Introduction
Schistosomiasis is a major infectious tropical
disease caused by blood flukes (trematode worms) of the genus
Schistosoma. Following malaria, schistosomiasis is the
second most socioeconomically devastating parasitic disease in the
world. The disease is prevalent in East and Southeast Asia, Africa
and Latin America and affects >200 million individuals each year
(1). There are three major species
of Schistosoma worms that parasitize the human body:
mansoni, japonicum and haematobium (2).
In East and Southeast Asia, including China, where
schistosomiasis is a serious problem, the prevalent species of the
parasite is Schistosoma japonicum (3). The first drug that was shown to be
effective against schistosomiasis, in 1918, was antimony potassium
tartrate. Praziquantel (PZQ) was discovered in the mid-1970s and
has effectively been the only drug used for the large-scale
treatment of schistosomiasis since its discovery (4). Due to this, however, parasites with
low susceptibility to PZQ have begun to emerge (5,6),
thus making the development of new drugs for the treatment of
schistosomiasis an urgent necessity.
Thioredoxin glutathione reductase (TGR) plays a
crucial role in maintaining redox homeostasis in the parasite
(7). TGR is a homodimeric
flavoprotein in which each subunit comprises a glutaredoxin (Grx)
domain fused to a typical thioredoxin reductase (TR) domain. The TR
domain is analogous to the glutathione reductase (GR) domain: Both
the TR and GF enzymes belong to the same superfamily of dimeric
flavoenzymes, and share similar global folds, cofactors (FAD),
substrate binding sites and active site residues, and have similar
catalytic mechanisms. Schistosoma has lost the genes
encoding TR and GR (two of the main detoxification pathways in
mammals) and depends on the single TGR enzyme, which combines the
enzymatic activities of GR, TR and Grx, to control redox
homeostasis. Adult parasites are killed by RNA interference gene
silencing of TGR, confirming TGR as a potential drug target for the
treatment of schistosomiasis (8).
The overall structure of TGR from Schistosoma
mansoni (smTGR) is a fusion of two domains: Grx (residues
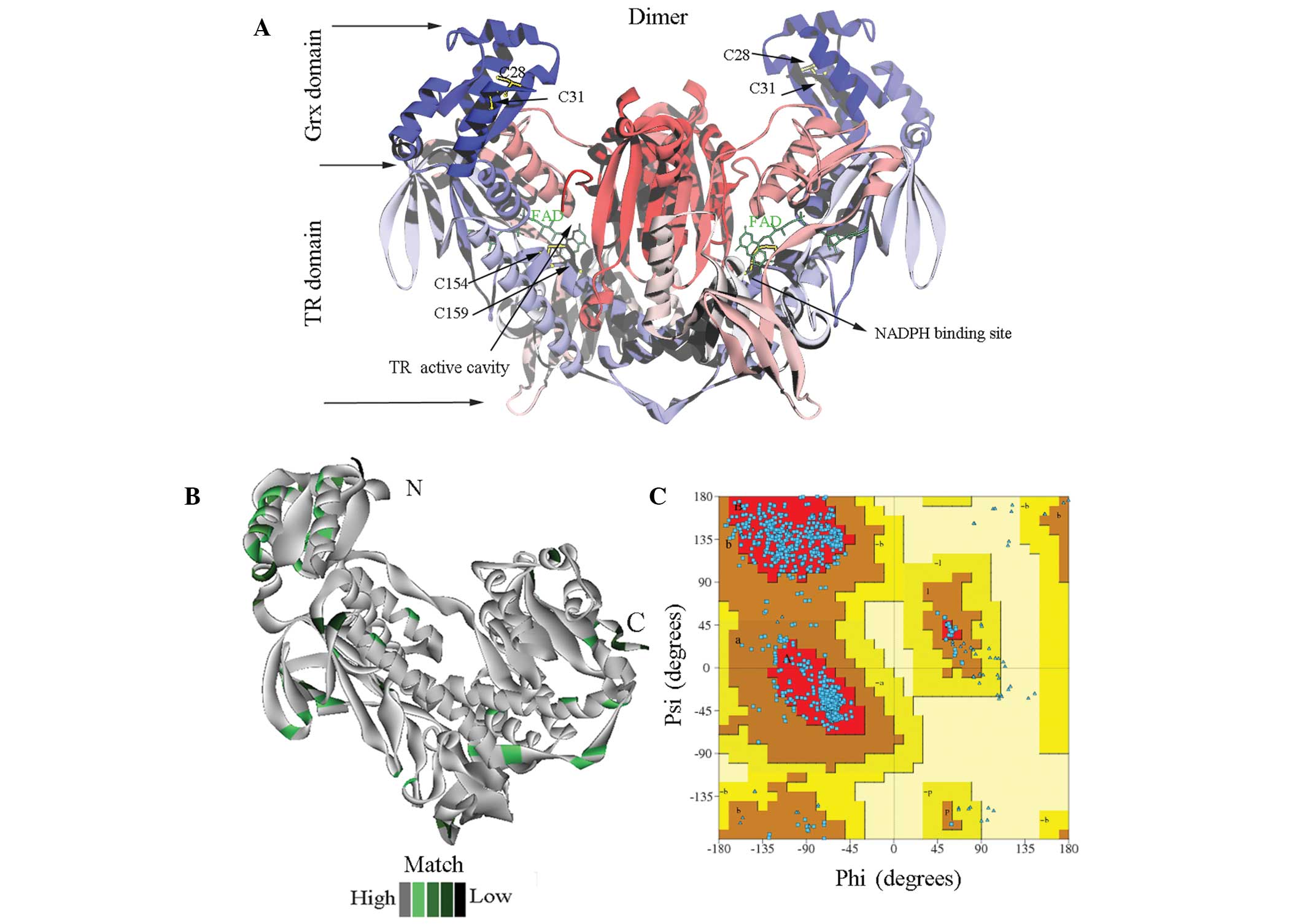
1–106) and TR (residues 107–598) (9). The active cavity of the TR domain
comprises residues from both subunits: An FAD-binding motif and a
redox-active Cys154-Cys159 pair from one subunit, and a C-terminal
domain containing a conserved redox-active four peptide fragment
tail (-Gly595′-Cys596′-Sec597′-Gly598′) from the adjacent subunit.
The NADPH binding site is located in the middle of the TR domain,
close to the FAD-binding site and the thiol/disulphide redox active
centre Cys154-Cys159.
The proposed electron flow within the TGR protein is
from NADPH to the thiol/disulphide Cys154-Cys159 pair that forms
the redox-active centre, and then to the C-terminus and, finally,
from the C-terminus to the Grx active site (Cys28–Cys31) or
thioredoxin (10,11). Three binding site cavities within
the TGRs therefore appear to be important for electron delivery: i)
The GSH binding site in Grx; ii) the NADPH binding site and iii)
the TR active cavity, which contains the FAD-binding site and a
redox-active Cys154-Cys159 pair from one subunit, and a
redox-active C-terminus from the other subunit. Inhibitors
occupying these sites may disrupt electron delivery within the TGR
proteins.
It has been reported in studies by Doenhoff et
al (4), Simeonov et al
(12) and Sayed et al
(13) that several compound
groups, including oxadiazole 2-oxides, phosphinic acid amides,
isoxazolones and phosphoramidites, inhibit smTGR.
4-Phenyl-1,2,5-oxadiazole-3-carbonitrile-2-oxide (termed compound 1
in the present study) has been found to inhibit both the TR and GR
activities of smTGR in the low nanomolar range and has also been
shown to be active against TGR from Schistosoma japonicum
(sjTGR) (13). The binding sites
of these prototype inhibitors of TGR, however, remain unclear.
To explore how these inhibitors interact with the
TGRs, we docked six compounds from the four groups described above
into the Grx domain, the NADPH-binding site and the TR active site
cavity of sjTGR and smTGR using AutoDock 4.2.5.1. (The Scripps
Research Institute, La Jolla, CA, USA) (14). Knowledge of the possible binding
sites is likely to enhance the understanding of the inhibitory
mechanisms and facilitate the development of new
anti-schistosomiasis drugs.
Materials and methods
Homology modelling of sjTGR
The three-dimensional (3D) structure of sjTGR has
not yet been elucidated; however, since the sequence homology
between sjTGR and smTGR is 89%, the sjTGR structure can be modeled
on that of smTGR, for which an X-ray structure has been
determined.
The amino acid sequence of sjTGR was extracted from
UniProtKB/TrEMBL (B5THG7), and the smTGR X-ray structure [Protein
Data Bank (PDB) ID: 2X99, resolutions 2.3 Å] (9) was used as a template. Homology
modelling of the sjTGR subunit was performed using Discovery Studio
2.5 (Accelrys Software, Inc., San Diego, CA, USA). The sjTGR dimer
was assembled using PISA (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html)
(15).
Protein structure analysis and graphical display
were performed using Discovery Studio View 4.0 (Accelrys Software,
Inc.). The derived protein structure was then evaluated using the
web-based software PROCHECK (http://www.ebi.ac.uk/pdbsum/) (16,17).
Chemistry
The six inhibitor molecules selected for this study
could be divided into four groups: The oxadiazole 2-oxides
(compounds 1–3), a phosphinic acid amide (compound 4), an
isoxazolone (compound 5) and a phosphoramidite (compound 6)
(Table I). Two-dimensional
structures of all drug molecules were sketched using ISIS Draw 2.5
(MDL Information Systems, Inc., San Leandro, CA, USA) and converted
into optimised 3D structures using Discovery Studio 2.5 (Accelrys
Software, Inc.). All six compounds have been reported to inhibit
smTGR (4,12,13),
and 4-phenyl-1,2,5-oxadiazole-3-carbonitrile-2-oxide (compound 1)
has also been reported to inhibit sjTGR in vitro (4,12).
 | Table ISelected inhibitors for docking. |
The native substrates, glutathione (GSH), oxidised
glutathione (GDS) and NADPH, were also docked into the
corresponding sites as controls. GDS, the specific substrate of GR,
was extracted from human GR (PDB ID: 2GRT) (18). GSH and NADPH were extracted from
smTGR (PDB ID: 2X99) (9).
Molecular docking
Molecular docking was performed using AutoDockTools
4.2.5.1 (The Scripps Research Institute) (14). The AutoDockTools graphical
interface AutoDockTools 1.5.6 was used to preserve polar hydrogen
atoms and to add partial charges to the proteins using Gasteiger
charges (14). AutoGrid was used
to generate the grid maps. Each grid was centred at the binding
site of the protein. The grid dimensions were 40 points in each
dimension separated by 0.375 Å (except NADPH for 40×40×60). For all
ligands, random starting positions, orientations and torsions were
used. Default values in AutoDock were used for the translation,
quaternion and torsion steps. The Genetic Algorithm was used with
2,500,000 energy evaluations and a population of 300 individuals;
100 runs were carried out. Following docking, the results were
clustered into groups with root-mean-square deviation <2.0 Å.
The ranking of the clusters was performed based on the lowest
energy representative of each cluster. Interactive visualisation
and analysis of molecular structures and interactions between
protein and ligand were performed using Discovery Studio View 4.0
software (Accelrys Software, Inc.)
Results and Discussion
Homology modelling and structure
evaluation
There is 89% identity between the template smTGR
(smTGR 2×99) and the sjTGR sequences. Due to the fact that the
template sequence lacks the N- and C-terminal SeC tail peptide
fragments, the model of sjTGR that was used contained only residues
6–592 (Fig. 1A). The coincidence
degree between the sjTGR model and the template is shown in
Fig. 1B. The shades of the
colours, from dark green to grey, represent the model's degree of
coincidence, from low to high. The unmatched region was mainly
located at the C-terminal; other regions of the model showed high
coincidence with the template (Fig.
1B). The rationality of the stereochemistry in the newly
constructed model was evaluated using PROCHECK. The ϕ-ψ plot of the
sjTGR dimer is shown in Fig. 1C.
With the exception of the Gly and Pro residues, there were 916
amino acids (89.5%) in the most favoured regions and 104 amino
acids (10.2%) in additional allowed regions. G-factor values of all
dihedral angles were >−0.5, which suggested that the overall
structure was reasonably good (Fig.
1C).
Molecular docking
Six compounds, belonging to four different groups,
were docked separately into the Grx and TR active sites and the
NADPH binding site. The free energy of binding, comprising van der
Waals, electrostatic and hydrophobic interactions and solvation
energy, was used to evaluate the binding affinity. The docked
conformations were clustered by energy and the conformation with
the lowest free binding energy was selected for further analysis.
As controls, GSH (a native substrate of the Grx domain), GDS (a
native substrate that can bind in the GR/TR active site) and NADPH
were also docked/redocked into the appropriate sites for
comparison.
Native substrates in the corresponding
sites
The structure of a TGR in complex with a
substrate/inhibitor in the thiol/disulphide redox active pocket (TR
active cavity) has not yet been determined. The TR domain of TGR
has similarities with TRs and GRs and exhibits TR and GR enzymatic
activities. Since the GDS binding pocket close to the FAD-binding
site (thiol/disulphide redox active centre) has been proposed to
play a major role in the reduction of GDS in GR, TR and smTGR
(19,20), GDS was docked into the TR active
cavity of smTGR and sjTGR, based on the structure of the human
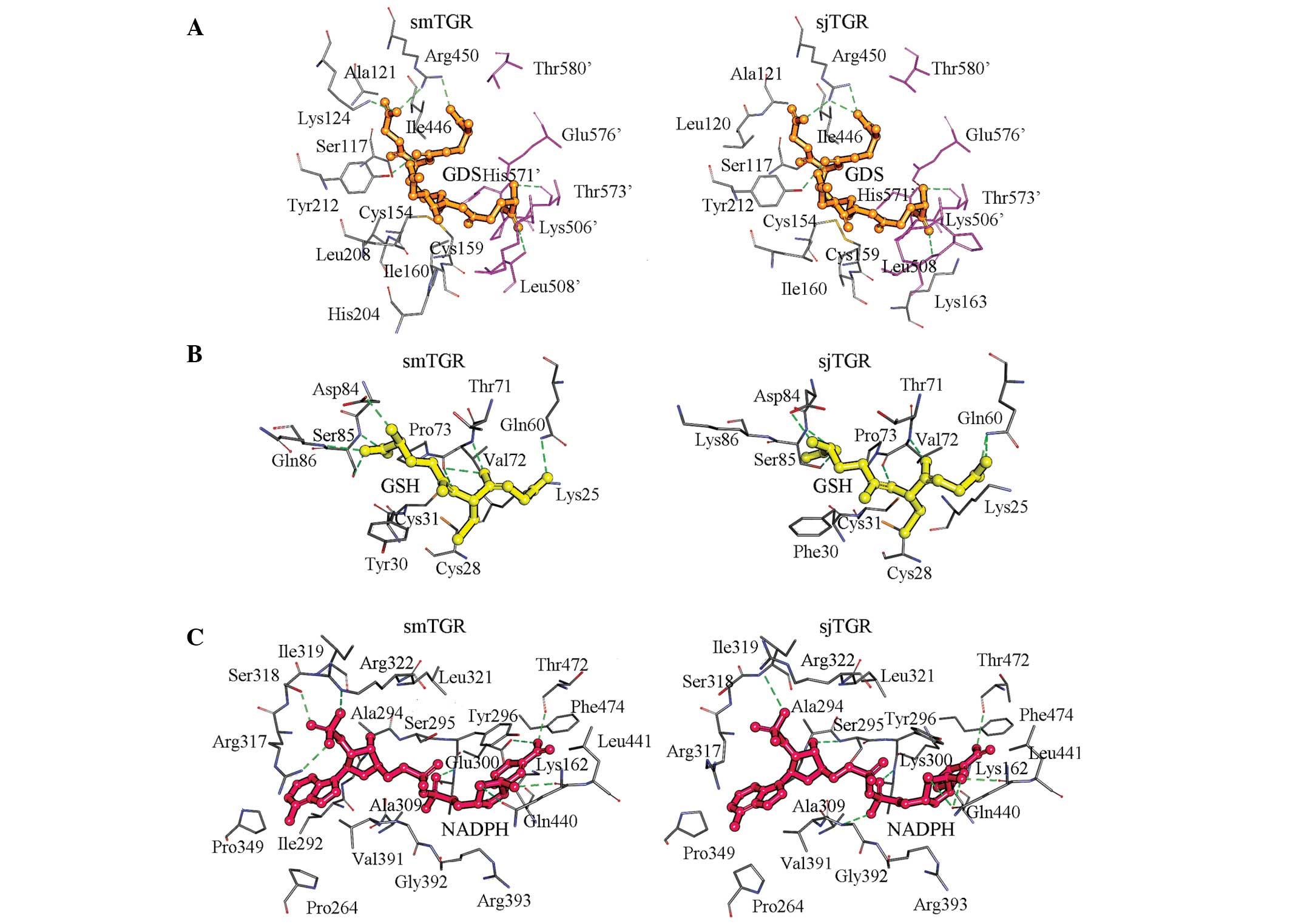
GR-GDS complex (PDB ID: 2GRT) (18). It was found that GDS was embedded
into the dimer interface, near the FAD-binding site and the
disulphide motif (Cys154-Cys159). The binding site for GDS in the
TR active cavity of TGR was observed to be composed of residues
from two subunits. In smTGR, GDS was surrounded by Ser117, Ala121,
Lys124, Val155, Ile160, Lys163, His204, Leu208, Tyr212, Ile446 and
Arg450 from one subunit and Lys506′, Leu508′, His571′, Pro572′,
Thr573′, Cys574′, Glu576′, Thr577′ and Thr580′ from another
subunit. As observed in docking results reported by Sharma et
al (20), GDS in the active
site formed hydrogen bonds with Tyr212, Lys124 and Arg450, residues
that are also conserved in GR. GDS also formed hydrogen bonds with
Leu508′, His571′, Thr573′ and Glu576′ from the other subunit.
In sjTGR, GDS occupies the same position as in
smTGR, except that Leu120 replaces the Lys124 that interacts with
GDS in smTGR (Fig. 2A). The
remaining residues that form contacts with GDS in sjTGR are the
same as those described for smTGR. The estimated free energy of
binding of GDS to smTGR is −2.80 kcal/mol, compared with −3.39
kcal/mol for binding to sjTGR (Tables
II and III), indicating that
GDS binding to sjTGR is more stable than GDS binding to smTGR.
| Table IIEstimated free energy of binding of
drugs and native substrates with thioredoxin glutathione from
Schistosoma mansoni. |
Table II
Estimated free energy of binding of
drugs and native substrates with thioredoxin glutathione from
Schistosoma mansoni.
| Ligand | Estimated free
binding energy (kcal/mol)
|
|---|
| Grx active
site | TR active
cavity | NADPH-binding
site |
|---|
| Compound 1 | −6.19 | −6.63 | −7.15 |
| Compound 2 | −5.42 | −7.75 | −7.86 |
| Compound 3 | −4.72 | −7.03 | −7.21 |
| Compound 4 | −5.80 | −7.23 | −7.34 |
| Compound 5 | −6.14 | −7.56 | −8.46 |
| Compound 6 | −6.68 | −7.66 | −7.02 |
| GDS | | −2.80 | |
| GSH | −4.14 | | |
| NADPH | | | −10.17 |
| Table IIIEstimated free binding energy of
drugs and native substrates with thioredoxin glutathione from
Schistosoma japonicum. |
Table III
Estimated free binding energy of
drugs and native substrates with thioredoxin glutathione from
Schistosoma japonicum.
| Ligand | Estimated free
binding energy (kcal/mol)
|
|---|
| Grx active
site | TR active
cavity | NADPH-binding
site |
|---|
| Compound 1 | −4.73 | −6.45 | −6.50 |
| Compound 2 | −3.98 | −7.04 | −7.67 |
| Compound 3 | −4.23 | −6.86 | −7.10 |
| Compound 4 | −4.57 | −6.92 | −6.81 |
| Compound 5 | −4.63 | −7.72 | −6.48 |
| Compound 6 | −4.55 | −7.44 | −6.35 |
| GDS | | −3.39 | |
| GSH | −3.17 | | |
| NADPH | | | −6.59 |
One reduced glutathione (GSH) was found to bind in
the active site of the Grx domain of the smTGR template (PDB ID
2×99) (9), surrounded by three
segments of the polypeptide, residues Lys25-Tyr30, Thr71-Pro73 and
Asp84-Glu86. It was also found that Gln60 could form a hydrogen
bond with GSH. As a control, GSH was re-docked into the Grx domain
of smTGR and it was found that the docked molecule completely
overlapped with the GSH conformation observed in the X-ray
structure. As with smTGR, GSH in sjTGR also made contacts with the
conserved residues Lys25, Cys28, Gln60, Thr71, Val72, Pro73, Asp84
and Ser85 (Fig. 2B). In sjTGR,
residues Phe30 and Lys86 replace Tyr30 and Gln86 of smTGR,
respectively. These replacements resulted in an increase in free
binding energy from −4.14 kcal/mol in smTGR to −3.17 kcal/mol in
sjTGR (Tables II and III), indicating that the Grx domains of
the two TGRs have different binding capacities for GSH.
NADPH functions as an electron donor in the electron
transport chain of TGRs. An X-ray structure of smTGR in complex
with NADPH (9) showed the NADPH
was surrounded by residues Lys162, Pro264, Ile292-Ala298, Glu300,
Met315, Arg317-Ile319, Leu321, Arg322, Pro349, Glu390-Arg393,
Gln440, Leu441, Thr472 and Phe474. The present docking results
showed that NADPH could re-dock into its initial position with an
estimated free binding energy of −10.17 kcal/mol. In sjTGR, the
docked NADPH interacts with the conserved residues Lys162, Pro264,
Gly293-Ala298, Lys300, Arg317-Ile319, Leu321, Arg322,
Ala390-Arg393, Gln440, Leu441 and Thr472-Phe474 (Fig. 2C). Although the NADPH binding site
appears to be conserved among TGRs, the estimated free binding
energy with sjTGR (−6.59 kcal/mol) was significantly higher than
that with smTGR.
The present data suggested that sjTGR and smTGR have
different binding capacities for their native substrates, despite
the fact that they share high sequence identity and conserved
residues in their binding sites. Our previous kinetic analysis
(21) showed that the Km value for
sjTGR with GDS (49.55 µM) was lower than that for smTGR
(71.5 µM), whereas the Km values for sjTGR with NADPH
(21.426 µM) and GSH (1,698 µM) were higher than those
for smTGR (13.7 and 248.6 µM, respectively). This indicates
that sjTGR has a higher binding capacity for GDS than does smTGR,
while the binding capacities of sjTGR for GSH and NADPH were lower
than those of smTGR, in agreement with our conclusion. These
results are concordant with those of the present study, which
suggest that sjTGR and smTGR have different binding capacities for
their native substrates, despite sharing high sequence identity and
conserved residues in their binding sites.
TR active site cavity as the effective
binding site for inhibitors
Although the six selected compounds inhibit smTGR
in vitro (4,12,13,22),
the mechanism by which that happens remains unknown. The present
study has focused on whether these molecules can directly gain
access to the three main functional sites of TGR. The binding
scores of the best-docked conformations of ligands in smTGR and
sjTGR are listed in Tables II and
III, respectively.
In smTGR, the NADPH-binding site and TR active
cavity (GDS binding site) exhibit lower estimated free binding
energies for the compounds than does the Grx domain (Table II). The free binding energy shown
by one of the controls, NADPH, was much lower than the free binding
energy shown by all the test compounds.
In sjTGR, the NADPH-binding site and the TR active
cavity exhibited better binding capacities for the test compounds
than the Grx domain (Table III).
In each site, the binding capacity of sjTGR for the test compounds
was generally marginally weaker than that of smTGR, apart from
compound 5, which exhibited a better free binding energy for the TR
active cavity of sjTGR. The most marked differences in binding
capacities were observed in the Grx active site, with an increase
of 1.21 kcal/mol in average binding energy for each test
compound.
It is not sufficient, however, to estimate the free
binding energy of single test molecules alone. In order to clarify
whether compounds can competitively inhibit the enzymatic activity
of TGR, the free binding energy between test molecules and native
substrates was compared. The ratios of the free binding energies of
inhibitors to those of native substrates (docking ratios) were used
to evaluate the inhibitory efficiency of the compounds.
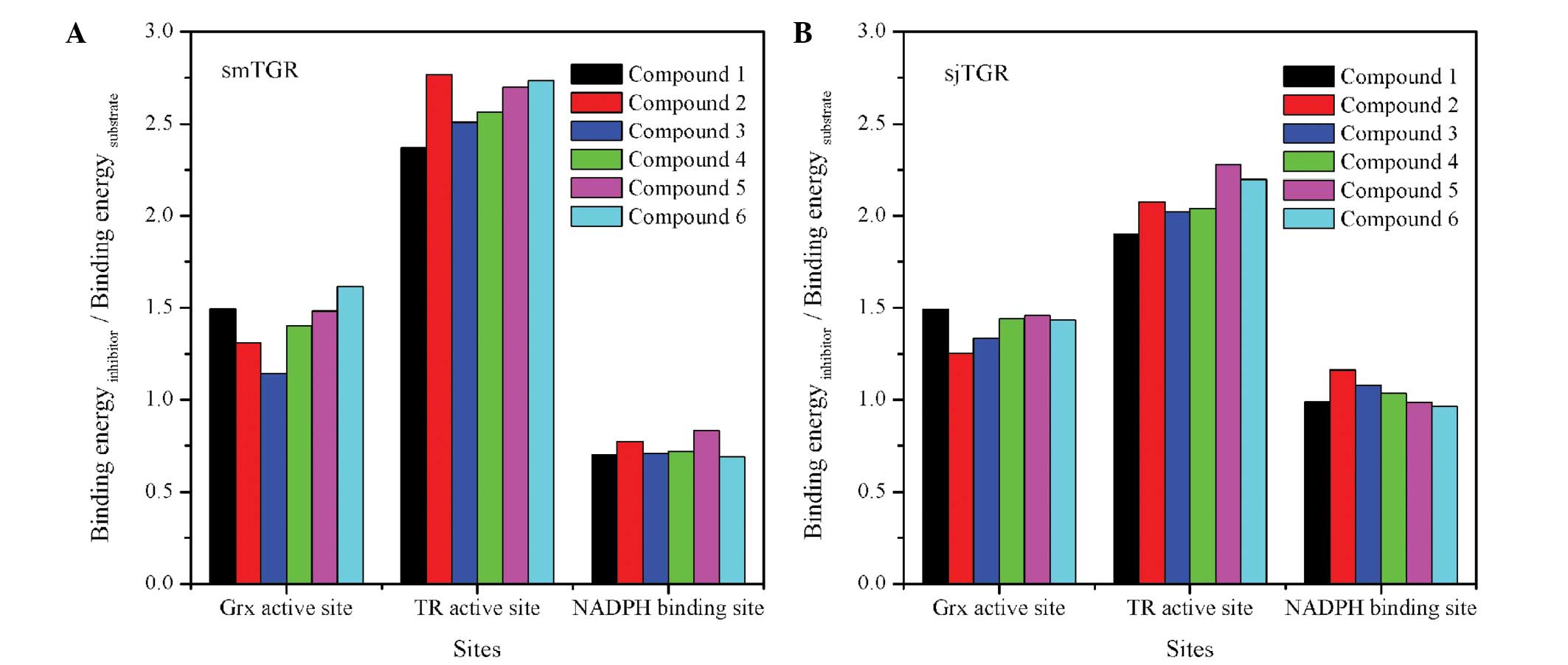
The docking ratios of the test molecules in smTGR
are shown in Fig. 3A. All docking
ratio values in the TR active cavity were >2.3, higher than
those in the Grx domain or the NADPH-binding site. For sjTGR, the
values of the docking ratio in the TR active centre were >1.9
(Fig. 3B). These values were
generally lower than those for smTGR, indicating that the compounds
were less effective sjTGR inhibitors. The data suggested that it is
necessary to design effective inhibitors for the specific TGRs from
different Schistosoma species.
The docking ratios in the TR active cavity of sjTGR
and smTGR were both higher than those in the Grx domain or the
NADPH-binding site, suggesting that these compounds may exert their
inhibitory effects mainly through interactions in the TR active
cavity, with smaller effects on the Grx domain and NADPH-binding
site. The TR active cavity may thus be the favoured binding site
for these compounds, which may effectively prevent the native
substrate from accessing this binding site and disrupting the
electron transfer in the TGRs. The following section focuses on the
binding mode of compounds in the TR active cavity.
Binding mode of compounds in the active
site of the GR/TR domain
Oxadiazole 2-oxides
Simeonov et al (12) identified 39 molecules with an
IC50 <10 µM from a quantitative
high-throughput screen (qHTS) of 71,028 compounds. The
IC50 values of the oxadiazole 2-oxides (compounds 1 and
2) against smTGR in the initial qHTS test were 8.7 and 9.2
µM, respectively, although compound 2 was much more active
(IC50=0.02 µM) in the TGR assays than compound 1
(IC50=1.8 µM) (11). According to Sayed et al
(13), however, the
IC50 values of compounds 1, 2 and 3 against GR activity
were 0.32, 0.51 and 12.5 mM, respectively.
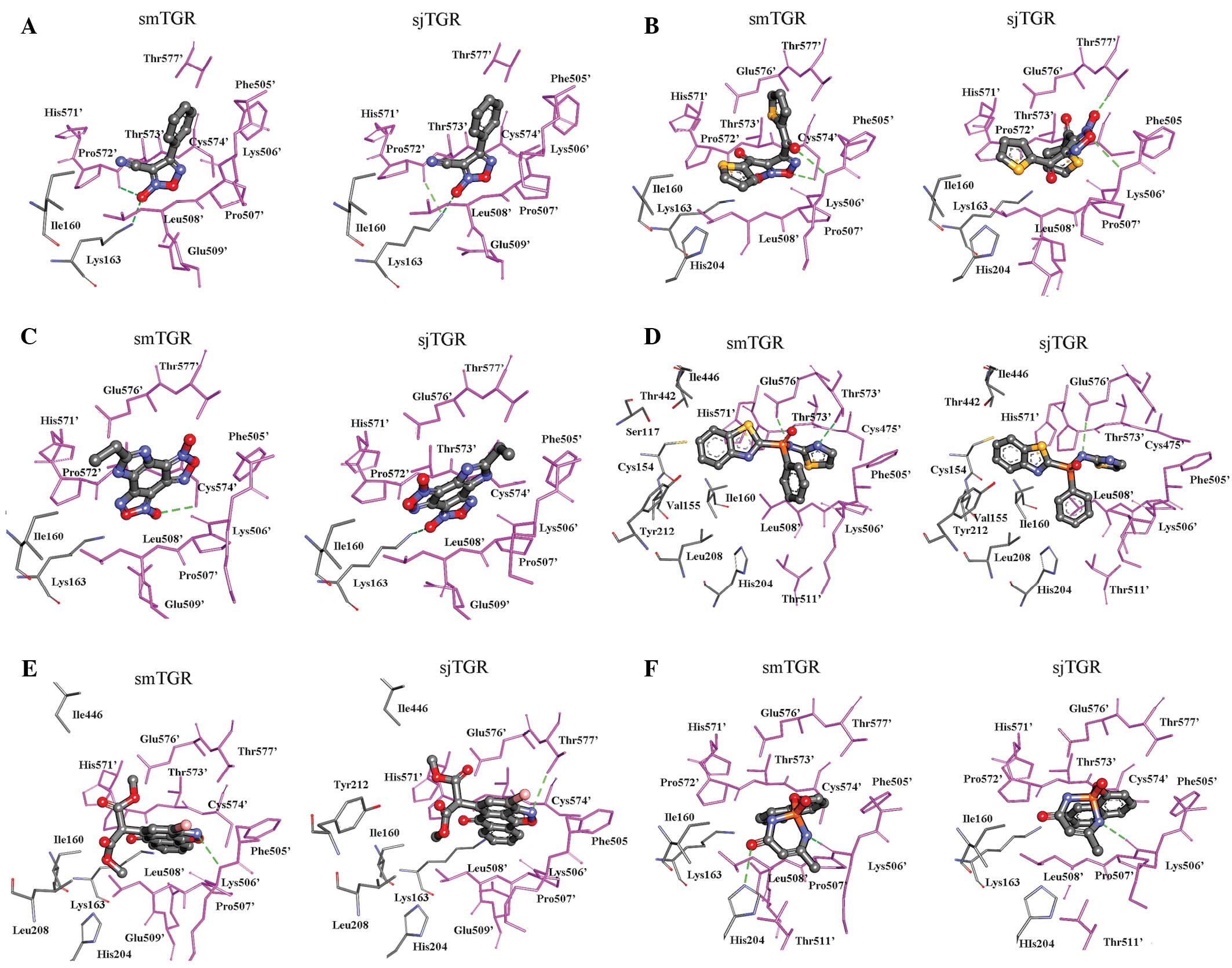
The present docking results showed that the
oxadiazole 2-oxides occupy a similar position at the dimer
interface in the two TGRs (Fig.
4A–C), making contacts with Ile160, Lys163 His204 (from one
subunit) and Phe505′, Lys506′, Leu507′, Glu508′, His571′-Cys574′
and Thr577′ (from another subunit).
It was found that compound 1 adopted the same
conformation in both smTGR and sjTGR (Fig. 4A) and had similar free binding
energies (6.63 and 6.45 kcal/mol, respectively). The cyano group
was in close proximity to Pro572′ (both 2.8 Å in the two TGRs), the
N-atom of the oxadiazole formed a hydrogen bond with Leu508′ (both
3.0 Å) and the phenyl ring made an arene-H interaction with Lys506′
(both 4.2 Å). These results indicate that compound 1 may also
inhibit sjTGR and are consistent with the conclusion of Sayed et
al (13) that this compound is
also active against Schistosoma japonicum.
Compound 2 has two large acetylthiophene groups in
the oxadiazole ring. In smTGR, the carbonyl O of one
acetylthiophene group and the O-atom of the oxadiazole formed
hydrogen bonds with Lys506′ and Cys574′, respectively, stabilising
its conformation in the active site. The best docking conformation
of compound 2 in sjTGR, however, was different from that in smTGR.
The O-atom of the oxadiazole ring formed a hydrogen bond with
Lys506′ and N-O with Thr577′ of sjTGR (Fig. 4B).
The compound 3 molecule exists in a rigid plane. It
was found to bind in the same position at the dimer interface of
the TGRs by interacting with the Leu160 and Lys163 residues and
peptide fragments 505′–508′ and 571′–577′ in the other chains
(Fig. 4C). We cannot explain why
Sayed et al (13) found
compound 3 to be less active against GR and TR than compound 1,
since the free binding energy of compound 3 was better than that of
compound 1 in both TGRs.
Phosphinic acid amide
N-(benzothiazol-2-yl-phenyl-phosphoryl)-1,3-thiazol-2-amine
(compound 4) has been identified as an inhibitor of smTGR and has
also been found to be effective against the parasite (11,12).
Simeonov et al (12)
indicated that the role of the benzothiazole group is critical for
the inhibitory activity.
In the present case, the phosphinic acid amide
accessed the same pocket as the oxadiazole 2-oxides (Fig. 4A–C). Both in smTGR and sjTGR, the
phosphinic acid amide formed hydrogen bonds with the carboxyl group
of Glu576′. The benzothiazole ring reached out to the redox couple
(Cys154-Cys159) and was surrounded by Cys154, Val155, Ile160,
Leu208, Thr442 and Ile446. Two peptide segments of the TGRs,
505′–508′ and 572′–577′, stabilised the thiazole group of compound
4 in the TGRs through van der Waals forces and hydrophobic
interactions (Fig. 4D).
Isoxazolone
Compound 5, the isoxazolone dimethyl
2-[3-bromo-6-oxo-6H-anthra (1,9-cd)isoxazol-5-yl] malonate, adopted
a similar conformation in both smTGR and sjTGR. The binding pocket
for isoxazolone is mainly composed of residues from two subunits
and involves residues 160–163, 204, 208, 212, 442, 446, 505′–509′
and 571′–577′ (Fig. 4E). The
ligand is stabilised in the active site of the proteins, mainly
through van der Waals forces and hydrophobic interactions. In the
TGRs, the isoxazolone exhibited a high binding score, indicating
that it may be an effective inhibitor. This finding is in agreement
with the data presented by Simeonov et al (12), in which the isoxazolone exhibited a
low IC50 against smTGR in the qHTS. The isoxazolone
exhibited the lowest free binding energy of all the compounds with
sjTGR and its binding score for docking into sjTGR was even higher
than that for docking into smTGR, therefore suggesting that
isoxazolone may also be active against sjTGR.
Phosphoramidite
Compound 6, the phosphoramidite
6-methyl-2-(naphthalen-1-yloxy)-2,3-dihydro-1,3,2-diaza-phosphinin-4(1H)-one
2-oxide, was identified as an inhibitor of smTGR by both the qHTS
and the TGR assay (12). As with
the other compounds, the best binding site of the phosphoramidite,
both in smTGR and in sjTGR, involved Ile160, Lys163, His204,
Phe505′–Leu508′ and Pro572′–Thr577′. Lys506′ and His571′ were
within hydrogen bonding distance of the ligand (Fig. 4F). The estimated free binding
energies of the phosphoramidite were −7.66 kcal/mol with smTGR and
−7.44 kcal/mol with sjTGR (Tables
II and III). These data
suggest that the phosphoramidite may also be active against
Schistosoma japonicum.
Conclusion
In order to explore the interaction mechanism of
these compounds, six previously identified TGR inhibitors were
docked into the NADPH binding site, the TR active cavity and the
Grx active site of smTGR, as determined by the X-ray structure
(Schistosoma mansoni), and into the structure of sjTGR
(Schistosoma japonicum), as constructed by homology
modelling. The present results indicate that the favourite binding
site of inhibitors is the TR active cavity surrounded by an
FAD-binding site, a redox-active Cys154/Cys159 pair from one
subunit and a C-terminal peptide from the other subunit. Although
these inhibitors are structurally quite different, the best binding
positions of all the compounds in smTGR and sjTGR were similar in
the GDS binding site. Two peptide fragments from another subunit,
Phe505′–Leu508′ and Pro572′–Thr577′, played a critical role in the
interactions with the inhibitors. The data also suggest that the
inhibitors are generally marginally less effective against sjTGR
than against smTGR, implying that it would be necessary to design
inhibitors specific for the TGRs from different Schistosoma
species. An enhanced understanding of the binding mechanisms of
potential inhibitors of Schistosoma TGRs would facilitate
structure-based ligand design.
Acknowledgments
This study was supported by grants from the Jiangsu
Provincial Nature Science Foundation (grant no. BK2011573), the
Ministry of Science and Technology (grant nos. 2012AA020304 and
2012ZX09401012), the National Undergraduate Innovation Program
(grant no. J1103512) and the National Natural Science Foundation of
China (grant no. J1210026).
References
|
1
|
Engels D, Chitsulo L, Montresor A and
Savioli L: The global epidemiological situation of schistosomiasis
and new approaches to control and research. Acta Trop. 82:139–146.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mbanefo EC, Huy NT, Wadagni AA, Eneanya
CI, Nwaorgu O and Hirayama K: Host determinants of reinfection with
schistosomes in humans: A systematic review and meta-analysis. PLoS
Negl Trop Dis. 8:e31642014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hung YW and Remais J: Quantitative
detection of Schistosoma japonicum cercariae in water by real-time
PCR. PLoS Negl Trop Dis. 2:e3372008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Doenhoff MJ, Cioli D and Utzinger J:
Praziquantel: Mechanisms of action, resistance and new derivatives
for schistosomiasis. Curr Opin Infect Dis. 21:659–667. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang W, Dai JR, Li HJ, Shen XH and Liang
YS: Is there reduced susceptibility to praziquantel in Schistosoma
japonicum? Evidence from China. Parasitology. 137:1905–1912. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Melman SD, Steinauer ML, Cunningham C,
Kubatko LS, Mwangi IN, Wynn NB, Mutuku MW, Karanja DM, Colley DG,
Black CL, et al: Reduced susceptibility to praziquantel among
naturally occurring Kenyan isolates of Schistosoma mansoni. PLoS
Negl Trop Dis. 3:e5042009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alger HM and Williams DL: The disulfide
redox system of Schistosoma mansoni and the importance of a
multifunctional enzyme, thioredoxin glutathione reductase. Mol
Biochem Parasitol. 121:129–139. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuntz AN, Davioud-Charvet E, Sayed AA,
Califf LL, Dessolin J, Arnér ES and Williams DL: Thioredoxin
glutathione reductase from Schistosoma mansoni: An essential
parasite enzyme and a key drug target. PLoS Med. 4:e2062007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Angelucci F, Dimastrogiovanni D, Boumis G,
Brunori M, Miele AE, Saccoccia F and Bellelli A: Mapping the
catalytic cycle of Schistosoma mansoni thioredoxin glutathione
reductase by X-ray crystallography. J Biol Chem. 285:32557–32567.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Williams DL, Bonilla M, Gladyshev VN and
Salinas G: Thioredoxin glutathione reductase-dependent redox
networks in platyhelminth parasites. Antioxid Redox Signal.
19:735–745. 2013. View Article : Google Scholar :
|
|
11
|
Prast-Nielsen S, Huang HH and Williams DL:
Thioredoxin glutathione reductase: Its role in redox biology and
potential as a target for drugs against neglected diseases. Biochim
Biophys Acta. 1810:1262–1271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simeonov A, Jadhav A, Sayed AA, Wang Y,
Nelson ME, Thomas CJ, Inglese J, Williams DL and Austin CP:
Quantitative high-throughput screen identifies inhibitors of the
Schistosoma mansoni redox cascade. PLoS Negl Trop Dis. 2. pp.
e1272008, View Article : Google Scholar
|
|
13
|
Sayed AA, Simeonov A, Thomas CJ, Inglese
J, Austin CP and Williams DL: Identification of oxadiazoles as new
drug leads for the control of schistosomiasis. Nat Med. 14:407–412.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morris GM, Huey R, Lindstrom W, Sanner MF,
Belew RK, Goodsell DS and Olson AJ: AutoDock4 and AutoDockTools4:
Automated docking with selective receptor flexibility. J Comput
Chem. 30:2785–2791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krissinel E and Henrick K: Inference of
macromolecular assemblies from crystalline state. J Mol Biol.
372:774–797. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
de Beer TA, Berka K, Thornton JM and
Laskowski RA: PDBsum additions. Nucleic Acids Res. 42:D292–D296.
2014. View Article : Google Scholar :
|
|
17
|
Laskowski RA, Hutchinson EG, Michie AD,
Wallace AC, Jones ML and Thornton JM; PDBsum: A Web-based database
of summaries and analyses of all PDB structures. Trends Biochem
Sci. 22:488–490. 1997. View Article : Google Scholar
|
|
18
|
Stoll VS, Simpson SJ, Krauth-Siegel RL,
Walsh CT and Pai EF: Glutathione reductase turned into
trypanothione reductase: Structural analysis of an engineered
change in substrate specificity. Biochemistry. 36:6437–6447. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Angelucci F, Miele AE, Boumis G,
Dimastrogiovanni D, Brunori M and Bellelli A: Glutathione reductase
and thioredoxin reductase at the crossroad: The structure of
Schistosoma mansoni thioredoxin glutathione reductase. Proteins.
72:936–945. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sharma M, Khanna S, Bulusu G and Mitra A:
Comparative modeling of thioredoxin glutathione reductase from
Schistosoma mansoni: A multifunctional target for antischistosomal
therapy. J Mol Graph Model. 27:665–675. 2009. View Article : Google Scholar
|
|
21
|
Song L, Li J, Xie S, Qian C, Wang J, Zhang
W, Yin X, Hua Z and Yu C: Thioredoxin glutathione reductase as a
novel drug target: Evidence from Schistosoma japonicum. PLoS One.
7:e314562012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rai G, Thomas CJ, Leister W and Maloney
DJ: Synthesis of oxadiazole-2-oxide analogues as potential
antischistosomal agents. Tetrahedron Lett. 50:1710–1713. 2009.
View Article : Google Scholar : PubMed/NCBI
|