Introduction
Increasing attention has been focussed on
investigations into the genes and microRNAs (miRNAs) of acute
lymphoblastic leukemia (ALL). However, elucidation of the
pathogenesis of ALL is likely to remain challenging if a
comprehensive view is not established in analyzing this disease.
ALL is an acute form of leukemia or cancer of the white blood
cells, characterized by the overproduction of cancerous, immature
white blood cells, termed lymphoblasts (1,2). It
is a heterogeneous type of cancer, which is characterized by the
rapid and uncontrolled proliferation of immature B- or T-lymphoid
precursors. It represents the most common type of childhood
malignant neoplasia and, despite significant progress in current
treatment methods, 20–30% of affected children relapse and the
causes remain to be elucidated (3).
Transcription factors (TFs) and micro (mi)RNAs are
prominent regulators of gene expression (4). TFs are specific proteins, which
activate or repress the transcription of genes by binding to
cis-regulatory elements located in the upstream regions of
genes (5). TFs regulate gene
expression either alone or in combination with other proteins at
the transcriptional level (6).
miRNAs are small, non-coding RNAs, which are pivotal in several
cellular functions, including proliferation, differentiation and
apoptosis. There are certain genes, which are targeted by miRNAs
and there are numerous databases supplying a substantial quantity
of experimentally validated data to investigate the associations
between miRNAs and their targets (7).
The genes, on which miRNAs are located are termed
the host genes of these miRNAs. Rodriguez et al indicated
that miRNAs are transcribed in parallel with their host
transcripts, and identified two transcription classes of miRNAs,
exonic and intronic (8).
Baskerville et al indicated that intronic miRNAs and their
host genes are closely associated (9). Intronic miRNAs and their host genes
are usually coordinately expressed in biological progression. They
usually act as a potential partner to achieve biological functions
and affect the alteration of pathways (10).
Numerous experiments have been performed to
investigate differentially expressed genes and miRNAs, which has
enabled a deeper understanding of ALL (11–15).
However, in practice, the majority of these experiments focused on
a single element, gene or miRNA, which limits the ability to
establish the general pathogenesis of ALL. The present study aimed
to focus on all associations between genes and miRNAs, including
how genes regulate miRNAs, how miRNAs target genes and how miRNAs
locate on host genes. Each of these elements were analyzed in a
comprehensive investigation and focuses on all the ALL-associated
elements obtained from databases. The present study then aimed to
construct the associated networks, containing the global network,
the related network and the network of differentially expressed
factors, to determine the differentially expressed genes and miRNAs
involved in the pathogenesis of ALL, and the involvement of the
global and related networks. By analyzing and comparing the
similarities and differences among these networks, a number of key
pathways may be highlighted, which may be of significance in the
pathogenesis of ALL. In addition, the present study aimed to
construct a transcriptional network to enable long-term
investigations using predicted transcriptional factors, which may
provide a novel direction in future investigations.
Materials and methods
Material collection and data
processing
The experimentally validated dataset of human miRNAs
(has-mir or has-let) and their target genes were obtained from
Tarbase 5.0 (http://diana.cslab.ece.ntua.gr/tarbase/) and
miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/). The symbols used
in the present study to unify each gene and miRNA were the official
symbols from the National Center for Biotechnology Information
(NCBI) database (http://www.ncbi.nlm.nih.gov/gene/).
A human experimentally-validated dataset of TFs and
miRNAs was extracted from TransmiR (http://www.cuilab.cn/transmir) (16), the data of which is obtained from
publicly available literature and biological experiments.
The host gene of human miRNA was manually extracted
from the miRBase (http://www.mirbase.org/) (17) and NCBI databases. The official
symbol and official ID was used to describe each host gene.
The differentially expressed genes, composed of
mutated genes, aberrantly-expressed protein, overexpressed genes
and single nucleotide polymorphisms (SNPs) were obtained from the
Cancer Genetics Web (http://www.cancerindex.org/geneweb/index.html) and
NCBI SNP database (http://www.ncbi.nlm.nih.gov/snp/) and the relevant
literature (11–15). The ALL-associated genes were
obtained from the GeneCards database (www.genecards.org) (18) and relevant literature, which
included genes affecting tumor growth and migration, and genes
involved in radiation therapy, prevention, diagnosis, development
and the clinical outcome of ALL. The differentially-expressed
genes, mentioned above, were also considered to be a part of the
associated genes. In addition, 28 important TFs were extracted
using the P-match method. Briefly, 1,000 nt promoter region
sequences of the targets of differentially expressed genes were
downloaded from the UCSC database (19), and the P-match method, which
combines pattern matching and weight matrix approaches, was used to
identify transcription factor binding sites (TFBSs) in the 1,000 nt
promoter region sequences. The TFBSs were then mapped onto the
promoter region of targets. The matrix library of P-match also
contains sets of known TFBSs, which are collected in TRANSFAC
(http://www.biobase-international.com/), providing the
possibility to identify a variety of TF binding sites. The
vertebrate matrix was used with restricted high-quality criterion
for the matrix. This method enables the identification of the
possible TFs that may have effects in ALL. In the present study,
only human elements were selected, and the corresponding genes were
determined, which were also considered to be associated-genes.
However, the TFs examined in the study were only those that
appeared in transmiR.
Subsequently, the differentially-expressed miRNAs
were extracted from the mir2Disease database (http://www.mir2disease.org/) (20) and relevant literature. The ALL
miRNAs were obtained manually from the relevant literature, and the
differentially expressed miRNAs were added to these.
Construction of the networks
All the regulatory associations between the genes
regulating miRNAs, miRNA target genes and miRNA location on the
host genes, were analyzed. Following combining all these
associations, Ctyoscype software version 3.1.1 (U.S. National
Institute of General Medical Sciences; award number GM070743) was
used to construct the global network. The network of differentially
expressed factors was extracted from the global network, however,
only the associations between the differentially expressed genes
the and miRNAs with their host genes were selected from the global
network. Following combining of these associations, the network of
differentially expressed factors was constructed. The same method
was used to constructing the ALL-related network.
Results
Association between differentially
expressed factors of ALL
The significant regulatory association between
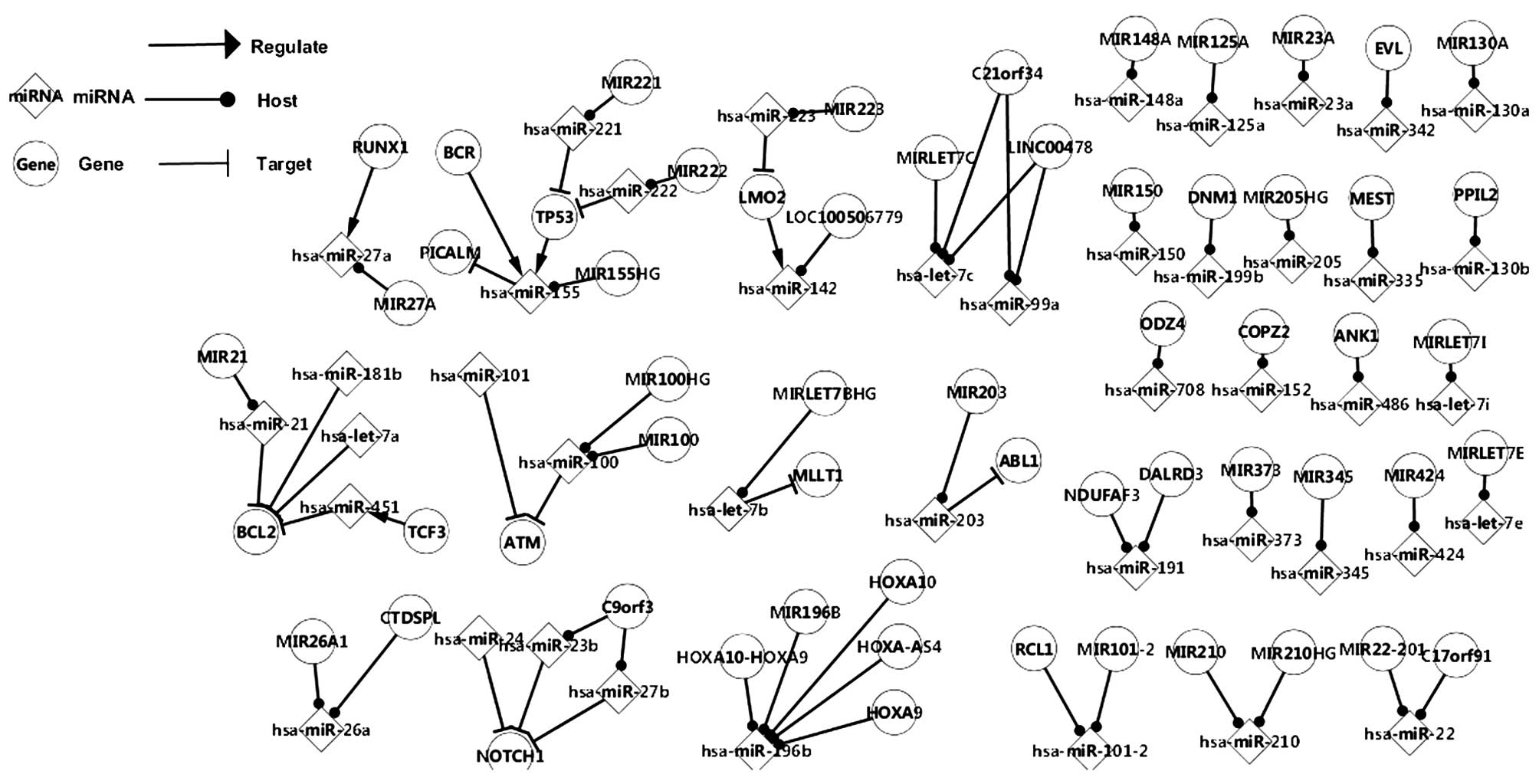
differentially expressed genes and miRNAs in ALL is shown in
Fig. 1. The network is comprised
of mirRNAs and their host genes, five TFs (TP53, TF3, RUNX1, LMO2
and BCR) and other genes, which were aberrantly expressed in ALL
and targeted by at least one differentially expressed miRNA. In
terms of the functions of the TFs, certain important pathways were
identified, for example, the mir-222-TP53-mir155 and the
TCF3-mir451-BCL2 pathways were observed in the
differentially-expressed network, and they may represent a deeper
association between genes and miRNAs. Although, there is no
specific data to indicate a direct association between miRNAs and
miRNAs, they may affect on each other in an indirect manner.
For example, since miR-142 is regulated by LOM2, and
LOM2 is targeted by miR-223, it was suggested that hsa-mir-223
affects has-mir-142 indirectly via LOM2. In addition, genes were
observed to have similar associations, for example TF3 regulates
miR-451 directly, while has-mir-451 targets the BCL2 gene.
Therefore, it was hypothesized that there may be an association
between TCF3 and BCL2, with hsa-mir-451 as a transition. It was
also observed that miRNAs were regulated by more than one TF, and
that the genes may be targeted by several miRNAs. For example,
miR-27a was observed to be regulated by RUNX1 while TP53 and BCR
exhibited co-regulation with miR-155. In addition, ATM was found to
be targeted by two miRNAs (miR-101 and miR-100) simultaneously,
BCL2 was targeted by four miRNAs and ABL1 was only targeted by
miR-203. As for the miRNAs and their host genes, although the host
genes themselves were not differentially-expressed in ALL, their
miRNAs were aberrantly expressed. The C9orf3 contained hsa-mir-23b
and hsa-mir-27b, which targeted NOTCH1 with hsa-mir-24, and
C21orf34 and LINC00478 contained hsa-mir-99a and hsa-let-7c, whilst
hsa-let-7c located on the MIRLET7C gene. Several of the differently
expressed miRNAs in ALL were found to locate to only one gene,
including miR-708 or miR-502. However, exceptions included
hsa-mir-101-2, which located to RCL1 and MIR101-2, while
hsa-mir-196 was found on 5 host genes in total.
The identification of additional genes and miRNAs in
the network of differentially expressed factors is likely in future
studies of this disease, however, the network of differentially
expressed factors established in the present study identifies and
offers insight into the regulatory pathogenesis of ALL.
Network of genes and miRNAs involved in
ALL
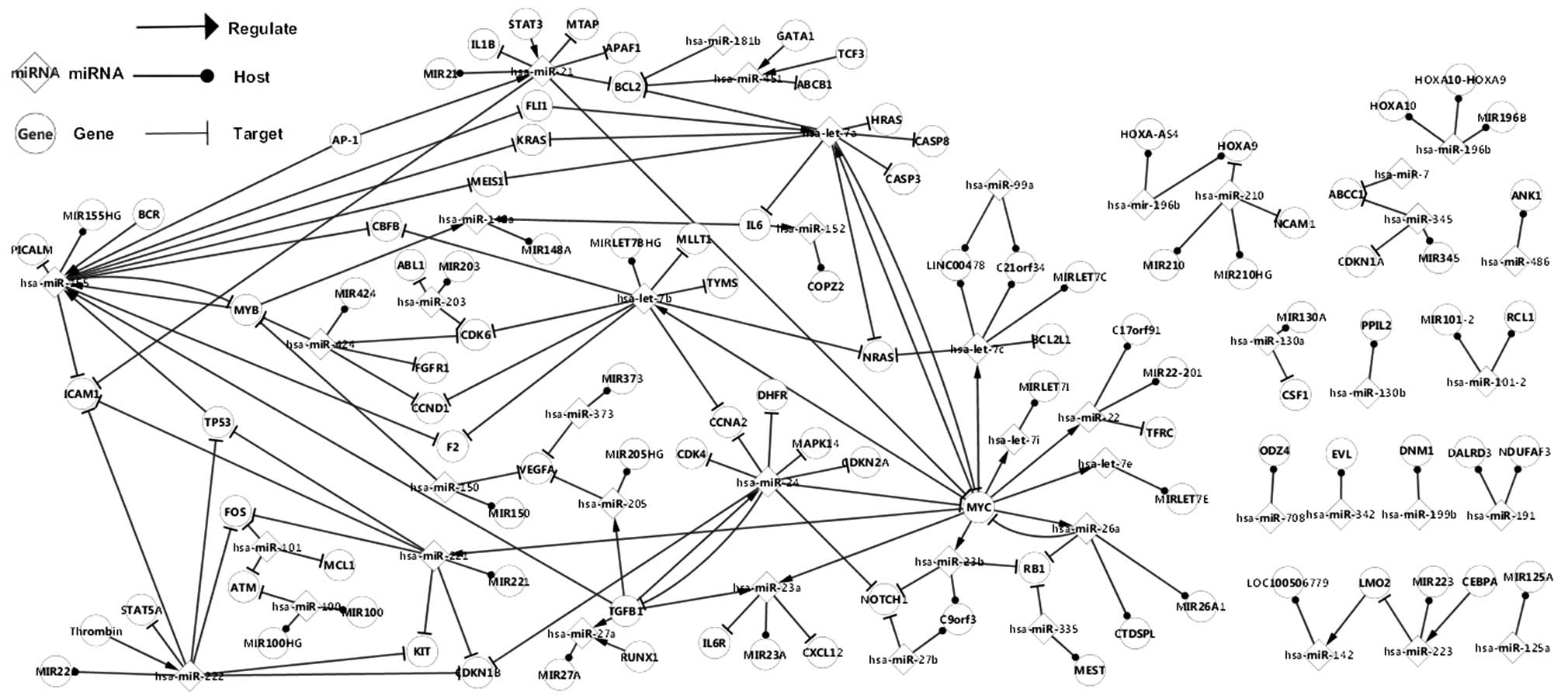
The second network constructed (Fig. 2) represents a more complex
association, compared with the network of differentially expressed
factors, since it contains a larger number of genes and miRNAs, and
more comprehensive pathways. In particular, the differentially
expressed genes and miRNAs are included in this network. As a
result, this network of differentially expressed factors forms the
related network. In the present study, certain specific pathways
were observed in this network. A self-adaptation association was
identified between MYC and has-let-7a, MYC and hsa-mir-26b, TGFB1
and hsa-mir-24, and MYB and hsa-mir-255, meaning that these genes
may have an effect on themselves via the miRNAs they regulate.
The related network elucidated additional
topological associations of differentially expressed elements, for
instance, the network of the TP53 was immensely expanded with the
addition of the relative nodes, contributing to the understanding
of the progression of ALL.
Regulation by predicted TFs and
differentially expressed genes and miRNAs in ALL
The regulatory network of predicted TFs,
differentially expressed genes and miRNAs, termed the global
network, in ALL covers more comprehensive regulatory associations
between these three datasets. It is an experimentally validated
biological network in the human body, and it contains all the
confirmed genes, miRNAs and associations between humans. Notably,
it is too complex to present in the current study. Additionaly, it
includes the network of differentially expressed factors and the
related network.
Transcriptional network of predicted
TFs
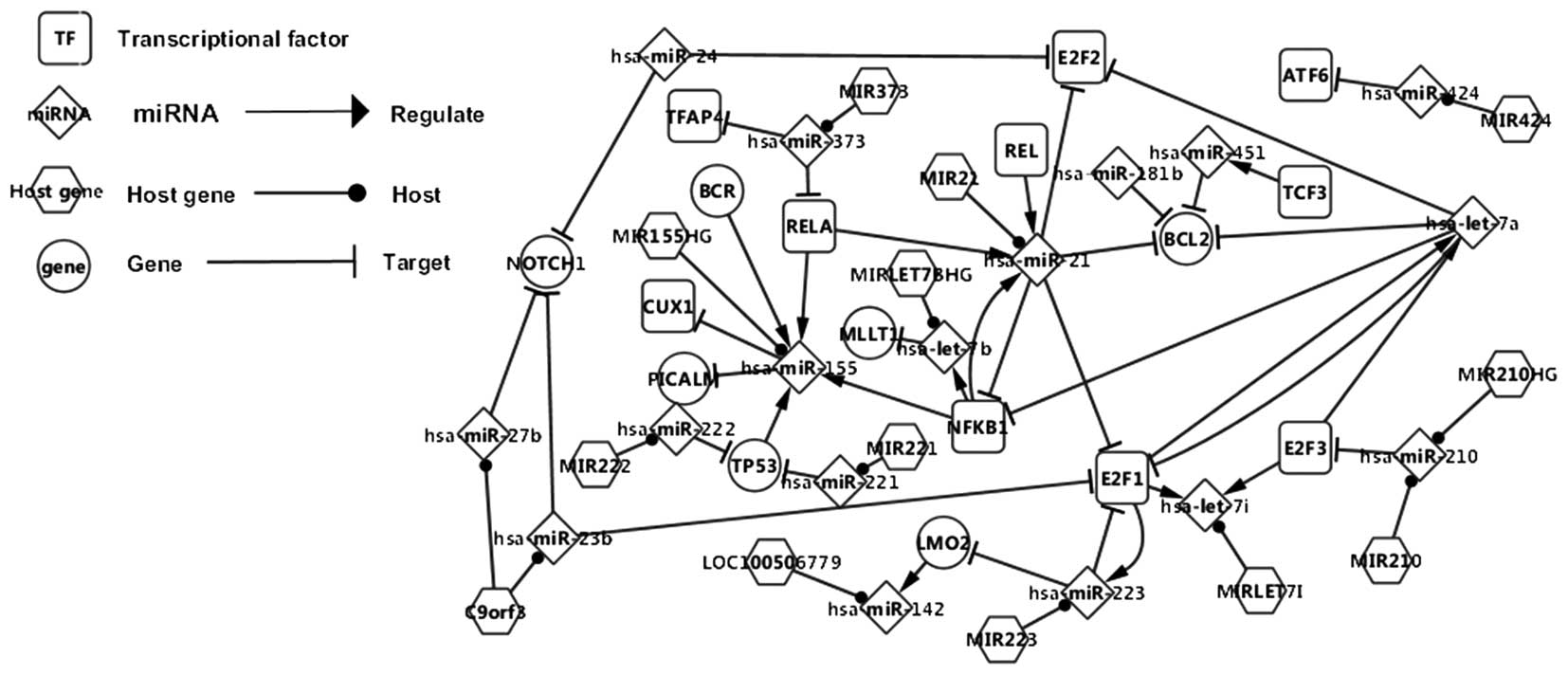
A total of 14 differentially expressed miRNAs, which
were regulated by predicted TFs or targeted these TFs, identified
in the transcriptional network, were analyzed further. Fig. 3 shows the regulatory associations
of predicted TFs and the differentially expressed miRNAs in ALL.
These elements sequentially affect their successors by targeting or
regulating them. The differentially expressed genes, which were
observed to be indirectly or directly associated with the predicted
TFs were also included in the transcription network. For the
predicted TFs, the p-match method was used to identify genes, which
were predicted to be involved in the process of ALL, a number of
which have already been confirmed as differentially expressed,
including TCF3 and PAX5 (21). The
TFs were combined with the differentially expressed genes and
mirRNAs, as well as their host genes, to obtain an overall view of
how TFs affect the differentially expressed miRNAs. The
transcriptional network revealed a potential mechanism involved in
the pathogenesis of ALL, and requires further investigation in the
future. Determination of whether the associations among the network
are involved in ALL may assist in further investigations aiming to
cure the disease. For example, a self-adaptation association
between hsa-miR-223 and E2F1, which exhibits the same association
with hsa-let-7a, was observed (Fig.
3). Therefore, E2F1 may be involved in ALL for the special
position it has with the differentially expressed miRNAs and the
predicted associations require confirmation. Similar associations
were also identified in NFKB1.
Regulation by differentially expressed
genes
The upstream and downstream factors of the elements
identified as important were also identified in the present study,
including the differentially expressed genes, differentially
expressed miRNAs and the TFs obtained using P-match method, in
order to examine the network of ALL more clearly.
In addition, the successor and precursor nodes of
the differentially expressed genes in the three networks were
extracted in order to analyze and compare the regulatory pathways.
Among these genes, TP53 and LMO2 may exhibit the typical
characteristics with their precursor and successor nodes,
indicating the specific regulatory association between the gene and
miRNAs, and the associations between the miRNAs.
The data presented in Table I shows TP53 and the precursors and
successors of the gene, as well as their regulatory associations.
TP53 was significantly featured in the three networks. hsa-miR-221
and hsa-miR-222 were observed to target TP53, which itself
regulates hsa-miR-155 in these three networks (Table I). Therefore, the hsa-miR-221 and
hsa-miR-222 miRNAs indirectly affect hsa-miR-155 through TP53. A
mutation in the TP53 gene may be implicated in the pathogenesis of
ALL via constitutive activation of the TP53 pathway.
 | Table IRegulatory associations between miRNAs
and the TP53 gene in the three networks. |
Table I
Regulatory associations between miRNAs
and the TP53 gene in the three networks.
|
Differentially-expressed miRNA | Related network | Global network |
|---|
| Targeting TP53 | | |
| hsa-miR-221 | hsa-miR-221 | hsa-miR-125b-1 |
| hsa-miR-222 | hsa-miR-222 | hsa-miR-125b |
| | hsa-miR-125b-2 |
| | hsa-miR-1285 |
| | hsa-miR-1285-1 |
| | hsa-miR-1285-2 |
| | hsa-miR-15a |
| | hsa-miR-16 |
| | hsa-miR-16-1 |
| | hsa-miR-16-2 |
| | hsa-miR-221 |
| | hsa-miR-222 |
| | hsa-miR-25 |
| | hsa-miR-30d |
| | hsa-miR-612 |
| Regulated by
TP53 | | |
| hsa-miR-155 | hsa-miR-155 | hsa-miR-107 |
| | hsa-miR-125b |
| | hsa-miR-125b-1 |
| | hsa-miR-125b-2 |
| | hsa-miR-143 |
| | hsa-miR-145 |
| | hsa-miR-155 |
| | hsa-miR-192 |
| | hsa-miR-194 |
| | hsa-miR-194-1 |
| | hsa-miR-194-2 |
| | hsa-miR-200a |
| | hsa-miR-200b |
| | hsa-miR-200c |
| | hsa-miR-215 |
| | hsa-miR-29 |
| | hsa-miR-29a |
| | hsa-miR-29b-1 |
| | hsa-miR-29b-2 |
| | hsa-miR-29c |
| | hsa-miR-34 |
| | hsa-miR-34a |
| | hsa-miR-34b |
| | hsa-miR-34c |
| | hsa-miR-519d |
| | hsa-miR-29a |
| | hsa-miR-29b-1 |
| | hsa-miR-29b-2 |
| | hsa-miR-29c |
| | hsa-miR-34 |
| | hsa-miR-34a |
| | hsa-miR-34b |
| | hsa-miR-34c |
| | hsa-miR-519d |
Regulation by differentially expressed
microRNAs
The same method used to compare and analyze
differentially expressed genes was also applied to the
differentially expressed miRNA pathways, which had been previously
extracted. Of all these miRNAs, three miRNAs, including
hsa-miR-27a, hsa-miR-155 and hsa-miR-451, with their corresponding
genes, were observed to exhibit a specific association between
miRNAs and genes, as well as associations between themselves.
Table II presents
the data of hsa-miR-155 and its predecessor and successor nodes,
which were extracted from the three networks. The results (Table II) indicated that TP53 and BCR
regulate hsa-miR-155, which targeted PICALM in the network of
differentially expressed factors, which indicate that TP53 and BCR
may either co-regulate or individually regulate PICALM indirectly
by hsa-miR-155. It was also demonstrated that MYB and hsa-miR-155
form self-adaptation associations separately.
| Table IIRegulatory associations between genes
and hsa-miR-155 in the three networks. |
Table II
Regulatory associations between genes
and hsa-miR-155 in the three networks.
|
Differentially-expressed miRNA | Related network | Global network |
|---|
| Targeting
hsa-miR-155 | | |
| TP53 | MYB | IRF1 |
| BCR | TGFB1 | JUN |
| TP53 | MYB |
| AP-1 | NFKB1 |
| BCR | RELA |
| | SMAD4 |
| | SMG1 |
| | SPI1 |
| | TGFB1 |
| | TP53 |
| | AKT1 |
| | BCR |
| | CAMP |
| | FOXP3 |
| Regulated by
hsa-miR-155 | | |
| PICALM | CBFB | AGTR1 |
| F2 | AMIGO2 |
| FLI1 | ANKFY1 |
| ICAM1 | APC |
| KRAS | ARFIP1 |
| MEIS1 | ARFIP2 |
| MYB | ARID2 |
| PICALM | ARL10 |
| | ARL5B |
| | ATG3 |
| | ATP6V1C1 |
| | BACH1 |
| | BCAT1 |
| | BET1 |
| | BRPF3 |
| | C3orf58 |
| | C5orf41 |
| | CAMTA1 |
| | CBFB |
| | CDK5RAP3 |
| | CEBPB |
| | CHAF1A |
| | C5orf41 |
| | CAMTA1 |
| | CBFB |
| | CDK5RAP3 |
| | CEBPB |
| | CHAF1A |
Regulation by popular TFs
With the same method used for the differentially
expressed miRNAs, data of the predicted TFs data, using the P-match
method, were processed. The precursor and successor nodes from the
three networks were extracted, classed and listed. Specific
regulatory associations were highlighted in the list, two predicted
TFs, E2F1 and NFKB1 exhibited an important characteristic, which
was common to the precursor and successor nodes among these TFs, as
self-adaption associations were identified between these TFs and
their corresponding miRNAs. For example, four differentially
expressed miRNAs, including hsa-let-7a, hsa-miR-21, hsa-miR-223 and
hsa-miR-23b, were observed to target E2F1, and three differentially
expressed miRNAs, including hsa-let-7a, hsa-miR-223 and hsa-let-7i,
were regulated by E2F2. Furthermore, the hsa-let-7a and hsa-miR-223
miRNAs formed separate self-adaption associations with E2F1.
The predicted TFs in a global network were
subsequently analyzed, and all the TFs were classified into four
categories. The first class has six types of adjacent nodes,
comprising three types of precursor and three types of successor.
TFs, including E2F1, E2F3, NFKB1 and RELA belonged to this
class.
The data in Table
III is that of NFKB1, which contains the precursor and
successor elements of the network of differentially expressed
factors, the related network and the global network. The data
demonstrated that hsa-miR-21 separately forms a self-adaption
association with NFKB1. Notably, no mutation of NFKB1 in ALL has
been reported previously, to the best of our knowledge. As
has-miR-21 was identified from the network of differentially
expressed factors, this miRNA may affect the aberrant expression of
other miRNAs through NFKB1.
| Table IIIRegulatory associations between
miRNAs and the NFKB1 gene. |
Table III
Regulatory associations between
miRNAs and the NFKB1 gene.
|
Differentially-expressed miRNA | Related
network | Global network |
|---|
| Targeting
NFKB1 | | |
| hsa-let-7a | hsa-let-7a | hsa-let-7a |
| hsa-miR-21 | hsa-miR-21 | hsa-let-7a-1 |
| | hsa-let-7a-2 |
| | hsa-let-7a-3 |
| | hsa-miR-146a |
| | hsa-miR-146b |
| | hsa-miR-15a |
| | hsa-miR-9 |
| | hsa-miR-9-1 |
| | hsa-miR-9-2 |
| | hsa-miR-9-3 |
| Regulated by
NFKB1 | | |
| hsa-let-7b | hsa-let-7b | hsa-let-7a-3 |
| hsa-miR-155 | hsa-miR-155 | hsa-let-7b |
| hsa-miR-21 | hsa-miR-21 | hsa-miR-10b |
| | hsa-miR-125b |
| | hsa-miR-125b-1 |
| | hsa-miR-125b-2 |
| | hsa-miR-146a |
| | hsa-miR-155 |
| | hsa-miR-16 |
| | hsa-miR-16-1 |
| | hsa-miR-16-2 |
| | hsa-miR-17 |
| | hsa-miR-199a-2 |
| | hsa-miR-21 |
| | hsa-miR-214 |
| | hsa-miR-224 |
| | hsa-miR-29a |
| | hsa-miR-29b |
| | hsa-miR-29b-1 |
| | hsa-miR-29b-2 |
| | hsa-miR-29c |
| | hsa-miR-34 |
| | hsa-miR-34a |
| | hsa-miR-21 |
| | hsa-miR-365 |
| | hsa-miR-365-1 |
| | hsa-miR-365-2 |
| | hsa-miR-365a |
| | hsa-miR-365b |
| | hsa-miR-448 |
| | hsa-miR-9 |
| | hsa-miR-91 |
| | hsa-miR-9-1 |
| | hsa-miR-9-2 |
| | hsa-miR-9-3 |
| | hsa-miR-199a-2 |
| | hsa-miR-214 |
| | hsa-miR-224 |
| | hsa-miR-29a |
| | hsa-miR-29b |
| | hsa-miR-29b-1 |
| | hsa-miR-29b-2 |
| | hsa-miR-29c |
| | hsa-miR-34 |
| | hsa-miR-34a |
| | hsa-miR-365 |
| | hsa-miR-365-1 |
| | hsa-miR-365-2 |
| | hsa-miR-365a |
| | hsa-miR-365b |
| | hsa-miR-448 |
| | hsa-miR-9 |
| | hsa-miR-91 |
| | hsa-miR-9-1 |
| | hsa-miR-9-2 |
| | hsa-miR-9-3 |
The second class of TFs were those TFs, which
contained only three types of precursors or successors. E2F2, REL,
CUX1, TFAP4, TCF3 and ATF6 were identified in this class.
The third class of TFs had only one adjacent node,
either a precursor or successor, and included CREB1 and STAT1.
The final class of TFs had neither a precursor node
nor a successor, and contained E2F4, NR2F2, HLF, ZEB, ELK1, PAX5,
RORA, ZBTB6, RREB1 and the ATF family (ATF1-5 and 7). The reason
for the identification of TFs with no adjacent nodes may be due to
the limited quantity of data that can be obtained to analyze ALL.
This indicates that additional experimentally validated data are
required and may offer a new direction in investigating ALL.
Discussion
The present study constructed and examined the
network of differentially expressed factors and the transcription
network of predicted transcription factors. In the present study,
pathways contain three or more elements were identified, for
example, hsa-miR-221 targets TP53, and TP53 regulates hsa-miR-155,
which targets PICALM. The TCF3 pathway was also observed to contain
three elements (TCF3, hsa-miR451 and BCL2). These pathways may have
a biological function in ALL.
TP53 has been identified as a typical gene that is
abnormally expressed in other types of cancer, including
retinoblastoma and human Hodgkin's lymphoma, which suggests that
TP53 requires attention in further investigations of ALL, and may
assist in defining the similarities among types of cancer. The
resulting understanding of the associations between genes may be
extended from one type of carcinoma to another, and a systematic
view in analyzing diseases may be obtained.
Another area, which requires further investigation
is the TFs, which were identified using the P-match method, and
suggested a potential association between the differentially
expressed miRNAs and TFs. These predicted associations require
experimental validation to improve understanding of the pathogenic
mechanism of ALL.
The present study constructed three regulatory
topological networks of the genes, miRNAs involved in ALL, and data
were extracted from the network in order to highlight specific
pathways, genes and transcription factors, which may be important
in the investigation of ALL. Focus on the successors and precursors
of the nodes in these three networks may assist in analyzing the
network. In addition, construction of a network using predictions
with the P-match method may providing important data for the
further investigation of ALL.
Abbreviations:
|
miRNA
|
microRNA
|
|
TFs
|
transcription factors
|
|
targets
|
target genes
|
|
ALL
|
acute lymphoblastic leukemia
|
|
NCBI
|
national center for biotechnology
information
|
|
TFBSs
|
transcription factor binding sites
|
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant. no. 60973091) and the
Science and Technology Development Plan of Jilin Province (grant.
no. 20130101166JC).
References
|
1
|
Weinblatt ME: Pediatric acute myelocytic
leukemia. Sakamoto KM, Windle ML, Cripe TP and Arceci RJ: Medscape
Reference. WebMD. Accessed 17 April 2014.
|
|
2
|
Seiter K: Acute lymphoblastic leukemia.
Sarkodee-Adoo C, Talavera F, Sacher RA and Besa EC: Medscape
Reference. WebMD. Accessed 17 April 2014.
|
|
3
|
Pui CH: Recent research advances in
childhood acute lymphoblastic leukemia. J Formos Med Assoc.
109:777–787. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hobert O: Gene regulation by transcription
factors and microRNAs. Science. 319:1785–1786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tran DH, Satou K, Ho TB and Pham TH:
Computational discovery of miR-TF regulatory modules in human
genome. Bioinformation. 4:371–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martin KJ: The interactions of
transcription factors and their adaptors, coactivators and
accessory proteins. BioEssays. 13:499–503. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang BH; Pan XP; Cobb GP, et al: Plant
microRNA: A small regulatory molecule with big impact. Dev Biol.
289:3–16. 2006. View Article : Google Scholar
|
|
8
|
Rodriguez A, Griffiths-Jones S, Ashurst JL
and Bradley A: Identification of mammalian microRNA host genes and
transcription units. Genome Res. 14:1902–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baskerville S and Bartel D: Microarray
profiling of microRNAs reveals frequent coexpression with
neighboring miRNAs and host genes. RNA. 11:241–247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ramalingam P, Palanichamy JK, Singh A, Das
P, Bhagat M, Kassab MA, Sinha S and Chattopadhyay P: Biogenesis of
intronic miRNAs located in clusters by independent transcription
and alternative splicing. RNA. 20:76–87. 2014. View Article : Google Scholar :
|
|
11
|
Armstrong SA, Mabon ME, Silverman LB, et
al: FLT3 mutations in childhood acute lymphoblastic leukemia.
Blood. 103:3544–3546. 2004. View Article : Google Scholar
|
|
12
|
Tartaglia M, Martinelli S, Cazzaniga G, et
al: The PTPN11 gene is mutated in pediatric acute lymphoblastic
leukemia. Blood. 102:65A. 2003.
|
|
13
|
Grossmann V, Haferlach C, Weissmann S, et
al: The molecular profile of adult T-cell acute lymphoblastic
leukemia: Mutations in RUNX1 and DNMT3A are associated with poor
prognosis in T-ALL. Genes Chromosomes Cancer. 52:410–422. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zenatti PP, Ribeiro D, Li W, Zuurbier L,
Silva MC, Paganin M, Tritapoe J, Hixon JA, Silveira AB, Cardoso BA,
et al: Oncogenic IL7R gain-of-function mutations in childhood
T-cell acute lymphoblastic leukemia. Nat Genet. 43:932–939. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Malyukova A, Dohda T, von der Lehr N,
Akhoondi S, Corcoran M, Heyman M, Spruck C, Grandér D, Lendahl U
and Sangfelt O: The tumor suppressor gene hCDC4 is frequently
mutated in human T-cell acute lymphoblastic leukemia with
functional consequences for Notch signaling. Cancer Res.
15:5611–5616. 2008.
|
|
16
|
Wang J, Lu M, Qiu C and Cui Q: TransmiR: A
transcription factor-microRNA regulation database. Nucleic Acids
Res. 38:D119–D122. 2010. View Article : Google Scholar
|
|
17
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–D157. 2011. View Article : Google Scholar :
|
|
18
|
Safran M, Dalah I, Alexander J, Rosen N,
Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, et al:
GeneCards Version3: The human gene integrator. Database (Oxford).
2010:baq0202010. View Article : Google Scholar
|
|
19
|
Fujita PA, Rhead B, Zweig AS, Hinrichs AS,
Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A,
et al: The UCSC Genome Browser database: Update 2011. Nucleic Acids
Res. 39:D876–D882. 2011. View Article : Google Scholar :
|
|
20
|
Bao J, Li D, Wang L, et al: MicroRNA-449
and microRNA-34b/c function redundantly in murine testes by
targeting E2F transcription factor-retinoblastoma protein (E2F-pRb)
pathway. J Biol Chem. 287:21686–21698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Familiades J, Bousquet M,
Lafage-Pochitaloff M, et al: PAX5 mutations occur frequently in
adult B-cell progenitor acute lymphoblastic leukemia and PAX5
haploinsufficiency is associated with BCR-ABL1 and TCF3-PBX1 fusion
genes: A GRAALL study. Leukemia. 23:1989–1998. 2009. View Article : Google Scholar : PubMed/NCBI
|