Introduction
Head and neck squamous cell carcinoma is the sixth
most common type of cancer, with laryngeal squamous cell carcinoma
becoming more common with an increasing occurrence of new cases and
mortality annually (1,2). The highest incidence rates in males
are registered in Asia at 12 cases per 100,000 individuals per year
(3). Laryngeal cancer patients
usually lose their laryngeal function, which affects speech,
swallowing and breathing; thus, normal activities of the patients
are impaired (4). At present,
surgery and radiotherapy are the predominant treatments and are
often followed by chemotherapy, particularly in locally advanced
disease (5). Multiple approaches
have been applied to treat this type of cancer; however, the
survival rate of patients has not significantly improved.
Therefore, laryngeal carcinoma represents a target for developing
novel therapeutic approaches. An improved understanding of the
mechanisms involved in the regulation of cell proliferation
requires the identification of novel genes associated with
laryngeal carcinoma. Additionally, the upregulation or
downregulation of these genes may be important in the treatment of
this severe disease.
Nemo-like kinase (NLK) has been identified as an
important regulator of cell growth, patterning and cell death in a
variety of organisms (6,7,8). NLK
was originally isolated as a murine orthologue of the
Drosophila Nemo gene by reverse transcription polymerase
chain reaction (RT-PCR) from neural tissue mRNA of the embryonic
mouse using degenerate primers designed for the conserved kinase
domains I, VI, VII and IX of the extracellular-signal regulated
kinase/mitogen-activated protein kinase (MAPK) family. NLK, an
evolutionarily conserved serine/threonine kinase, belongs to the
proline directed protein kinase superfamily, consisting of MAPK and
cyclin-dependent protein kinases (CDKs) (9). Genetic studies of the homologs of NLK
in animal models have demonstrated that NLK has a suppressive role
in Wnt/β-catenin signaling (10,11).
RNA silencing, also known as RNA interference (RNAi)
is a form of post-transcriptional gene silencing mediated by a
short double-stranded small interfering RNA (siRNA). This method
has been demonstrated to be highly effective for gene knockdown and
holds great promise in the field of cancer therapy (12). A number of gene products involved
in carcinogenesis have already been examined as targets for RNAi
and RNAi-targeting of molecules crucial for tumor-host interactions
and tumor resistance to chemo or radiotherapy has also been
investigated. Long-lasting RNAi-based gene silencing can be
achieved with the aid of lentivirus-based expression systems, which
drive the production of short hairpin RNA (shRNA) species.
Lentiviral vectors are an appealing tool for transgenesis in part
due to their ability to incorporate into genomic DNA with high
efficiency. In addition, they can also be maintained for long
periods of time (13). Lentiviral
vectors have become a promising tool for the establishment of
transgenic animals and the manipulation of the mammalian
genome.
Materials and methods
Cell culture
Hep-2 cells were obtained from the CEll Bank of the
Chinese Academy of Sciences (Shanghai, China) maintained in
Dulbecco's modified Eagle's medium (DMEM; HyClone, GE Healthcare,
Little Chalfont, UK) containing 10% fetal bovine serum (Biowest,
Loire Valley, France), 1% l-glutamine and 1% (v/v)
penicillin-streptomycin (Sigma-Aldrich, St. Louis, MO, USA). The
cells were subcultured following trypsinization once or twice per
week in a 1:5 split ratio. The cell lines were maintained as
monolayers in 75 cm2 cell culture flasks at 37°C in a
humidified atmosphere of 5% CO2.
Lentivirus infection
Cells were incubated with lentivirus in a small
volume of serum-free DMEM at 37°C for 4 h. Subsequently, 10% DMEM
was added and the cells were placed in an incubator for 4–5 days
for the following experiments. Lentivirus was used to infect Hep-2
cells at a multiplicity of infection (MOI) of 10, and the infection
efficiency was evaluated by green fluorescent protein (GFP) on the
premise that no virus toxic effect was observed. Thus, the
following experiments were performed using viruses at an MOI of 10,
unless otherwise indicated. The lentivirus packaged with GFP was
used for the cell proliferation assays and the lentivirus without
GFP was used for the cell apoptosis propidium iodide (PI) staining
to exclude interference with the GFP signal. The Hep-2 cells
transfected with the NLK-siRNA (5′-GATAGACCTATTGGATATG-3′) and the
negative control siRNA (5′-TTCTCCGAACGTGTCACGT-3′) were designated
Lv-shNLK and Lv-shCon, respectively. Furthermore, to determine the
infection efficiency, cells expressing GFP protein were observed
using fluorescence microscopy (CKX41; Olympus, Tokyo, Japan) 5 days
after infection.
Quantitative polymerase chain reaction
(qPCR)
At 5 days after infection, total RNA was extracted
from control, Lv-shNLK and Lv-shCon using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA). The following
primer sequences were used for the detection of NLK: NLK, sense
5′-ATCATCAGCACTCGCATCATC-3′ and antisense
5′-GACCAGACAACACCAAAGGC-3′; GAPDH, sense
5′-TGACTTCAACAGCGACACCCA-3′ and antisense
5′-CACCCTGTTGCTGTAGCCAAA-3′. GAPDH was used as an internal control.
The PCR mixture was prepared using SYBR Green master mix (Takara
Bio Inc., Ohtsu, Japan) in accordance with the manufacturer's
instructions. All RT-qPCR experiments were performed using an
Mx3000P thermal cycler (Agilent Technologies, Santa Clara, CA,
USA).
Western blot analysis
At 5 days after infection, the proteins from Hep-2
cells were analyzed using western blot analysis. Standard
procedures were used for western blotting. Briefly, Hep-2 cells
were washed with ice-cold phosphate-buffered saline (PBS;
Sigma-Aldrich), scraped and then lysed in radioimmunoprecipitation
assay buffer. Cell debris was removed by centrifugation at 10,300 ×
g for five minutes followed by flash freezing of the supernatants.
The protein concentrations were determined according to the
bicinchoninic acid assay (BCA Protein Assay reagent, 23227; Pierce,
Thermo Fisher Scientific, Rockford, IL, USA). Total protein (30
µg) was subjected to a 10% SDS-PAGE and electroblotted onto
a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). The membranes were blocked with 5% bovine serum albumin for 2
h and subsequently incubated with different antibodies, which were
used to detect respective proteins using an
electrochemiluminescence assay kit (Pierce) according to the
manufacturer's instructions. The antibodies used were as follows:
Rabbit polyclonal anti-NLK (1:500; ab116715; Abcam, Cambridge, UK);
mouse monoclonal anti-Cyclin B1 (1:1,000; cat. no. K0128-3; Medical
& Biological Laboratories, Aichi, Japan); mouse monoclonal
anti-Cyclin D1 (1:1,000; cat. no. MD-17-3; Medical & Biological
Laboratories); rabbit polyclonal anti-P53 (1:1,000; cat. no. 9282;
Cell Signaling Technology, Inc., Danvers, MA, USA); rabbit
polyclonal anti-Phospho-p53 (Ser315) (1:1000; cat. no. 2528; Cell
Signaling Technology, Inc.); rabbit polyclonal anti-GAPDH
(1:60,000; cat. no. 10494-1-AP; Proteintech Group, Inc, Chicago,
IL, USA). The secondary antibodies were goat anti-rabbit
immunoglobulin G (IgG) (1:5,000; cat. no. SC-2054; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and goat anti-mouse IgG
(1:5,000; cat. no. SC-2005; Santa Cruz Biotechnology, Inc.).
Cell viability assay
Cells were cultured in 96-well culture dishes at a
density of 1×104 cells/well. Following infection for 1,
2, 3, 4 and 5 days, the media in each well were removed and the
cell viability value was measured using an MTT colorimetric assay.
To determine the cell viability, 20 µl MTT (5 mg/ml) was
added to each well and cells were incubated for an additional 4 h.
The supernatant was then removed and the insoluble formazan product
was dissolved in acidified isopropanol. The optical density (OD) of
the 96-well culture plates was measured using an enzyme-linked
immunosorbent assay reader (2550; BioTek Instruments, Inc.,
Winooski, VT, USA) at 595 nm. The OD of formazan formed in
untreated control cells was interpreted as 100% viability.
Cell cycle analysis by flow
cytometry
At each indicated time point, the treated cells were
trypsinized and pooled with the floating cells in the culture
medium. The cells were collected by centrifugation at 300 × g for 5
min. The cell pellet was mildly resuspended in a solution
containing 584 µg/ml NaCl, 1,000 µg/ml sodium
citrate, 10 µg/ml RNAase A, 0.3 µg/ml Nonidet P-40
and 50 µg/ml PI. The cell suspensions were incubated for 30
min in the dark at room temperature, followed by the addition of a
solution containing 15 mg/ml citric acid, 0.25 mM sucrose and 50
µg/ml PI. The suspension of PI-stained isolated nuclei were
mixed and kept in the dark at 4°C prior to flow cytometric
measurement. The cell cycle distributions were analyzed on a
FACScan (Becton-Dickinson, Franklin Lakes, NJ, USA).
Colony formation assay
Briefly, 0.5 ml base layers consisting of 0.8% agar
medium were prepared in six-well plates. Uninfected and infected
cells were trypsinized, centrifuged at 105 × g for 5 min,
resuspended in 0.4% agar medium (equal volumes of 0.8% noble agar
and culture medium) and plated onto the top agar at 200 cells/well.
The cells were cultured for 14 days at 37°C. Following culturing
for 14 days, adherent cells were washed twice with PBS and then
fixed with 4% paraformaldehyde for 30 min at room temperature.
Colonies were visualized using a cell staining Giemsa solution
(Chemicon, Temecula, CA, USA) and counted under a fluorescence
microscope.
Statistical analysis
The results of all assays are expressed as the mean
± standard deviation. All assays were performed independently in
triplicate. Statistical analyses were performed using Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression of NLK in Hep-2 cells is
suppressed by infection with si-NLK
In order to knock down the NLK gene in Hep-2 cells,
GFP containing lentivirus-mediated infection of NLK siRNA was
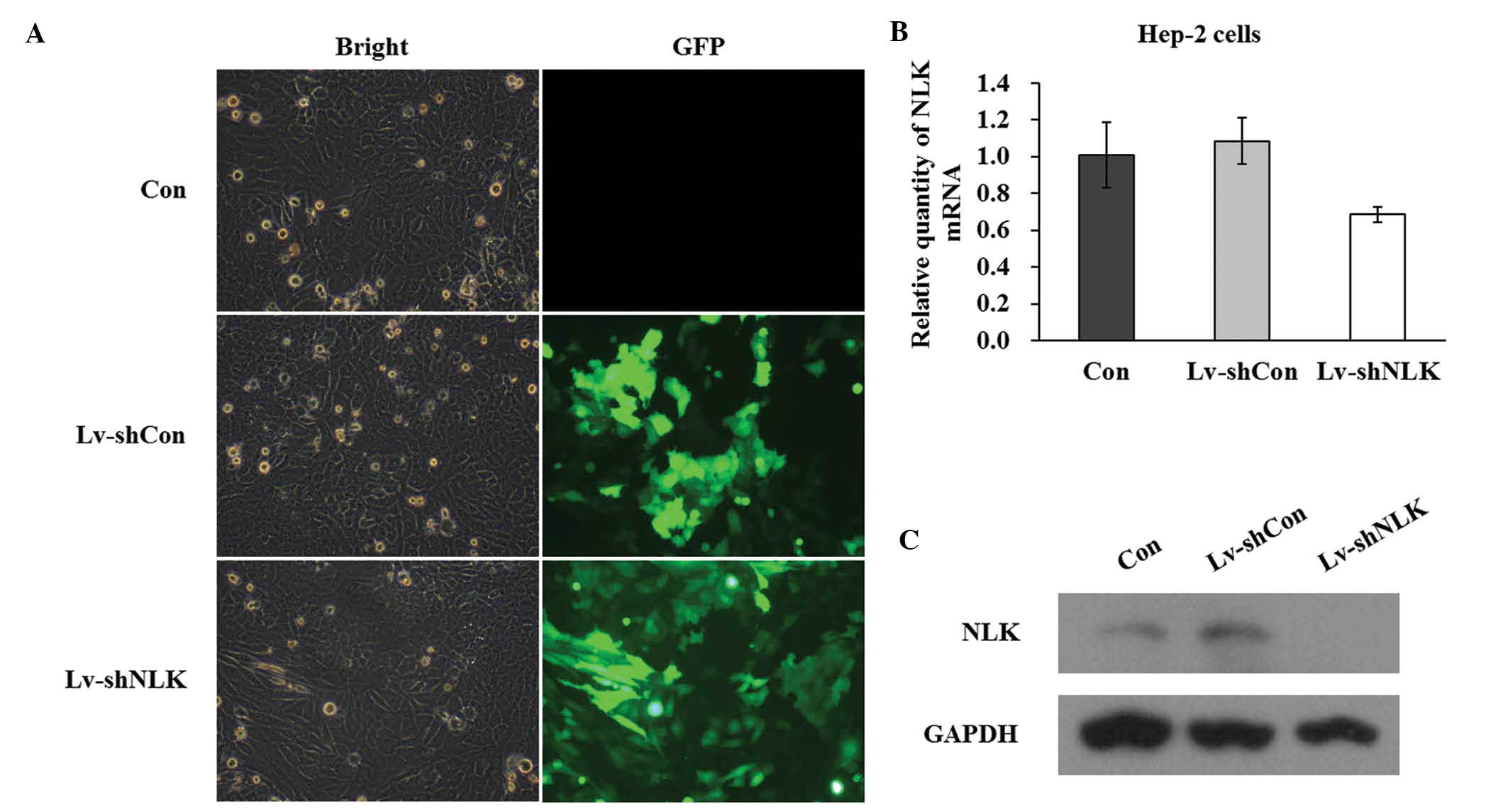
performed. The successful infection was confirmed by evaluating the
expression level of GFP. As shown in Fig. 1A, 5 days after NLK infection,
>90% of cells expressed GFP, indicating a high infection
efficiency. The mRNA expression level of NLK following infection
with siRNA was evaluated by qPCR. Quantitative analysis of NLK mRNA
level using densitometry revealed a significant decrease in
Lv-shNLK (36.6%) compared with the control (Fig. 1B). In addition, western blot assays
demonstrated that the protein expression of NLK in the two Hep-2
cell lines was significantly suppressed by NLK-specific siRNA
expression (Fig. 1C).
Collectively, these data suggested that lentivirus-mediated siRNA
successfully infected Hep-2 cells and in addition, may be used to
investigate NLK loss of function effects in Hep-2 cells.
Suppression of cell proliferation and
tumorigenesis in Hep-2 cells by si-NLK
To elucidate the role of NLK in cellular
proliferation, the proliferative property of Lv-shNLK was analyzed
using an MTT assay. The cell proliferation assay was performed for
5 days. The data revealed that there was a significant reduction in
Hep-2 cell proliferation as a result of NLK knockdown compared with
the uninfected cells (con) and Lv-shCon (Fig. 2A). Notably, 4 and 5 days
post-infection, si-NLK-infected cells demonstrated a significantly
lower cell viability of 0.58±0.00 and 0.60±0.01 fold, respectively,
compared with the control group (P<0.01). These findings suggest
that NLK is important in Hep-2 cell proliferation.
The results of the colony formation assay also
revealed that downregulation of NLK caused a decrease (83.34%) in
the number of colonies compared with uninfected cells (P<0.01;
Fig. 2B). In addition, as
evaluated by Giemsa staining and fluorescence microscopy, Hep-2
cells with NLK suppression exhibited a marked reduction in colony
size (Fig. 2C and D), suggesting
that the tumorigenesis of Hep-2 cells was impaired following
suppression of NLK expression.
NLK knockdown arrests the cell cycle at
the S phase in Hep-2 cells
The present study also aimed to examine the effects
of NLK knockdown on cell cycle progression. Hep-2 cells were
labeled with PI and analyzed by DNA flow cytometry. A
representative histogram for the Hep-2 cells is shown in Fig. 3A. NLK knockdown caused an increase
in the percentage of cells in the S phase and a corresponding
decrease in the percentage of cells in the
G0/G1 and G2-M phases (Fig. 3B). In addition, as depicted in
Fig. 3C, the percentage of
sub-G1 apoptotic cells of Lv-shNLK were significantly
higher compared with Lv-shCon and Con.
NLK regulates the expression of
cell-cycle regulatory proteins in Hep-2 cells
In addition to the possible molecular mechanism for
cell growth inhibition and cell cycle arrest in Hep-2 cells,
pro-apoptotic protein expression was also investigated. The protein
expression of cell cycle regulatory proteins, including cyclin B1,
cyclin D1, p53 and pS315-p53 were examined (Fig. 4). Protein levels of cyclin D1,
whose function is important for the G1-S transition,
were significantly upregulated in Lv-shNLK. In addition, cyclin B1
expression, which controls G2-M transition was also
upregulated in Lv-shNLK. These results demonstrated that NLK may be
involved in translational modification of cell cycle regulatory
proteins in Hep-2 cells. Furthermore, Lv-shNLK markedly
phosphorylated p53 (Fig. 4), while
uninfected cells and Lv-shCon did not exhibit phosphorylated p53.
These results confirm that Lv-shNLK specifically phosphorylates p53
in vitro.
Discussion
Treatment indications in cancer of the larynx are
often controversial, as there are few comparative studies of the
different available therapeutic approaches (14). At present, surgery and radiotherapy
are widely used and the choice between these two procedures is a
common therapeutic decision. Laryngeal function preservation has
gained more and more interest in previous decades and chemotherapy
is also a significant component of several curative approaches
(15). In addition, novel targeted
therapeutic methods, including gene therapies are opening a new way
to improve the treatment of patients diagnosed with laryngeal
cancer.
siRNA is now widely used in cancer studies and may
provide a promising method for laryngeal cancer therapy. Use of the
lentivirus for gene disruption is popular due to its high
efficiency and safety in humans (16). In the present study, the lentivirus
technique was employed to specifically inhibit NLK gene expression
and demonstrated that loss of NLK function significantly impaired
the proliferative and tumorigenic properties of Hep-2 cells. NLK is
an MAPK-like kinase, which belongs to the proline-directed
serine/threonine protein kinase superfamily. NLK has been revealed
to markedly alter the activity of other transcription factors,
including nuclear factor-κB, Smads, activator protein 1 and p53
(17). The function of NLK in
several cancer cell lines has been examined. For example, Jung
et al (6) demonstrated that
NLK inhibited cell growth and arrested cell cycle transition at the
G1/S phase in a Hep3B cell line. By contrast,
overexpression of NLK in colon and ovarian cancer cell lines
suppressed cell growth and increased programmed cell death
(18,19). It has been suggested that these
controversial results were at least partially due to the different
cancer types investigated, reflecting the complicated function of
NLK in the biological progression of different types of cancer
(20).
In the present study, NLK downregulation in human
laryngeal cancer cells were examined. Suppression of NLK inhibits
cell growth and tumorigenesis of Hep-2 cells, as elucidated using
MTT, colony formation and flow cytometric analysis. NLK
downregulation induces activation of the p53 protein. As a tumor
suppressor, p53 is essential for preventing inappropriate cell
proliferation, cell cycle arrest, apoptosis, DNA repair in a
variety of cell types and maintaining genomic integrity following
genotoxic stress (21). The
importance of p53 in tumor suppression is highlighted by the
observation that approximately half of all types of human cancer
exhibit evidence for the loss of normal p53 function due to a
mutation within the p53 gene (22). Thus, activation of p53, may
partially explain the inhibition of tumorigenesis through NLK
silencing. Furthermore, it was found that NLK knockdown reduced the
cell survival rate by arresting the cell cycle at S phase.
Progression from the G1-S phase of the cell cycle
required activation of CDK4, and CDK4 activation is controlled, in
part, by a complex formation with its catalytic partner, cyclin D1
(23). The eukaryotic cell cycle
is regulated through the sequential activation and inactivation of
CDKs, which drive cell cycle progression through the
phosphorylation of key regulatory proteins (24). Thus, cyclin D1 is necessary and
rate-limiting for S phase progression. In the present study, the
decrease in cell viability may be attributed to the occurrence of S
phase cell cycle arrest following knockdown of NLK by RNAi in Hep-2
cells. However, further investigation is required to elucidate the
detailed mechanisms.
In conclusion, the present study demonstrated that
the RNAi-mediated targeting of NLK regulates Hep-2 cell
proliferation. These results also suggest that the siRNA-mediated
downregulation of the target gene expression is sufficiently stable
in laryngeal cancer cells. In addition, disruption of NLK
suppressed tumor cell proliferation. The current data suggested
that NLK may be associated with the development of aggressiveness
in Hep-2 cells. Furthermore, an improved understanding of NLK
function and processing may provide novel insights into the
clinical therapy of laryngeal cancer.
References
|
1
|
Wang F, Song G, Liu M, Li X and Tang H:
miRNA-1 targets fibronectin1 and suppresses the migration and
invasion of the HEp2 laryngeal squamous carcinoma cell line. FEBS
Lett. 585:3263–3269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu T, Zhang M, Zhang H, Sun C and Deng Y:
Inhibitory effects of cucurbitacin B on laryngeal squamous cell
carcinoma. Eur Arch Otorhinolaryngol. 265:1225–1232. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bezerra de Souza DL, Jerez Roig J and
Bernal MM: Laryngeal cancer survival in Zaragoza (Spain): a
population-based study. Clin Transl Oncol. 14:221–224. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dechaphunkul T: Epidemiology, risk
factors, and overall survival rate of laryngeal cancer in
Songklanagarind Hospital. J Med Assoc Thai. 94:355–360.
2011.PubMed/NCBI
|
|
5
|
Pignon JP, Bourhis J, Domenge C and
Designé L: Chemotherapy added to locoregional treatment for head
and neck squamous-cell carcinoma: three meta-analyses of updated
individual data. Lancet. 355:949–955. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jung KH, Kim JK, Noh JH, et al: Targeted
disruption of Nemo-like kinase inhibits tumor cell growth by
simultaneous suppression of cyclin D1 and CDK2 in human
hepatocellular carcinoma. J Cell Biochem. 110:687–696. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meneghini MD, Ishitani T, Carter JC, et
al: MAP kinase and Wnt pathways converge to downregulate an
HMG-domain repressor in Caenorhabditis elegans. Nature.
399:793–797. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Verheyen EM, Mirkovic I, MacLean SJ,
Langmann C, Andrews BC and MacKinnon C: The tissue polarity gene
nemo carries out multiple roles in patterning during Drosophila
development. Mech Dev. 101:119–132. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yasuda J, Yokoo H, Yamada T, Kitabayashi
I, Sekiya T and Ichikawa H: Nemo-like kinase suppresses a wide
range of transcription factors, including nuclear factor-κB. Cancer
Sci. 95:52–57. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sampson EM, Haque ZK, Ku MC, et al:
Negative regulation of the Wnt-beta-catenin pathway by the
transcriptional repressor HBP1. EMBO J. 20:4500–4511. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishitani T, Ninomiya-Tsuji J and Matsumoto
K: Regulation of lymphoid enhancer factor 1/T-cell factor by
mitogen-activated protein kinase-related Nemo-like kinase-dependent
phosphorylation in Wnt/beta-catenin signaling. Mol Cell Biol.
23:1379–1389. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Castanotto D and Rossi JJ: The promises
and pitfalls of RNA-interference-based therapeutics. Nature.
457:426–433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park F: Lentiviral vectors: are they the
future of animal trans-genesis? Physiol Genomics. 31:159–173. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Licitra L, Bernier J, Grandi C, et al:
Cancer of the larynx. Crit Rev Oncol Hematol. 47:65–80. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marioni G, Marchese-Ragona R, Cartei G,
Marchese F and Staffieri A: Current opinion in diagnosis and
treatment of laryngeal carcinoma. Cancer Treat Rev. 32:504–515.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng K and Qin B: RNA interference for
cancer therapy. Pharmaceutical Perspectives of Cancer Therapeutics.
Lu Y and Mahato RI: Springer; US: pp. 399–440. 2009, View Article : Google Scholar
|
|
17
|
Emami KH, Brown LG, Pitts TE, Sun X,
Vessella RL and Corey E: Nemo-like kinase induces apoptosis and
inhibits androgen receptor signaling in prostate cancer cells.
Prostate. 69:1481–1492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yasuda J, Tsuchiya A, Yamada T, Sakamoto
M, Sekiya T and Hirohashi S: Nemo-like kinase induces apoptosis in
DLD-1 human colon cancer cells. Biochem Biophys Res Commun.
308:227–233. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Peng C, Wu G, et al: Expression
of NLK and its potential effect in ovarian cancer chemotherapy. Int
J Gynecol Cancer. 21:1380–1387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tan Z, Li M, Wu W, et al: NLK is a key
regulator of proliferation and migration in gallbladder carcinoma
cells. Mol Cell Biochem. 369:27–33. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bai L and Zhu WG: p53: structure, function
and therapeutic applications. J Cancer Mol. 2:141–153. 2006.
|
|
22
|
Dey A, Verma CS and Lane DP: Updates on
p53: modulation of p53 degradation as a therapeutic approach. Br J
Cancer. 98:4–8. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sherr CJ and Roberts JM: Living with or
without cyclins and cyclin-dependent kinases. Genes Dev.
18:2699–2711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gorospe M, Liu Y, Xu Q, Chrest FJ and
Holbrook NJ: Inhibition of G1 cyclin-dependent kinase activity
during growth arrest of human breast carcinoma cells by
prostaglandin A2. Mol Cell Biol. 16:762–770. 1996.PubMed/NCBI
|