Introduction
Retinitis pigmentosa (RP) is a classical inherited
eye disorder, which predominantly involves damage to the function
and structure of the rod and cone cell photoreceptors and the
retina pigment epithelium (1).
Individuals affected by RP usually experience night blindness from
the early stage of R P, accompanied or followed by the loss of
peripheral visual field (1). The
typical signs usually present as bone spicule-like pigmentation
deposits and a reduced or absent electroretinogram. There are
several different genetic defects that could lead to the
degeneration of cone or rod cells or to degeneration of the
connections between these cells. To date, >70 genes have been
identified and seven loci have been mapped for R P. The majority of
these genes encode proteins, which are involved in a wide variety
of cellular processes, including phototransduction, transcriptional
regulation and membrane structure formation. Progress in gene
discovery and mutation screening in individuals affected by RP and
their families has contributed to personalized treatments for R P,
which uses various novel treatment approaches, including stem
cells, gene therapy and nutritional supplementation (2–4).
The LPCAT1 gene is located on chromosome 5
and it encodes a 65 kDa protein, which contains three putative
transmembrane domains (5). This
enzyme, lysophosphatidylcholine acyltransferase 1 (LPCAT1), is
involved in lysophosphatidylcholine (LPC) and lipid syntheses,
which are important in the formation of biological membranes,
endocytosis, signaling and neuroprotection. The outer segment of
retinal photoreceptors contains stacks of membranous disks, which
are filled with opsin, a light-absorbing protein in the visual
transduction process (6). The loss
of phosphatidylcholine (PC) can lead to the disruption of membrane
structure and homeostasis maintenance, which can ultimately
contribute to photoreceptor cell degeneration (6). In addition, LPCAT1 has been
identified to be involved in the non-inflammatory PAF remodeling
pathway and in the progression of cancer, including like colorectal
and prostate cancer (7–9).
A previous study by Friedman et al (6) identified a single nucleotide
insertion (c.420_421insG) in exon 3 of the LPCAT1 gene in
rd11 mice, and identified a seven-nucleotide deletion
(c.14-20delGCCGCGG) in exon 1 in mice of the B6-JR2845
strain, which leads to premature truncation of the LPCAT1
protein. These two mouse strains present with typical symptoms of R
P. Dai et al demonstrated that retinal function and
structure can be rescued in rd11 mice using gene replacement
therapy (10). This further
supports the hypothesis that LPCAT1 may be a possible
candidate disease-causing gene of RP in humans. However, the
specific genetic mutations in LPCAT1, which predispose
individuals to RP remain to be elucidated. With the exception of
LPCAT1, a number of genes (Table I) have been demonstrated to cause
retinal degeneration in animal models, but not in human subjects
(11–15).
 | Table IList of RD-associated genes in an
animal model and human subjects (8–12). |
Table I
List of RD-associated genes in an
animal model and human subjects (8–12).
| Gene | Animal model
| Human
|
|---|
| Mutant | Phenotype | Mutant | Phenotype |
|---|
| LPCAT1 | Fs/Fs | RD | NS | ND |
| ARL3 | -/- | RD | NS | ND |
| TMEM218 | -/- | RD | NS | ND |
| CRB2 | -/- | RD | S | ND |
| CCDC66 | -/- | RD | NS | ND |
| CCND1 | -/- | RD | NS | ND |
There are three major patterns of inheritance in R
P, including autosomal dominant (ad), autosomal recessive (ar) and
X-linked inheritance. Of the total cases of R P, ~30% are adRP, 20%
are arRP and 15% are X-linked RP (16). The remaining cases, at least 30%,
are isolated cases, the patterns of which are difficult to
distinguish as either recessive or dominant inheritance. Since the
LPCAT1 mutations, which have been identified in mice are
homozygous, the present study hypothesized that the mode of
inheritance of this gene is either recessive or sporadic.
Therefore, the present study recruited a cohort of patients who
were diagnosed with RP and exhibited a either one of these two
types of inheritance patterns. In the present study, the
LPCAT1 gene was screened in 50 Chinese patients with R P,
and mutations in all previously investigated genes were excluded
based on a targeted exome-sequencing panel, suggesting the
existence of novel causative genes.
Materials and methods
Patient recruitment
The present study was performed in accordance with
the Declaration of Helsinki. The study was supported by the Ethics
Committee of The Eye Hospital of Wenzhou Medical University
(Division of Opthalmic Genetics, Wenzhou, China). Written informed
consent was obtained from each patient prior to commencement of the
investigation. All the patients were natives of China and the
detailed family histories of the patients were collected. The
majority of the participants were affected by isolated or simple
cases of R P, without any obvious genetic predisposition. The
diagnoses of RP were made based on the presence of symptoms,
including night blindness and impaired visual acuity, observed
typical fundus, reduced peripheral visual field and abnormal
optical coherence tomography (OCT) results. The total number of
patients enrolled in the present study was 50, which included 24
females and 26 males, aged between 5 and 65 years.
The 50 patients recruited were comprehensively
screened for mutations in all the known RP genes through targeted
exome sequencing using an Illumina HiSeq 2000 sequencer, as
described previously (17). This
was performed with the intention of identifying genetic factors
that may have predisposed these patients to R P.
DNA extraction
Total genomic DNA was extracted from the peripheral
blood (3 ml) using a Tiangen DNA Extraction kit (Tiangen Biotech,
Co., Ltd., Beijing, China) according to the manufacturer's
instructions. DNA was quantified using a Nanodrop 2000
spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Gene screening and data analysis
The primers used in polymerase chain reaction (PCR)
analysis were designed to detect mutations and polymorphisms in the
entire coding region and exon-intron boundaries of the
LPCAT1 gene in the patients with R P. The PCR reactions were
performed using a 50 µl reaction volume, which contained 100
ng genomic DNA, 2 pmol of each of the primers and 25 µl 2X
Taq PCR Master Mix (Biotake, Beijing, China). The PCR process was
performed using an ABI Veriti thermocycler (Applied Biosystems Life
Technologies, Foster City, CA, USA). With the exception of exon 1,
for which the denaturation temperature was set at 98°C, all PCR
reactions were performed for 32 cycles with a denaturation
temperature set at 95°C for 30 sec, an annealing temperature set
58°C for 30 sec, an extension step at 72°C for 40 sec and a final
elongation step at 72°C for 5 min. Sequence analyses were performed
using a Mutation Surveyor software (Softgenetics, State College PA,
USA) and the suspected variants were assessed using the
polymorphism phenotype (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/) and Mutation
Taster (http://www.mutationtaster.org/) tools to predict the
pathogenicity. Detailed information on the PCR primers used are
listed in Table II.
| Table IIPrimer sequences for the LPCAT1
exons. |
Table II
Primer sequences for the LPCAT1
exons.
| Exon | Forward (5′–3′) | Reverse (5′–3′) |
|---|
| 1 |
GGGAGGCGAGGCTTCCAG |
CTCCCTGGCCCCAGCATC |
| 2 |
TCAGCCTTGGTCAGCTGTG |
CAGAAGGGAAGGACAGATGG |
| 3 |
TGTTCCGGAGTCTCATGTTG |
CACTCATTTCCAGCAAGTGG |
| 4 |
CCTGGGGCATCTGGAGTC |
TGAGTTCACGCACTCACTAGG |
| 5 |
CGTACTGAATATGATCCCAGTGTC |
TCTAAGAACCCCAGCACACAG |
| 6 |
CTGGGTGTTTACGGTCAGGA |
CTGTCCTGCCCTCTCCAG |
| 7 |
CTGACTGACCCTGCCTCTTC |
ACAATGGGCTGAACCTAACG |
| 8 |
CGTGGGAGCGTTGACTG |
CTGAGAAACGGAAAGATGGG |
| 9 |
TGAAGACGGTTCTAATGGGC |
ACATGCATGAAGCTGGTTCC |
| 10 |
ATCGGCGGTTATTCTGGTG |
TGCTCAAGGAAGAAGAACCA |
| 11 |
TTCTTGAAAACTAGCTTGCTGC |
TGGGTGGTTTTCCTTCTCTG |
| 12 |
TCCTTCCAAGATTCCCTTTTC |
TGGGCATTTTACAAACAACAG |
| 13 |
CCACATGGAAGTTCGAGTCC |
AGAACCTTCCTTCTCAGGGG |
| 14 |
ACGATTCTAACCCTCCCTGG |
CTGGAACTCGGGCTGAAGAC |
Gene expression of LPCAT1 in various
tissues and the developmental retina
A three-month-old female C57BL/6 mouse was supplied
by the Animal Resource Center at Wenzhou Medical University, where
it was fed a standard chow diet and kept under standard conditions.
Animal care and husbandry followed the Association for Research in
Vision and Ophthalmology (ARVO) guidelines (http://www.arvo.org/Journals_and_Publications/Toolkit_for_Biomedical_Researchers_Using_Laboratory_Animals/)
and the mouse was sacrificed by cervical dislocation. Mouse total
RNA was prepared from various tissues, including whole brain,
retina, lens, sclera, cornea, spinal cord, heart, lung, pancreas,
testis, epencephal, vascular and skeletal muscle, and spleen. Total
RNA was extracted from different tissues of mouse, then cDNA was
synthesized using for semi-quantitative PCR. Retinal RNA at various
stages of development were also extracted using the Tiangen RNA
Extraction kit (Tiangen). A 354 bp fragment of LPCAT1 was
generated by reverse transcription using the Tiangen RT-PCR kit
(Tiangen) with the following primers: Forward
5′-GACTCGCGAAGGAAGACAGT-3′ and reverse 5′-CATGACACGCCTCACATTGC-3′.
The RNA was also used as a template for PCR, using β-actin primers
as a control.
Results
Phenotype determination
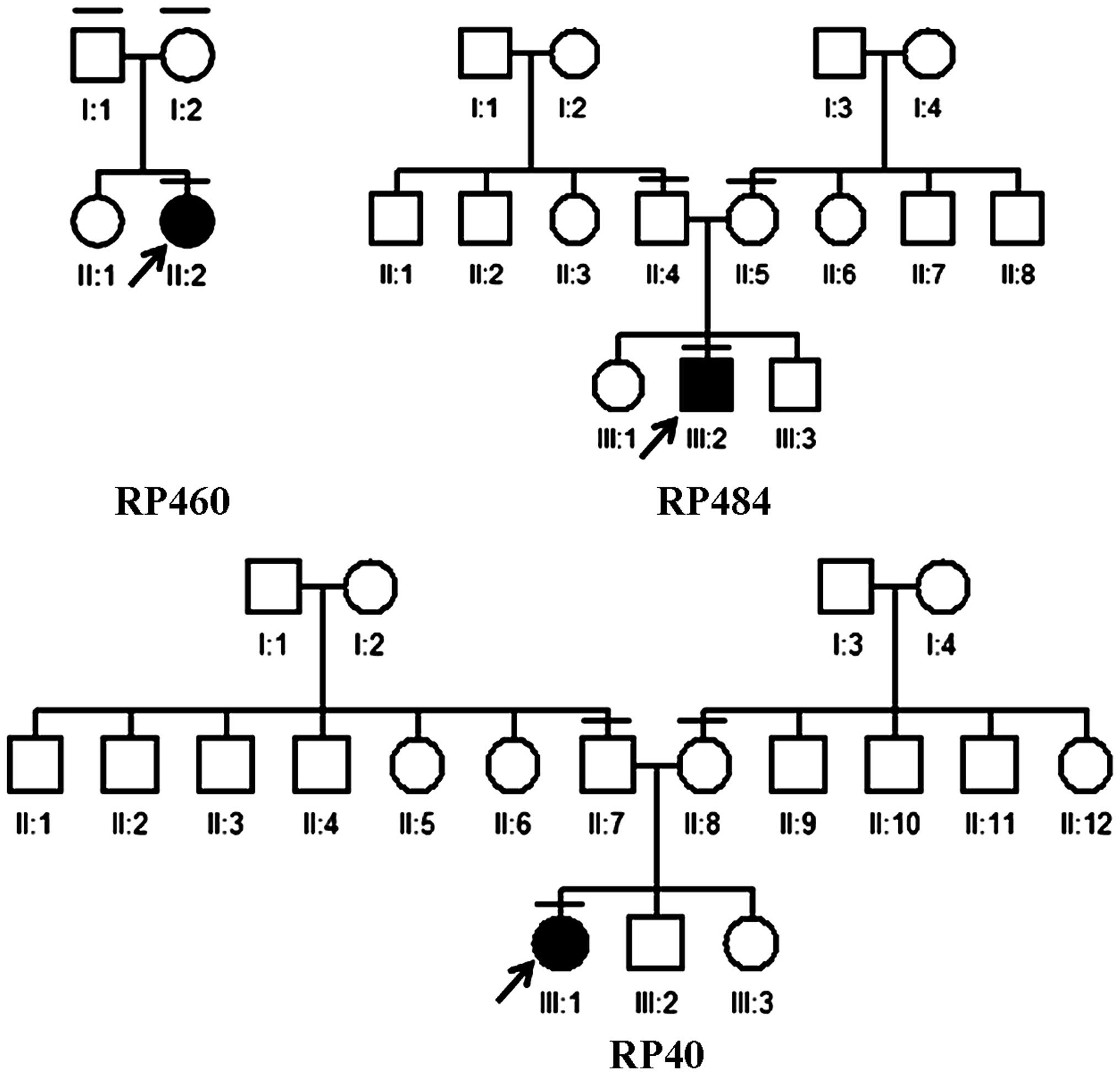
All patients recruited in the present study were
diagnosed with either recessive or sporadic RP (Fig. 1). The ages of disease onset varied
substantially, between childhood and late adult life. All patients
reported experiencing typical RP signs and symptoms, including
night blindness, visual field constriction, visual impairment, bone
spicule-like pigmentation, artery attenuation and waxy pallor of
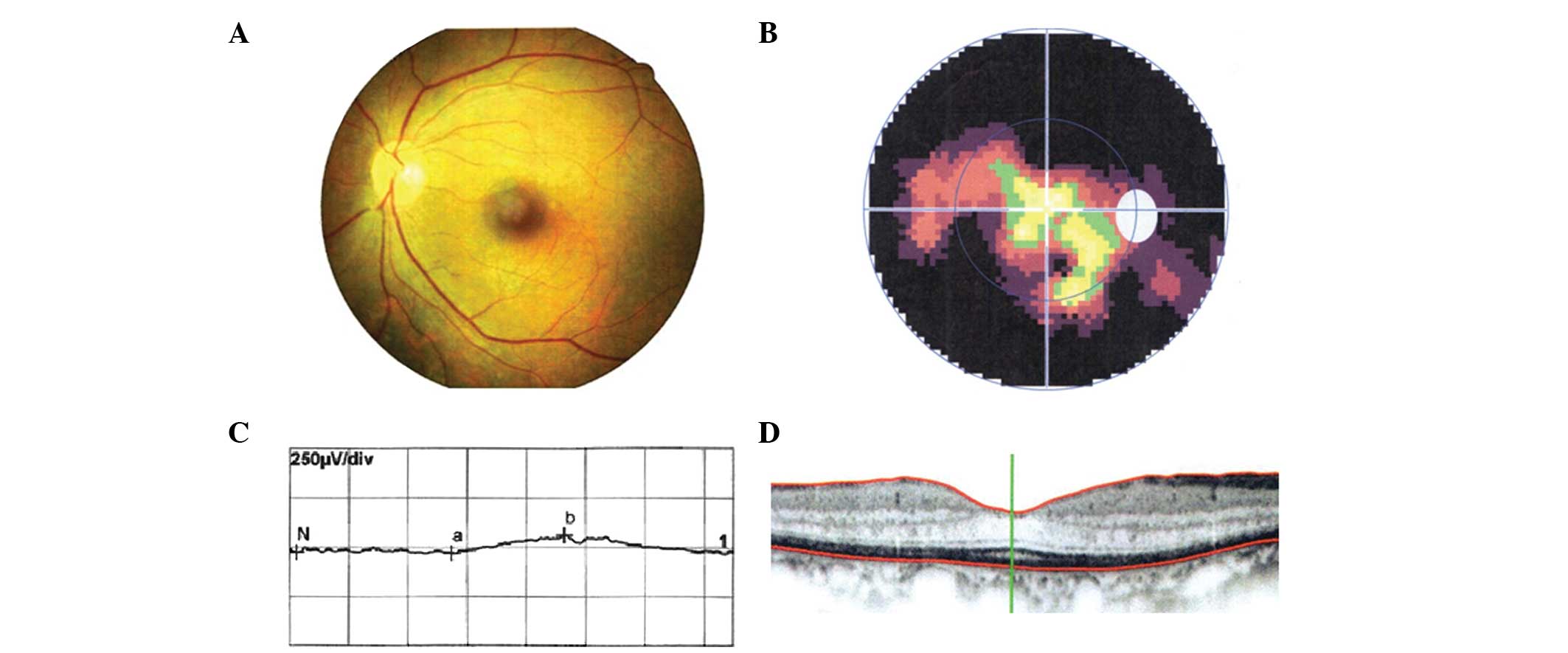
the optic nerve head in the fundi. Fig. 2 shows the representative clinical
results of patient RP40-III:1, which was from the family RP40.
Mutation screening of LPCAT1
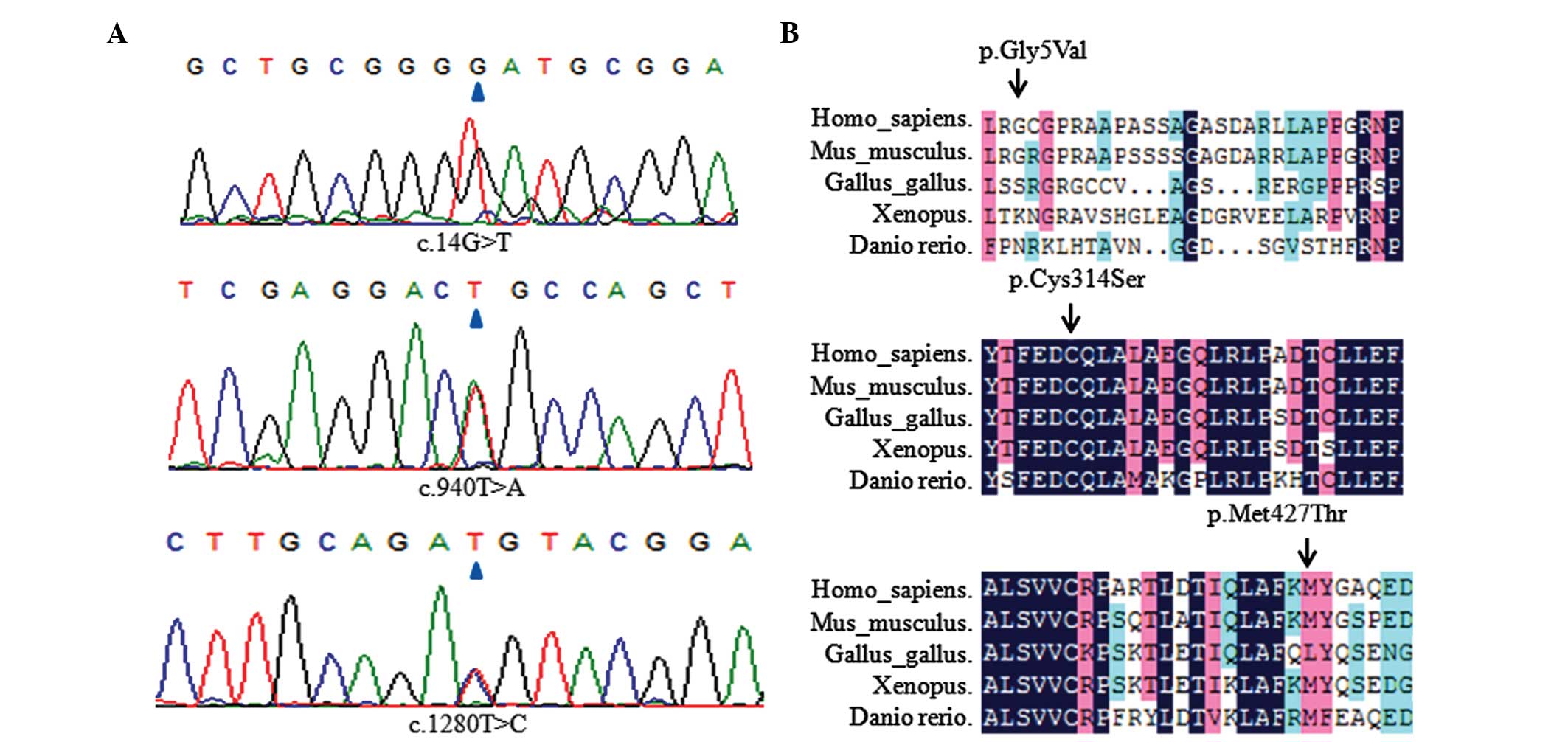
The results obtained from direct sequencing were
analyzed, from which three heterozygous missense variants
(p.Gly5Val, p.Met427Thr and p.Cys314Ser) and four synonymous
variants (c.399G>A, c.645A>C, c.657C>A and c.1365C>T)
were identified in the LPCAT1 gene in the patients. Of these
variants, two were predicted to be pathogenic, according to the
results of the computational prediction performed using PolyPhen-2
and Mutation Taster.
Co-segregation analysis
For the three possible heterozygous missense
mutations, their grade of conservation was analyzed and
co-segregation analysis was performed in each pedigree (Fig. 3). The three heterozygous mutation
sites in the parents of these patients were also screened. The
results demonstrated that the mutations were inherited from either
the paternal or the maternal allele. Since their parents did not
suffer from R P, the heterozygous variants were not considered to
predispose an individual to R P. Overall, no definite pathogenic
variant was identified in the LPCAT1 gene. In addition, the
frequencies of these three single nucleotide polymorphisms were
observed to be particularly high (>0.01)in the patients examined
(Table III).
| Table IIISummary of variants in patients with
retinitis pigmentosa. |
Table III
Summary of variants in patients with
retinitis pigmentosa.
| Exon | Mutation | Type | Amino acid | Frequency | Predictiona |
|---|
| 1 | c.14G>GT | Hetero | p.Gly5Val | 7/50 | Possibly
damaging |
| 3 | c.399G>A | Homo | – | 34/50 | Benign |
| 5 | c.645A>AC | Hetero | – | 20/50 | Benign |
| 5 | c.657C>AC | Hetero | – | 16/50 | Benign |
| 10 | c.940T>AT | Hetero | p.Cys314Ser | 12/50 | Possibly
damaging |
| 13 | c.1280t>ct | Hetero | p.Met427Thr | 21/50 | Benign |
| 13 | c.1365c>ct | Hetero | – | 7/50 | Benign |
Tissue distribution of LPCAT1 in
mice
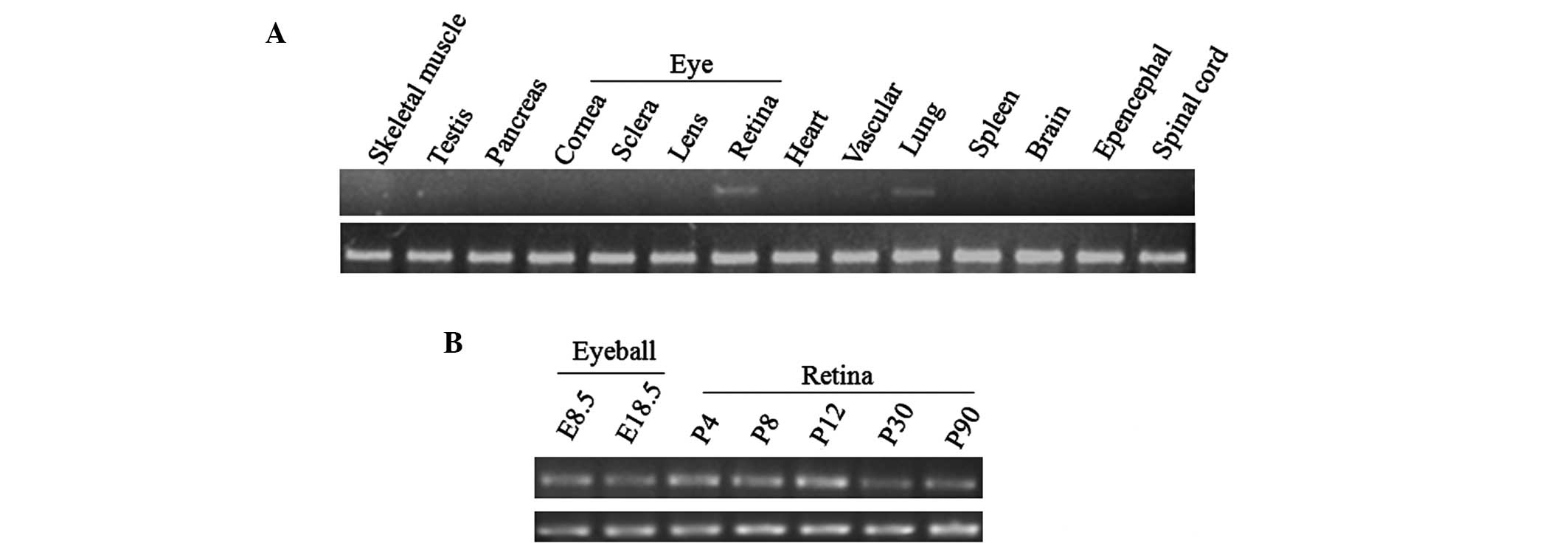
The tissue distribution of LPCAT1 was
analyzed using RT-PCR. The highest expression level of
LPCAT1 was observed in the retina, followed by the lung and
the spinal cord. Other tissues had particularly low expression
levels of LPCAT1 (Fig. 4).
These results suggested that LPCAT1 may be important in the
development and function of the mammalian retina.
Discussion
LPCAT, which is the most important enzyme in
membrane biogenesis and surfactant production, converts LPC to PC
(6). Based on previous studies on
LPCAT1 in mice and the availability of an animal model for
human R P, the present study aimed to investigate whether
LPCAT1 is involved in the development of RP in a Chinese
population (18).
In the present study, a total 50 patients diagnosed
with RP were enrolled for investigation. These patients were
previously screened for mutations in ~164 retinal-associated genes
using established targeted exome sequencing technology. Based on
the obtained family histories, the families selected for
investigation in the present study were affected by RP exhibiting
either recessive or sporadic inheritance patterns. All the coding
regions and exon-flanking regions of the LPCAT1 gene were
sequenced in the participants and, with the exception of certain
synonymous variants, three variants, which resulted in amino acid
changes, were identified. However, none of these missense variants
were determined as being pathogenic.
The results of the present study indicated that none
of the LPCAT1 variants identified were significantly
associated with RP in the examined group of Chinese patients. The
lack of correlation between the LPCAT1 variants and patients
with RP suggested that the possibility of these LPCAT1
genetic variations attributing to the pathogenesis of RP was low.
It is possible, however, that pathogenic mutations in this gene may
exist outside of the coding exons and flanking intron splice sites.
It is also possible that the pathogenic mutations, which occur in
LPCAT1, result in a form of retinal degeneration that was
absent in the patients included in the present study.
In conclusion, the results of the present study
suggested either that mutations in LPCAT1 do not confer a
genetic predisposition to R P, or that the incidence is rare in
patients with R P. An increase in sample sizes may enable the
screening of more patients with RP patients for pathogenic
mutations in LPCAT1. In addition, additional genetic
investigations in other ethnic populations are required to further
elucidate the potential association between LPCAT1 and R
P.
Acknowledgments
The authors would like to thank all patients for
their participation in the investigation. This study was supported
by the National Key Basic Research Program (grant. no.
2013CB967502) to Dr Zi-Bing Jin), the National Natural Science
Foundation of China (grant. no. 81371059) to Dr Zi-Bing Jin and the
Zhejiang Provincial Natural Science Foundation of China (grant.
nos. Y12H12003 and LR13H120001 to Dr Qin-Kang Lu and Dr Zi-Bing
Jin, respectively.
References
|
1
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dryja TP, McGee TL, Hahn LB, Cowley GS,
Olsson JE, Reichel E, Sandberg MA and Berson EL: Mutations within
the rhodopsin gene in patients with autosomal dominant retinitis
pigmentosa. N Engl J Med. 323:1302–1307. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Daiger SP, Sullivan LS and Bowne SJ: Genes
and mutations causing retinitis pigmentosa. Clin Genet. 84:132–141.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bessant DA, Ali RR and Bhattacharya SS:
Molecular genetics and prospects for therapy of the inherited
retinal dystrophies. Curr Opin Genet Dev. 11:307–316. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nakanishi H, Shindou H, Hishikawa D,
Harayama T, Ogasawara R, Suwabe A, Taguchi R and Shimizu T: Cloning
and characterization of mouse lung-type acyl-CoA:Lysophosphatidyl
choline acyltransferase 1 (LPCAT1). Expression in alveolar type II
cells and possible involvement in surfactant production. J Biol
Chem. 281:20140–20147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Friedman JS, Chang B, Krauth DS, Lopez I,
Waseem NH, Hurd RE, Feathers KL, Branham KE, Shaw M and Thomas GE:
Loss of lysophosphatidylcholine acyltransferase 1 leads to
photoreceptor degeneration in rd11 mice. Proc Natl Acad Sci USA.
107:15523–15528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harayama T, Shindou H, Ogasawara R, Suwabe
A and Shimizu T: Identification of a novel noninfammatory
biosynthetic pathway of platelet-activating factor. J Biol Chem.
283:11097–11106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grupp K, Sanader S, Sirma H, Simon R, Koop
C, Prien K, Hube-Magg C, Salomon G, Graefen M, Heinzer H, et al:
High lysophosphatidylcholine acyltransferase 1 expression
independently predicts high risk for biochemical recurrence in
prostate cancers. Mol Oncol. 7:1001–1011. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu B, Gao L, Wang L, Tang G, He M, Yu Y,
Ni X and Sun Y: Effects of platelet-activating factor and its
differential regulation by androgens and steroid hormones in
prostate cancers. Br J Cancer. 109:1279–1286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai X, Han J, Qi Y, Zhang H, Xiang L, Lv
J, Li J, Deng WT, Chang B, Hauswirth WW and Pang JJ: AAV-mediated
lysophosphatidylcholine acyltransferase 1 (Lpcat1) gene replacement
therapy rescues retinal degeneration in rd11 mice. Invest
Ophthalmol Vis Sci. 55:1724–1734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schrick JJ, Vogel P, Abuin A, Hampton B
and Rice DS: ADP-ribosylation factor-like 3 is involved in kidney
and photoreceptor development. Am J Pathol. 168:1288–1298. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gerding WM, Schreiber S,
Schulte-Middelmann T, de Castro Marques A, Atorf J, Akkad DA,
Dekomien G, Kremers J, Dermietzel R, Gal A, et al: Ccdc66 null
mutation causes retinal degeneration and dysfunction. Hum Mol
Genet. 20:3620–3631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma C, Papermaster D and Cepko CL: A unique
pattern of photoreceptor degeneration in cyclin D1 mutant mice.
Proc Natl Acad Sci U S A. 95:9938–9943. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alves CH, Sanz AS, Park B, Pellissier LP,
Tanimoto N, Beck SC, Huber G, Murtaza M, Richard F, Sridevi
Gurubaran I, et al: Loss of CRB2 in the mouse retina mimics human
retinitis pigmentosa due to mutations in the CRB1 gene. Hum Mol
Genet. 22:35–50. 2013. View Article : Google Scholar
|
|
15
|
Vogel P, Gelfman CM, Issa T, Payne BJ,
Hansen GM, Read RW, Jones C, Pitcher MR, Ding ZM, DaCasta CM, et
al: Nephronophthisis and retinal degeneration in tmem218-/-mice: A
novel mouse model for senior-loken syndrome? Vet Pathol.
52:580–595. 2015. View Article : Google Scholar
|
|
16
|
Daiger SP, Bowne SJ and Sullivan LS:
Perspective on genes and mutations causing retinitis pigmentosa.
Arch Ophthalmol. 125:151–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang XF, Huang F, Wu KC, Wu J, Chen J,
Pang CP, Lu F, Qu J and Jin ZB: Genotype-phenotype correlation and
mutation spectrum in a large cohort of patients with inherited
retinal dystrophy revealed by next-generation sequencing. Genet
Med. 17:271–278. 2015. View Article : Google Scholar
|
|
18
|
Dai X, Han J, Qi Y, Zhang H, Xiang L, Lv
J, Li J, Deng WT, Chang B, Hauswirth WW and Pang JJ: AAV-mediated
lysophosphatidylcholine acyltransferase 1 (Lpcat1) gene replacement
therapy rescues retinal degeneration in rd11 mice. Invest
Ophthalmol Vis Sci. 55:1724–1734. 2014. View Article : Google Scholar : PubMed/NCBI
|