.
Notably, cells cultured with AGEs and zinc combined (30 or
50µM) exhibited improved viability, compared with the AGE
group (Fig. 1B). The percentage of
apoptotic cells was markedly increased in the cells treated with
AGEs, whereas zinc pretreatment resulted in a significant decrease
in the percentage of apoptosis (Fig.
1C), as determined by flow cytometry. Similarly, cell survival
was higher when the cells were exposed to AGEs and zinc combined,
compared with the cells treated with AGEs alone, as observed in the
inverted phase contrast microscopy images (SZ51; Olympus, Tokyo,
Japan) (Fig. 1D). To further
investigate the protective effects of zinc, western blotting was
performed to detect the protein expression levels of caspase-3 and
-9, cyto-C, and Bax. Treatment with zinc significantly
downregulated the expression levels of mitochondrial
cleaved-caspase-3, cleaved-caspase-9 and Bax, and of cytosolic
cyto-C, compared with the AGE group. These data indicated that AGEs
induced MC3T3-E1 cell apoptosis, and that zinc may protect MC3T3-E1
cells from AGE-induced apoptosis.
 | Figure 1Effects of zinc on AGE-induced
apoptosis in MC3T3-E1 mouse preosteoblast cells. (A) MC3T3-E1 cells
were treated with AGEs (0–2,000 µg/ml) for 24, 48 and 72 h.
Following treatment, cell viability was detected using an MTT
assay. Results are presented as a percentage of the inhibition of
cell viability, which was calculated from the mean absorbance ±
standard deviation of triplicate samples. (B) MC3T3-E1 cells were
treated with BSA or 500 µg/ml AGEs, in the presence or
absence of 30 or 50 µM ZnSO4 for 48 h, and cell
viability was detected using an MTT assay. Values are presented as
the mean ± standard deviation of three independent experiments.
**P<0.01, vs. control; #P<0.01, vs. AGE
group. (C) MC3T3-E1 cells were treated with BSA or 500 µg/ml
AGEs, in the presence or absence of 50 µM ZnSO4,
for 48 h, and apoptosis was detected using flow cytometry following
Annexin V-PI double staining. (D) Cells were treated, as described
above, and cell morphology was observed under a microscope.
Magnification, ×400. (E) Cells were treated, as described above,
and the protein expression levels of cytosolic cyto-C,
mitochondrial Bax, and caspase-3 and caspase-9 were detected using
western blotting. (F) Values are presented as the mean ± standard
deviation of three independent experiments. β-actin was used as a
loading control. **P<0.01, vs. control;
#P<0.01 AGE + zinc group, vs. AGE group. AGEs,
advanced glycation end products; BSA, bovine serum albumin; PI,
propidium iodide; cyto-C, cytochrome c; Bax, B-cell lymphoma
2-associated X protein. |
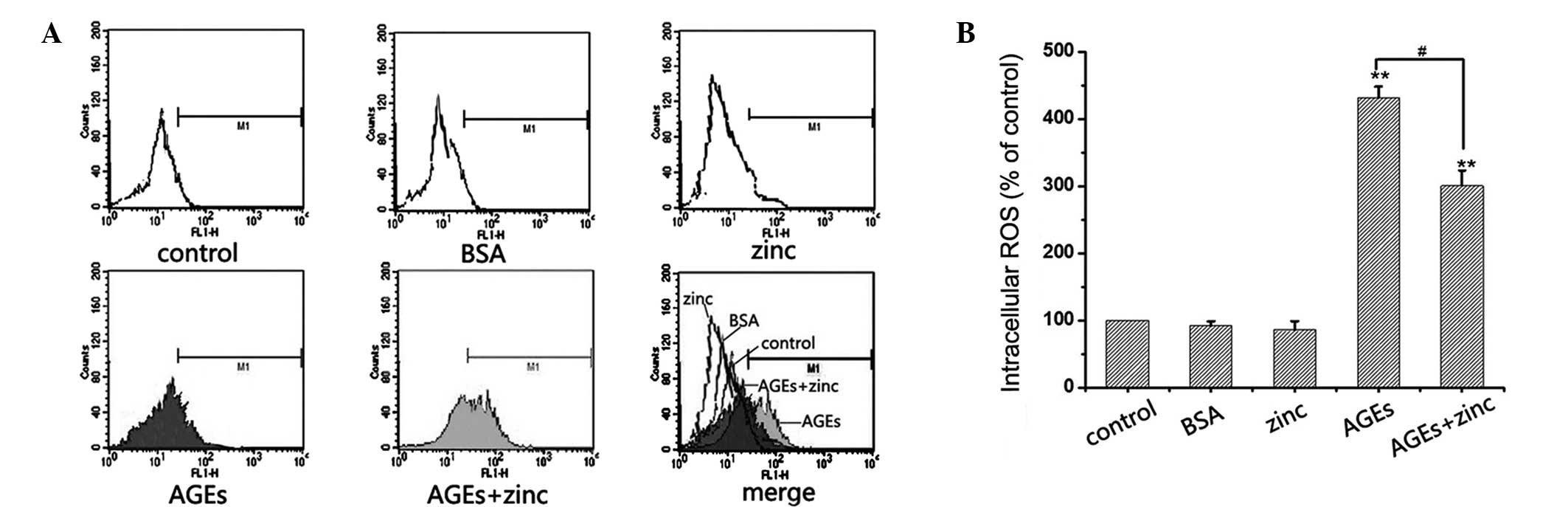
Zinc inhibits MC3T3-E1 cells from
AGE-induced ROS generation
A previous study demonstrated that high levels of
glucose and AGEs increase intracellular ROS levels in renal cells,
and contribute to the development and progression of diabetic renal
injury (15). Furthermore,
oxidative stress and apoptosis are closely associated. The present
study investigated the effects of zinc on AGE-induced ROS
generation. The MC3T3-E1 cells were treated with BSA, 50 µM
ZnSO4, or 500 µg/ml AGEs, in the presence or
absence of 50 µM ZnSO4, for 48 h, and
intracellular ROS levels were measured using flow cytometry. As
shown in Fig. 2, cellular ROS
levels were four-fold higher in the AGEs-treated cells, compared
with the control, whereas zinc significantly reduced of AGE-induced
DCF-sensitive cellular ROS (Fig.
2). These data indicated that zinc may inhibit AGE-induced
apoptosis in MC3T3-E1 cells by decreasing oxidative stress.
Zinc inhibits apoptosis through
mitogen-activated protein kinase (MAPK)/ERK signaling in MC3T3-E1
cells
Since MAPK/ERK is implicated in the triggering of
anti-apoptotic signal transduction pathways, the present study
investigated whether the integrity of the pathway was required for
the anti-apoptotic effects of zinc. To provide further insight into
this signaling pathway leading to the protection of apoptosis in
AGE-induced MC3T3-E1 cells, a specific MEK1 inhibitor, PD98059, was
used at a concentration stated in a previous study (16). Treatment with zinc effectively
protected the cells from AGE-induced apoptosis; however, when the
cells were pretreated with zinc and PD98059 together, the
protective effects of zinc were attenuated. Furthermore, zinc
significantly increased the expression levels of p-ERK in the
AGE-treated cells; however, co-treatment with 20 µM PD98059
effectively inhibited the upregulation of ERK phosphorylation.
However, the expression levels of total-ERK were not altered
significantly. Inhibition of the MEK pathway with PD98059
attenuated the protective effects of zinc, suggesting a role for
the MAPK/ERK pathway in this effect (Fig. 3). These results suggested that the
activation of ERK is a critical event in the anti-apoptotic effects
of zinc in MC3T3-E1 cells.
 | Figure 3Effects of zinc on the ERK pathway in
AGE-induced apoptosis. (A) MC3T3-E1 cells mouse preosteoblasts were
incubated with or without 20 µM PD98059 for 1 h, and were
then exposed to 500 µg/ml AGEs, in the presence or absence
of 50 µM ZnSO4, for 48 h. Apoptosis was
determined using flow cytometry following Annexin V-PI double
staining. (B) MC3T3-E1 cells were treated, as described above, and
the expression levels of total ERK and p-ERK were detected using
western blotting. Quantification of the expression levels of (C)
total ERK and (D) p-ERK. Values are presented as the mean ±
standard deviation of three independent experiments. β-actin was
used as loading control. **P<0.01, vs. control;
#P<0.01 AGE + zinc group, vs. AGE group. ERK,
extracellular signal-regulated kinases; p-ERK; phosphorylated-ERK;
AGEs, advanced glycation end products; PI, propidium iodide. |
Zinc inhibits apoptosis through
phosphoinositide 3-kinase (PI3K)/AKT signaling in MC3T3-E1
cells
The PI3K/Akt pathway is a zinc-sensing process,
which links extracellular survival signals with
apoptosis-associated pathways and has a critical role in cell
survival by inhibiting apoptotic pathways (17). In order to investigate the effects
of this signaling pathway on the protection of apoptosis in
AGE-induced MC3T3-E1 cells, a specific PI3K/AKT inhibitor,
LY294002, was used, at a concentration stated in a previous study
(18). Treatment with zinc
effectively protected the cells from AGE-induced apoptosis;
however, when the cells were pretreated with LY294002, the
protective effects of zinc were attenuated. Furthermore, zinc
significantly increased the expression levels of p-AKT in the
AGE-treated cells; whereas co-treatment with 10 µM LY294002
effectively inhibited the upregulation of AKT phosphorylation. The
expression levels of total-AKT were not altered significantly.
Furthermore, inhibition of the PI3K/AKT pathway by LY294002
attenuated the protective effects of zinc, suggesting a role for
the PI3K/AKT pathway in this effect (Fig. 4). These results suggested that zinc
protects cells from AGE-induced apoptosis through the PI3K/AKT
pathway.
Discussion
Zinc is an essential trace element, which increases
osteoblast proliferation and bone formation, and has been
considered an important factor in bone metabolism (19). Zinc deficiency can alter the
balance of bone remodeling, and low zinc intake has been reported
to be associated with low bone mass in females (20). Therefore, it has been suggested
that zinc supplementation may be a sustainable approach for the
improvement of bone health. Fung et al (21) demonstrated that zinc
supplementation results in a marked increase in total-body bone
mass, compared with a placebo. Furthermore, zinc as an antioxidant
may inhibit apoptosis. Kumar et al (22) demonstrated that zinc
supplementation significantly decreases apoptosis and reduces the
levels of ROS in the embryos of diabetic mice. The results of the
present study revealed that pretreatment of cells with zinc
markedly protected the cells from AGE-induced damage. Treatment
with zinc decreased the production of intracellular ROS and
significantly inhibited AGE-induced apoptosis of the MC3T3-E1
cells.
A previous study demonstrated that there are
decreased bone levels of Zn2+ in postmenopausal females
with osteoporosis, due to the reduction in osteoblast
differentiation as bone mineralizing capacity decreases (20). Furthermore, a positive correlation
has been detected between zinc levels and osteoprotegerin (OPG)
levels in the serum (20,23). Zinc increases osteogenic function
by stimulating osteoblast proliferation and OPG activity (23). In addition, zinc is required for
normal immune function and enhances the in vitro
effectiveness of insulin. Impaired immune function has previously
been reported in patients with diabetes, and decreased serum zinc
levels and hyperzincuria occur in a number of diabetic patients and
animals (24). Furthermore,
diabetes results in increased oxidative stress, which is important
in its pathogenesis. A previous study demonstrated that zinc
supplementation in healthy volunteers resulted in a reduction in
plasma levels of lipid peroxidation products, compared with a
control group; therefore, zinc supplementation may have beneficial
effects on glycemic control (25).
In addition, zinc has been demonstrated to have a stimulatory
effect on osteoblastic bone formation, and zinc compounds may be
considered a novel supplementation factor for the prevention and
treatment of diabetes-induced osteoporosis. A previous study
demonstrated that cell viability and zinc levels were reduced
following exposure to AGEs, and were improved with the
supplementation of zinc in the endothelial cells (26). The results of the present study
demonstrated that zinc inhibited the AGE-induced production of ROS
and apoptosis in MC3T3-E1 cells.
Previous studies have demonstrated that the ERK
and/or AKT pathways may contribute to the cell protective effects
of several growth factors via apoptotic inhibition (27,28).
The present study investigated the possible signaling pathways that
may mediate the protective effects of zinc in AGE-treated MC3T3-E1
cells. Western blot analysis demonstrated that zinc induced the
phosphorylation of AKT and ERK in the MC3T3-E1 cells. In addition,
treatment with PD98059 prevented the zinc-induced phosphorylation
of ERK, and inhibited zinc-mediated anti-apoptotic effects
(Fig. 3). Concordantly, treatment
with the PI3K/Akt inhibitor, LY294002, inhibited zinc-induced AKT
phosphorylation, suggesting that the PI3K/AKT pathway is involved
with zinc-induced cell protection (Fig. 4). Therefore, it was concluded that
zinc-mediated cell protection is associated with the ERK and AKT
signaling pathways.
In conclusion, the results of the present study
demonstrated that zinc exerted a cell protective effect against
AGE-induced apoptosis in the MC3T3-E1 cells. The MAPK/ERK and
PI3K/AKT signaling pathways were found to be involved in the potent
anti-apoptotic effects of zinc in AGE-treated cells. Although
further investigations are required to determine the therapeutic
effects of zinc in the protection of MC3T3-E1 viability and
diabetic osteoporosis in vivo, these results indicate the
potential of using zinc as a novel therapeutic strategy to treat
the complications of diabetes, particularly osteoporosis.
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
PI
|
propidium iodide
|
|
MTT
|
3-[4,5-dimethylthiazol-2-y]-2,5-diphenyltetrazolium bromide
|
|
BSA
|
bovine serum albumin
|
|
DMSO
|
dimethyl sulfoxide
|
|
AGEs
|
advanced glycation end products
|
|
DCF-DA
|
2,7-dichlorofluorescein-diacetate
|
|
TBS
|
tris-buffered saline
|
|
α-MEM
|
α-minimal essential medium
|
References
|
1
|
Hofbauer LC, Brueck CC, Singh SK and
Dobnig H: Osteoporosis in patients with diabetes mellitus. J Bone
Miner Res. 22:1317–1328. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Das S and Crockett JC: Osteoporosis - a
current view of pharmacological prevention and treatment. Drug Des
Devel Ther. 7:435–448. 2013.PubMed/NCBI
|
|
3
|
Schousboe JT, Tanner SB and Leslie WD:
Definition of osteoporosis by bone density criteria in men: Effect
of using female instead of male young reference data depends on
skeletal site and densitometer manufacturer. J Clin Densitom.
17:301–306. 2014. View Article : Google Scholar
|
|
4
|
Rachner TD, Khosla S and Hofbauer LC:
Osteoporosis: Now and the future. Lancet. 377:1276–1287. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Montagnani A, Gonnelli S, Alessandri M and
Nuti R: Osteoporosis and risk of fracture in patients with
diabetes: An update. Aging Clin Exp Res. 23:84–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lechleitner M, Pils K, Roller-Wirnsberger
R, Beubler E, Gasser R, Mrak P, Hoppichler F and Pietschmann P:
Diabetes and osteoporosis: Pathophysiological interactions and
clinical importance for geriatric patients. Z Gerontol Geriatr.
46:390–397. 2013.In German. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wongdee K and Charoenphandhu N:
Osteoporosis in diabetes mellitus: Possible cellular and molecular
mechanisms. World J Diabetes. 2:41–48. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Watanabe R and Okazaki R: Diabetes
mellitus and osteoporosis. Diabetes mellitus and bone and calcium
metabolism. Clin Calcium. 22:1307–1314. 2012.In Japanese.
PubMed/NCBI
|
|
9
|
Motyl KJ, McCauley LK and McCabe LR:
Amelioration of type I diabetes-induced osteoporosis by parathyroid
hormone is associated with improved osteoblast survival. J Cell
Physiol. 227:1326–1334. 2012. View Article : Google Scholar
|
|
10
|
Vlassara H and Uribarri J: Advanced
glycation end products (AGE) and diabetes: Cause, effect, or both?
Curr Diab Rep. 14:4532014. View Article : Google Scholar :
|
|
11
|
Yamagishi S: Role of advanced glycation
end products (AGEs) in osteoporosis in diabetes. Curr Drug Targets.
12:2096–2102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yamaguchi M: Role of nutritional zinc in
the prevention of osteoporosis. Mol Cell Biochem. 338:241–254.
2010. View Article : Google Scholar
|
|
13
|
Zhang X, Zhao Y, Chu Q, Wang ZY, Li H and
Chi ZH: Zinc modulates high glucose-induced apoptosis by
suppressing oxidative stress in renal tubular epithelial cells.
Biol Trace Elem Res. 158:259–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ruz M, Carrasco F, Rojas P, Codoceo J,
Inostroza J, Basfi-fer K, Valencia A, Vásquez K, Galgani J, Pérez
A, et al: Zinc as a potential coadjuvant in therapy for type 2
diabetes. Food Nutr Bull. 34:215–221. 2013.PubMed/NCBI
|
|
15
|
Ha H and Lee HB: Reactive oxygen species
and matrix remodeling in diabetic kidney. J Am Soc Nephrol. 14(8
Suppl 3): S246–S249. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan YX, Gong YW, Guo Y, Lv Q, Guo C,
Zhuang Y, Zhang Y, Li R and Zhang XZ: Mechanical strain regulates
osteoblast proliferation through integrin-mediated ERK activation.
PLoS One. 7:e357092012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bruinsma JJ, Jirakulaporn T, Muslin AJ and
Kornfeld K: Zinc ions and cation diffusion facilitator proteins
regulate Ras-mediated signaling. Dev Cell. 2:567–578. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park SJ, Kim SH, Choi HS, Rhee Y and Lim
SK: Fibroblast growth factor 2-induced cytoplasmic asparaginyl-tRNA
synthetase promotes survival of osteoblasts by regulating
anti-apoptotic PI3K/Akt signaling. Bone. 45:994–1003. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brzóska MM, Rogalska J, Galazyn-Sidorczuk
M, Jurczuk M, Roszczenko A, Kulikowska-Karpińska E and
Moniuszko-Jakoniuk J: Effect of zinc supplementation on bone
metabolism in male rats chronically exposed to cadmium. Toxicology.
237:89–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gurban CV and Mederle O: The OPG/RANKL
system and zinc ions are promoters of bone remodeling by osteoblast
proliferation in postmenopausal osteoporosis. Rom J Morphol
Embryol. 52(3 Suppl): 1113–1119. 2011.PubMed/NCBI
|
|
21
|
Fung EB, Kwiatkowski JL, Huang JN,
Gildengorin G, King JC and Vichinsky EP: Zinc supplementation
improves bone density in patients with thalassemia: A double-blind,
randomized, placebo-controlled trial. Am J Clin Nutr. 98:960–971.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumar SD, Vijaya M, Samy RP, Dheen ST, Ren
M, Watt F, Kang YJ, Bay BH and Tay SS: Zinc supplementation
prevents cardiomyocyte apoptosis and congenital heart defects in
embryos of diabetic mice. Free Radic Biol Med. 53:1595–1606. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang D, Yang M, Guo B, Cao J, Yang L and
Guo X: Zinc upregulates the expression of osteoprotegerin in mouse
osteo-blasts MC3T3-E1 through PKC/MAPK pathways. Biol Trace Elem
Res. 146:340–348. 2012. View Article : Google Scholar
|
|
24
|
Niewoehner CB, Allen JI, Boosalis M,
Levine AS and Morley JE: Role of zinc supplementation in type II
diabetes mellitus. Am J Med. 81:63–68. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jayawardena R, Ranasinghe P, Galappatthy
P, Malkanthi R, Constantine G and Katulanda P: Effects of zinc
supplementation on diabetes mellitus: A systematic review and
meta-analysis. Diabetol Metab Syndr. 4:132012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhuang X, Pang X, Zhang W, Wu W, Zhao J,
Yang H and Qu W: Effects of zinc and manganese on advanced
glycation end products (AGEs) formation and AGEs-mediated
endothelial cell dysfunction. Life Sci. 90:131–139. 2012.
View Article : Google Scholar
|
|
27
|
Wijesekara N, Krishnamurthy M,
Bhattacharjee A, Suhail A, Sweeney G and Wheeler MB:
Adiponectin-induced ERK and Akt phosphorylation protects against
pancreatic beta cell apoptosis and increases insulin gene
expression and secretion. J Biol Chem. 285:33623–33631. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
García-Pérez AI, Galeano E, Nieto E, Estañ
MC and Sancho P: Dequalinium induces cytotoxicity in human leukemia
NB4 cells by downregulation of Raf/MEK/ERK and PI3K/Akt signaling
pathways and potentiation of specific inhibitors of these pathways.
Leuk Res. 38:795–803. 2014. View Article : Google Scholar : PubMed/NCBI
|