Introduction
Diabetic retinopathy (DR) is the most common
microvascular complication in diabetes, and DR has emerged as a
leading cause of visual impairment and blindness in individuals
aged >50 years (1). Clinical
and experimental evidence has revealed that diabetic microvascular
and macrovascular complications persist in diabetic patients
regardless of whether blood glucose normalisation has occurred;
this phenomenon has been defined as 'metabolic memory' (2–5).
Although numerous studies have investigated the underlying
mechanisms of metabolic memory (5), this particular negative phenomenon
remains poorly understood and poses a major challenge in the
treatment of diabetes.
There is accumulating evidence that DR exhibits
certain characteristics of a low-grade inflammatory disease, in
which retinal inflammatory mediators and apoptosis of retinal cells
contribute to the process of metabolic memory (6–8).
Nuclear factor (NF)-κB is a master regulator of various genes
involved in inflammatory and immune responses, cellular
proliferation and apoptosis (9–11).
Diabetes-induced activation of NF-κB was shown to promote
expression of proinflammatory cytokines and various pro-apoptosis
regulators (4). This activation
also contributes to the apoptosis of retinal endothelial cells
(RECs), which are significant in the pathogenesis of DR (12). Further studies have demonstrated
that NF-κB is activated in the retina as early as two months after
the onset of diabetes (12).
Reinstitution of good blood glucose control after six months of
poor blood glucose control did not exhibit an effect on activated
NF-κB levels in the retinas of streptozotocin (STZ)-induced
diabetic rats, indicating that NF-κB-associated signalling pathways
remained activated, resulting in a cellular metabolic memory effect
(7). However, the mechanisms by
which hyperglycaemia induces the activation of NF-κB and its
dependent signalling pathways in diabetic metabolic memory have not
been elucidated.
Class III histone deacetylase, sirtuin 1 (SIRT1) is
a multifunctional deacetylase that is critically involved in
regulating inflammation, stress responses, metabolism, DNA repair
and cell survival via deacetylation of key transcription factors,
enzymes and proteins (13–15). Our previous study demonstrated that
SIRT1 conferred resistance to cellular metabolic memory, which had
been induced by high glucose (5).
Recently, both in vitro and in vivo studies indicated
that SIRT1 suppresses NF-κB signalling and results in the reduction
of the inflammatory responses mediated by NF-κB in endothelial
cells (16,17). Therefore, SIRT1 may be significant
in the pathogenesis of the metabolic memory phenomenon via the
NF-κB signalling pathway.
Fenofibrate, a peroxisome proliferator-activated
receptor α (PPARα) agonist, is an effective lipid-lowering
therapeutic agent that is widely administered in the clinical
setting. In addition to its lipid effects, fenofibrate affects
various signalling pathways involved in inflammation, angiogenesis
and cell survival, and has received attention as a novel medical
treatment for DR and other diabetes-induced microvascular
complications. A previous study indicated that fenofibrate inhibits
high glucose-induced metabolic memory in Schwann cells in diabetic
neuropathy (18). Additional
previous studies indicated that fenofibrate activates SIRT1 and
suppresses cellular inflammation by activation of PPARα (19,20).
These findings resulted in the current investigation
to establish whether fenofibrate may suppress high glucose-induced
metabolic memory via its anti-inflammatory effect in endothelial
cells during DR. Furthermore, the association between fenofibrate
and the SIRT1-dependent signalling pathway was assessed.
Materials and methods
Cell culture and treatment
Human RECs (HRECs) and attachment factor were
purchased from the Applied Cell Biology Research Institute
(Kirkland, WA, USA) and maintained in EGM2-MV media (Lonza Group
AG, Basel, Switzerland) with 5% fetal bovine serum (Lonza Inc.,
Allendale, NJ, USA) in flasks coated with the attachment factor.
Cultured HRECs of three to four passages were used in the
experiments. The cells were incubated with a normal concentration
of glucose (normal glucose; 5 mmol/l) for three weeks, a high
concentration of glucose (high glucose; 30 mmol/l) for 3 weeks, or
high glucose for one week followed by normal glucose for two
weeks.
For pharmacological prevention of glucose-induced
metabolic memory, after 1 week, cells were switched from high
glucose to normal glucose with fenofibrate at various
concentrations (25, 50 and 100 µM) for 48 h and subsequently
maintained without fenofibrate for 12 days. To equalize the
osmolarity and rule out the influence of increased osmolarity on
the cellular memory of high glucose-induced stress, cells were
incubated in 25 mmol/l mannitol along with normal glucose at 5
mmol/l, which served as osmotic controls. Additionally, PPARα
antagonist, GW6471 (1 µM) was added to the media for 1 h and
incubated with fenofibrate to investigate whether the protective
effect of fenofibrate on SIRT1 expression is mediated by PPARα
activation in diabetic metabolic memory.
RNA interference and transfection
The human small interfering (si)RNA targeting SIRT1
(SIRT1 siRNA) and negative control (NC) siRNA were chemically
synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China)
using the following sequences: 5′-GAUGCUGUGAAAUUACUGC-3′for SIRT1
siRNA and 5′-GGATCATAAGGCGCATAGC-3′ for NC siRNA. Transfection was
performed with Lipofectamine 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer's
instructions. A final concentration of 50 nM RNA (for SIRT1 siRNA)
or 100 nM RNA (for NC siRNA) and their respective NCs was used for
each transfection in the subsequent experiments.
Quantitative polymerase chain reaction
(qPCR)
Total RNA was extracted from cultured cells using
TRIzol reagent (Invitrogen Life Technologies). The expression level
of SIRT1 mRNA was quantified by qPCR using a QuantiTect
SYBR® Green PCR kit (Qiagen GmbH, Hilden, Germany) and
normalized to β-actin using the following primers: Forward
5′-AGTACTGGGGAGAAAAATGAAAGA-3′ and reverse
5′-CTGCCACAAGAACTAGAGGATAAG-3′ for SIRT1; forward
5′-CCCAAGGCCAACCGCGAGAAGATG-3′ and reverse
5′-GTCCCGGCCAGCCAGGTCCAGA-3′ for β-actin. The cycling conditions
were as follows: Denaturation at 95°C for 10 sec, followed by
annealing at 58°C for 20 sec and extension at 58°C for 20 sec, for
40 cycles. The changes in expression were calculated using the
2−ΔΔCt method (21).
Western blot analysis
Total protein was extracted from the cultured cells
or tissues using a Total Protein Extraction kit (cat. no. 2140; EMD
Millipore, Billerica, MA, USA) according to the manufacturer's
instructions. The protein content was determined using a
Bicinchoninic Acid Protein Assay kit (cat. no. 23225; Invitrogen
Life Technologies) with bovine serum albumin (Gibco Life
Technologies, Carlsbad, CA, USA) serving as the standard. Proteins
(20 µg) were separated by 10% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes (EMD Millipore). The membranes
were blocked in 5% non-fat milk and Tris-buffered saline with 0.05%
Tween-20 (Invitrogen Life Technologies) at room temperature for 2
h, then probed with antibodies as follows: Mouse anti-SIRT1
monoclonal antibody [1:6,000; Abcam, Cambridge, MA, USA (cat. no.
ab110304)], rabbit anti-PPARα polyclonal antibody [1:1,000; Santa
Cruz Biotechnology, Dallas, TX, USA (cat. no. sc-2772)], human
anti-NF-κB antibody [1:400; Enzo Life Sciences, Inc., Farmingdale,
NY, USA (cat. no. 7971)] or β-actin [1:1,000, Sigma-Aldrich, St.
Louis, MO, USA (cat. no. A2103)] and developed with an enhanced
chemiluminescence kit (cat. no. RPN2132; GE Healthcare Life
Sciences, Chalfont, UK).
SIRT1 deacetylase activity assay
SIRT1 deacetylase activity was assessed in the
nuclear fraction using a commercial fluorometric assay kit (cat.
no. CS1040; Sigma-Aldrich). Protein (30–40 µg) was incubated
with the substrate (coupled to the fluorophore and quencher) and
NAD+ for 3 min at room temperature. The fluorescence
emitted, due to deacetylation of the substrate by SIRT1, was
measured at 345 nm excitation and 450 nm emission wavelengths using
a fluorescence microplate reader (SpectraMax® M5;
Molecular Devices, LLC, Sunnyvale, CA, USA).
TUNEL assay
Cells treated with normal glucose, high glucose,
high glucose followed by normal glucose, or high glucose followed
by normal glucose plus fenofibrate at different concentrations (10,
25 and 50 µM) were grown on glass coverslips in 24-well
plates. The cells were then fixed with 4% paraformaldehyde, which
was followed by permeabilization with 0.1% Triton X-100
(Sigma-Aldrich). The apoptotic cells were stained with a
fluorometric TUNEL assay kit (DeadEnd™ Fluorometric TUNEL System;
Promega Corporation, Madison, WI, USA) and visualized under a
fluorescence microscope (Olympus BX41; Olympus Corporation, Tokyo,
Japan) according to the manufacturer's instructions. TUNEL-positive
cells were scored in a minimum of five fields per coverslip, and
≥1,000 cells were counted for each coverslip.
Statistical analysis
Data were presented as the mean ± standard deviation
from at least three independent experiments. Group means were
compared by one-way analysis of variance using GraphPad Prism 4.0
software system (GraphPad, San Diego, CA, USA) and the statistical
software program, SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA).
Correlations between NF-κB expression and the expression and
activity of SIRT1 were calculated using Spearman's rank
correlation. All P-values were two-sided and P<0.05 was
considered to indicate a statistically significant difference.
Results
Fenofibrate suppressed the expression of
NF-κB induced by high glucose after glucose normalization in
HRECs
To investigate the inhibitory effect of fenofibrate
on NF-κB expression in high glucose-induced metabolic memory in
HRECs, the cells were exposed to normal (5 mmol/l) or high (30
mmol/l) glucose concentrations, or high glucose followed by normal
glucose with different concentrations of fenofibrate (10, 25 and 50
µM).
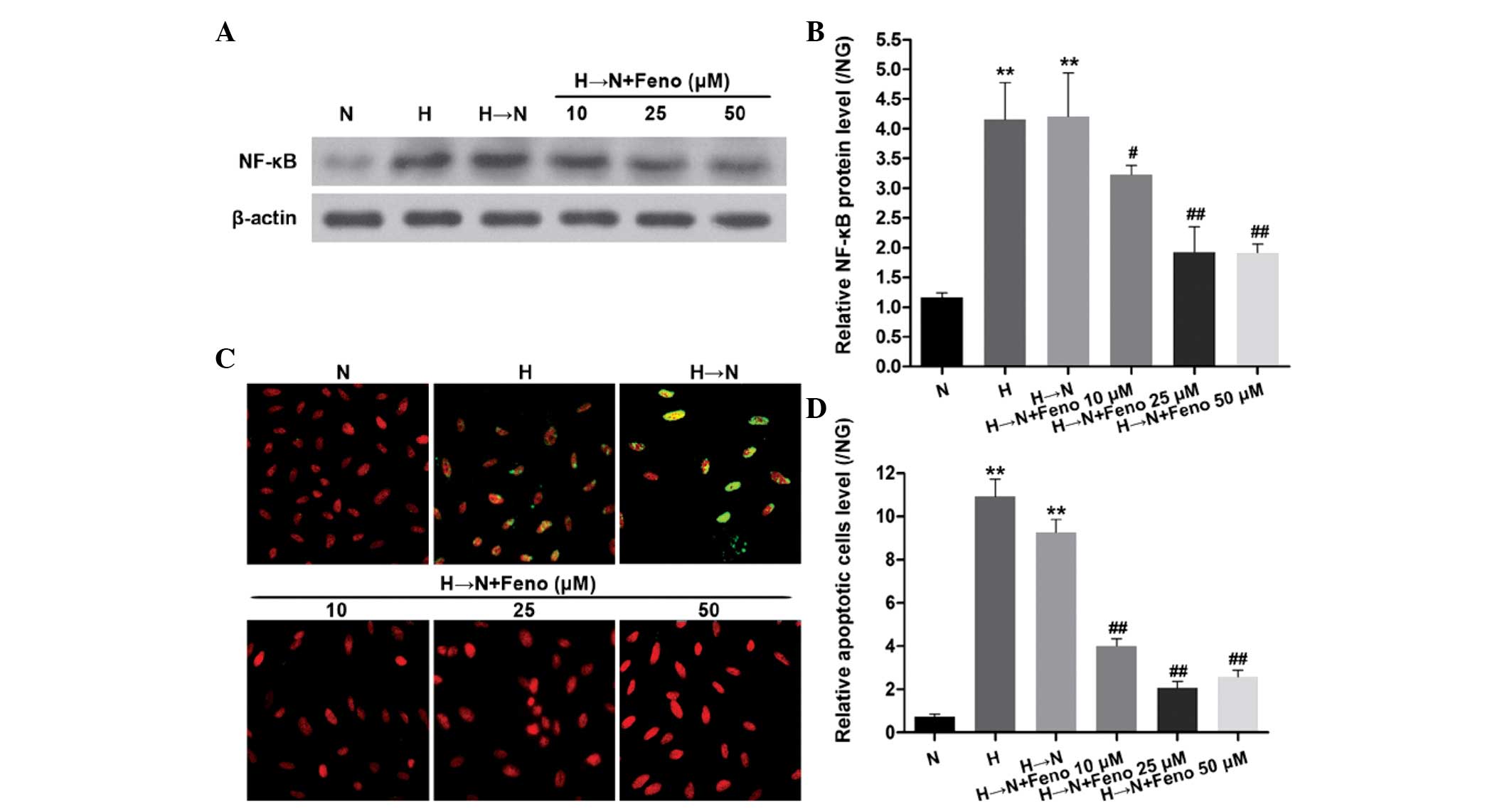
As shown in Fig. 1A
and B, chronic exposure to high glucose resulted in significantly
increased protein levels of NF-κB. Compared with exposure to
continuous normal glucose, NF-κB remained increased in the cells
that were treated with high glucose for 1 week followed by normal
glucose for 2 weeks. Fenofibrate downregulated the protein
expression of NF-κB, indicating that fenofibrate suppresses high
glucose-induced NF-κB expression following glucose normalization in
HRECs.
To equalize the osmolarity with the high glucose
treatment at 30 mmol/l and rule out the influence of increased
osmolarity on the cellular memory of high glucose-induced stress,
25 mmol/l mannitol was added along with 5 mmol/l normal glucose to
cells for 3 weeks, and no effects on memory were observed (data not
shown).
Furthermore, the level of cellular apoptosis was
observed using a TUNEL assay. Chronic exposure to high glucose
caused a significant increase in cellular apoptosis. Apoptosis also
remained increased in the cells exposed to high glucose for 1 week
followed by normal glucose for 2 weeks. Fenofibrate suppressed the
cellular apoptosis in high glucose-induced metabolic memory in
HRECs (Fig. 1C and D). These
results support the hypothesis that NF-κB is involved in
fenofibrate's suppression of high glucose-induced cellular
metabolic memory.
Fenofibrate inhibits high glucose-induced
NF-κB expression through SIRT1 in HRECs
To investigate the potential synergistic roles of
SIRT1, NF-κB and fenofibrate in modulating high glucose-induced
metabolic memory in HRECs, qPCR and western blot analysis were
conducted to examine the expression levels of SIRT1 in the cells
exposed to normal glucose, high glucose, or high glucose followed
by normal glucose with different concentrations of fenofibrate (10,
25 and 50 µM). As shown in Fig.
2A–D, chronic exposure to high glucose resulted in
significantly decreased levels of SIRT1. SIRT1 levels remained
decreased in cells treated with high glucose for 1 week followed by
normal glucose for 2 weeks when compared with exposure to
continuous normal glucose. However, fenofibrate significantly
suppressed the inhibition of SIRT1, which was induced by high
glucose following glucose normalization in HRECs. Furthermore, a
significant negative correlation between NF-κB and SIRT1 protein
expression and activity levels was revealed in HRECs (Fig. 2E and F).
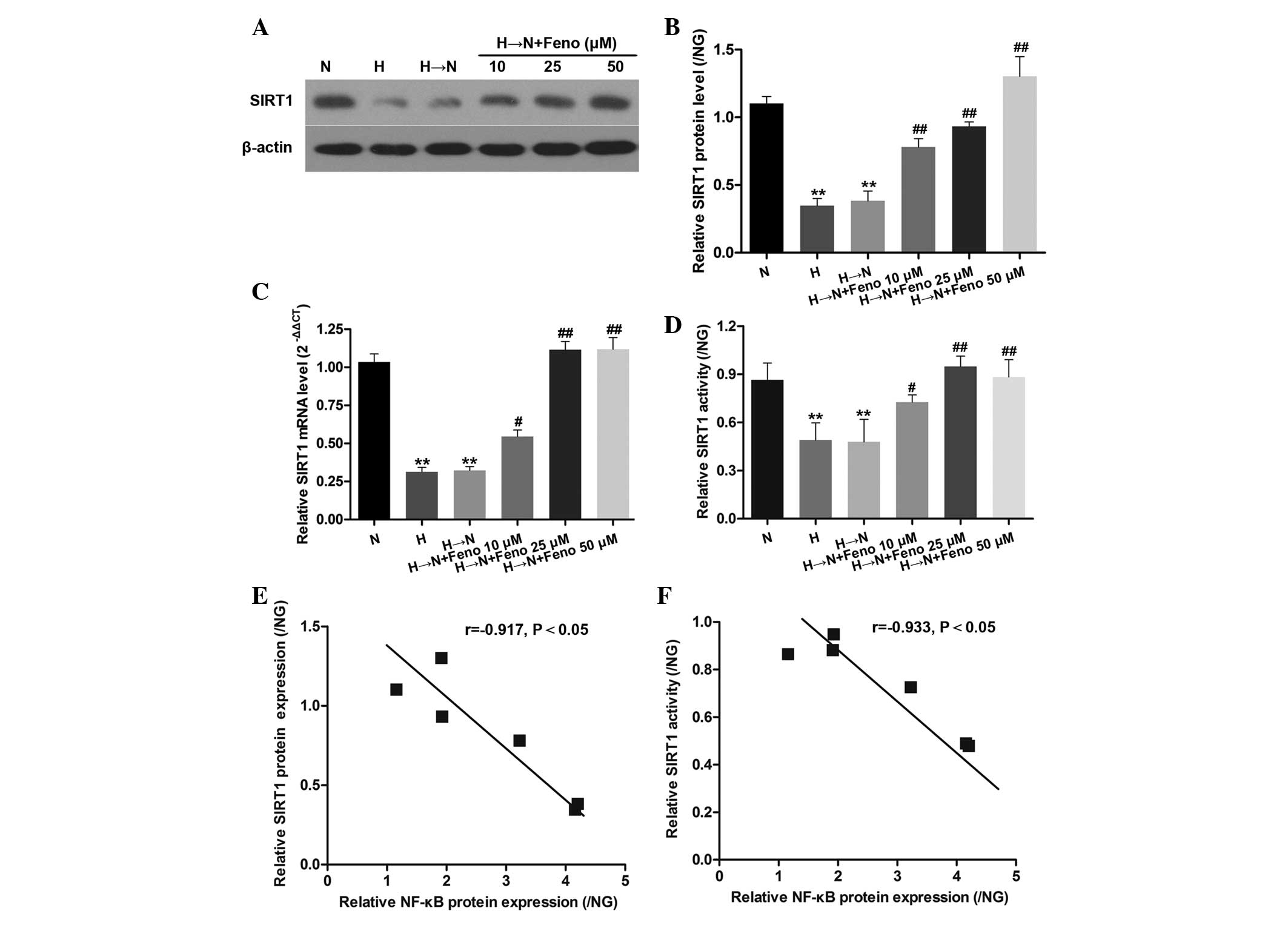 | Figure 2Inverse correlation between NF-κB and
SIRT1 protein expression and activity levels in HRECs. (A) Relative
expression of SIRT1 mRNA in the cell treatment groups: Normal
glucose (N), high glucose (H), high glucose followed by normal
glucose (H→N), and H→N plus Feno at different concentrations (10,
25 and 50 µM). SIRT1 (B) protein, (C) mRNA and (D) activity
levels in the six HRECs groups. Results are presented as the means
+ standard deviations of three representative experiments. (E and
F) Inverse correlation was calculated by Spearman's correlation:
(E) r=−0.523 (P=0.018) for NF-κB and SIRT1 protein expression and
(F) r=−0.397 (P=0.043) for NF-κB and SIRT1 activity level. The
results of all the groups are displayed as the ratio of the
control, NG (normal glucose). **P<0.01 vs. N;
#P<0.05 vs. H→N; ##P<0.01 vs. H→N. The
relative expression data were analyzed using the 2−ΔΔCt
method. SIRT1, sirtuin 1; HRECs, human retinal vascular endothelial
cells; Feno, fenofibrate; NF-κB, nuclear factor-κB. |
To confirm the regulatory role of SIRT1 in
fenofibrate-mediated inhibition of NF-κB expression, HRECs were
transfected with SIRT1-specific siRNA to decrease SIRT1 expression
prior to incubation with fenofibrate. The results showed that the
inhibitory effect of fenofibrate on high glucose-induced NF-κB
protein expression and cellular apoptosis was abolished by
knockdown of SIRT1 (Fig. 3).
Therefore, these results indicate that fenofibrate inhibits high
glucose-induced metabolic memory in HRECs via SIRT1-dependent
suppression of NF-κB.
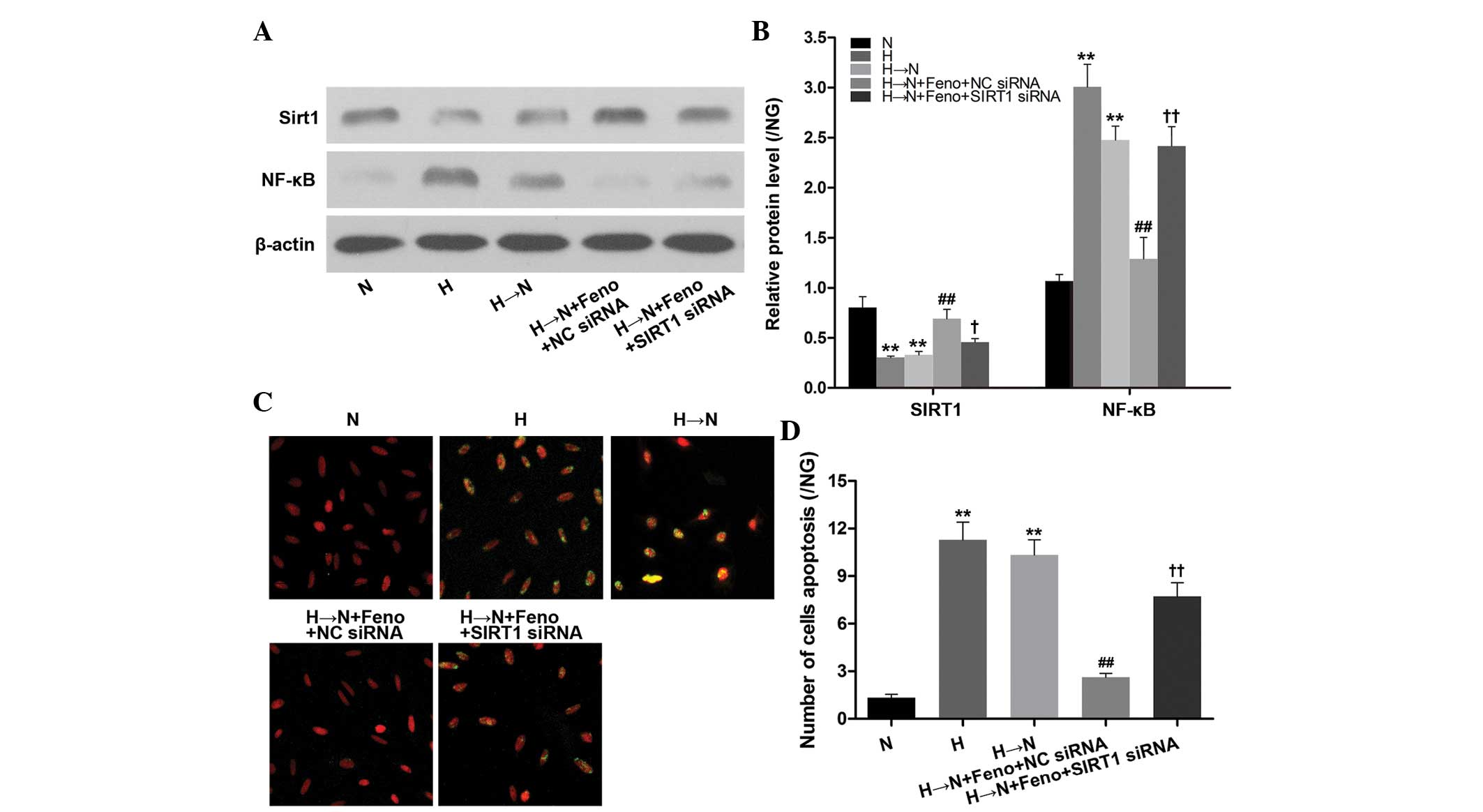 | Figure 3Feno inhibits NF-κB expression and
cellular apoptosis through SIRT1 in human retinal vascular
endothelial cells following culture in high glucose followed by
normal glucose (H→N). (A) Western blotting and (B) quantification
of SIRT1 and NF-κB protein expression in the cell treatment groups:
Normal glucose (N), high glucose (H), high glucose followed by
normal glucose (H→N), H→N plus Feno and NC siRNA (H→N + Feno + NC
siRNA) or SIRT1 siRNA (H→N + Feno + SIRT1 siRNA). (C) Analysis of
cellular apoptosis levels conducted in the five groups by TUNEL.
(D) Analysis of apoptotic cell number. (B,D) Results are presented
as the means + standard deviations of three representative
experiments. The results of all the groups are displayed as the
ratio of the control, NG (normal glucose). **P<0.01
vs. N; #P<0.05 vs. H→N; ##P<0.01 vs.
H→N; ††P<0.01 vs. H→N + Feno + NC siRNA. SIRT1,
sirtuin 1; Feno, fenofibrate; NF-κB, nuclear factor-κB; NC,
negative control; siRNA, small interfering RNA. |
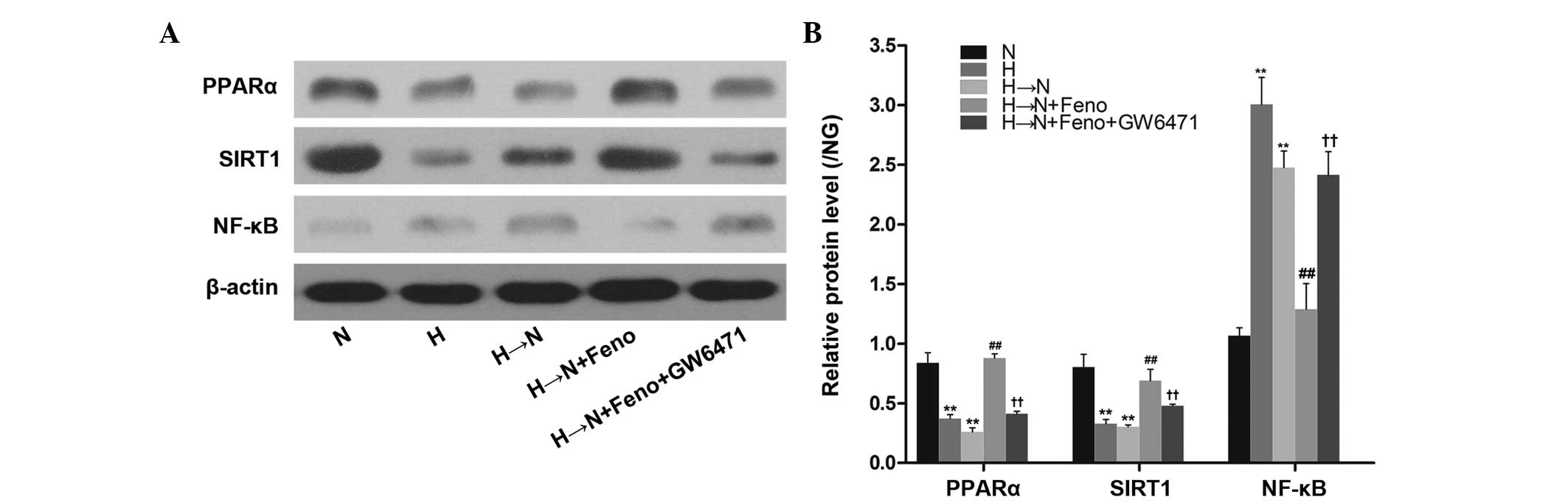
Fenofibrate upregulates SIRT1 expression
through PPARα activation in HRECs
To investigate whether the protective effect of
fenofibrate on SIRT1 expression is mediated by PPARα activation in
diabetic metabolic memory, HRECs were pretreated with the PPARα
antagonist, GW6471 (1 µM) for 1 h and incubated with
fenofibrate. As shown in Fig. 4,
chronic exposure to high glucose decreased the expression of PPARα
even after glucose normalization in HRECs. Pretreatment of the
cells with fenofibrate increased the PPARα expression and this
effect was abolished by treatment with GW6471. Furthermore,
pretreating the cells with 1 µM GW6471 reversed the effect
of fenofibrate on SIRT1 protein expression. These results indicate
that fenofibrate upregulates SIRT1 expression through activation of
PPARα in HRECs.
Discussion
Our previous study demonstrated that SIRT1 confers
resistance to cellular metabolic memory induced by high glucose
(5). In the current study, further
evidence is presented that fenofibrate upregulates SIRT1 expression
and activity via PPARα activation, and downregulates NF-κB
expression to suppress the memory of hyperglycaemic stress in
HRECs.
A major challenge in treating diabetic microvascular
complications, such as DR, is that the molecular and pathological
features resulting from high glucose are maintained despite
subsequent effective control of blood glucose (22,23).
The prolonged impact of the early metabolic environment on the
development and progression of microvascular complications is
referred to as 'metabolic memory' (24).
Inflammation is significant early and throughout the
pathogenesis of microvascular complications (25). NF-κB is a rapid response
transcription factor involved in inflammatory reactions, as well as
the expression of cytokines, chemokines, cell adhesion molecules
and growth factors. It is considered to be a key signalling factor
by which high glucose concentrations trigger a pro-apoptotic
program in diabetes (26). A study
by Kowluru et al (7) using
STZ-induced diabetic rats indicated that chronic exposure to high
glucose caused a significant increase in the levels of activated
caspase-3 and NF-κB, which remained at the increased levels six
months later (7), suggesting that
the activated inflammation-associated signalling pathways had
remained activate. In our previous study (5), using an established cell model of
metabolic memory induced by high glucose, the reinstitution of
normal glucose levels after 1 week of high glucose was observed to
have no effect on the levels of activated NF-κB expression and cell
apoptosis. This finding indicated that the activated
inflammation-associated signalling pathways remained activated.
There is accumulating evidence that SIRT1 inhibits
NF-κB signalling, and the activation of SIRT1 may alleviate a
multitude of NF-κB-driven inflammatory and metabolic disorders
(27–29). In contrast to NF-κB, the present
study showed an adverse tendency of SIRT1 expression in HREC cells.
The results were consistent with our previous study, which
demonstrated that SIRT1 activation reduced high glucose-induced
cellular metabolic memory in RECs by suppressing production of the
cellular inflammatory gene, NF-κB and attenuating the expression of
the cellular apoptosis gene, Bax (5). These data implied that SIRT1
activators may exert significant protective effects against
metabolic memory in diabetic microvascular complications, such as
DR.
Recently, fenofibrate, a specific PPARα agonist, has
displayed marked and robust efficacy in arresting the progression
of microvascular complications in type 2 diabetes in FIELD and
ACCORD studies (30,31). Furthermore, various studies have
demonstrated that fenofibrate activates SIRT1 and suppresses
cellular inflammation by activation of PPARα (19,20).
However, its function in the retina has rarely been investigated.
In the present study, it was found that fenofibrate
dose-dependently reversed the changes to SIRT1 and NF-κB expression
in high glucose-induced cellular metabolic memory in HRECs.
Knockdown of SIRT1 attenuated the inhibitory effect of fenofibrate
on NF-κB expression, suggesting that fenofibrate inhibits high
glucose-induced metabolic memory in HRECs via SIRT1-dependent
suppression of NF-κB.
A recent study found that the inhibitory effect of
SIRT1 on monocyte chemoattractant protein-1 mRNA expression was
attenuated by the PPARα antagonist, GW6471 in cardiomyocytes,
indicating that SIRT1 acted in association with PPARα to protect
cardiomyocytes from inflammation (32). Thus, whether the protective effect
of fenofibrate on SIRT1 expression was mediated by PPARα activation
in diabetic metabolic memory was investigated in the current study.
It was demonstrated that exposure to high glucose levels reduced
PPARα expression, whereas treatment with fenofibrate activated
PPARα and exerted an anti-inflammatory effect via SIRT1-dependent
suppression of NF-κB.
Various studies indicate that fenofibrate may
inhibit NF-κB-mediated cellular inflammation by suppressing the
AMPK/eNOS/NO (33,34) and Toll-like receptor signalling
pathways (35). However, the
mechanism by which fenofibrate may inhibit NF-κB signalling
pathways in diabetic microvascular dysfunction remains unclear. To
the best of our knowledge, the present study is the first to link
SIRT1 with the inhibitory effect of fenofibrate on NF-κB-mediated
cellular inflammation in high glucose-induced cellular metabolic
memory. Notably, a recent study indicated that the PPARα agonist,
fenofibrate inhibits tumour necrosis factor α-induced cluster of
differentiation 40 expression and regulates the inflammatory
response in 3T3-L1 adipocytes via the SIRT1-dependent signalling
pathway (20). Our results are
consistent with this, and support the hypothesis that
PPARα/SIRT1/NF-κB may be a commonly used signalling pathway during
the cellular inflammation of different pathological processes.
In conclusion, the present study demonstrates that
high glucose levels activate the NF-κB-associated inflammation
signalling pathway and induce metabolic memory, which prolong the
impact of early metabolic dysfunction on the progression of retinal
endothelial injury. Fenofibrate was observed to activate PPARα and
inhibit high glucose-induced metabolic memorial injury via
SIRT1-dependent suppression of NF-κB in HRECs. These findings may
provide a promising strategy for suppressing the development of DR
and other associated complications of diabetes.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81271032,
81070739 and 30872828) and the National Key Basic Research
Programme (grant nos. 2010CB535006).
References
|
1
|
Lutty GA: Effects of diabetes on the eye.
Invest Ophthalmol Vis Sci. 54:ORSF81–87. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
No authors listed. Retinopathy and
nephropathy in patients with type 1 diabetes four years after a
trial of intensive therapy. The Diabetes Control and Complications
Trial/Epidemiology of Diabetes Interventions and Complications
Research Group. N Engl J Med. 342:381–389. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Writing Team for the Diabetes Control and
Complications Trial/Epidemiology of Diabetes Interventions and
Complications Research Group: Effect of intensive therapy on the
microvascular complications of type 1 diabetes mellitus. JAMA.
287:2563–2569. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ceriello A, Ihnat MA and Thorpe JE:
Clinical review 2: The 'metabolic memory': is more than just tight
glucose control necessary to prevent diabetic complications? J Clin
Endocrinol Metab. 94:410–415. 2009. View Article : Google Scholar
|
|
5
|
Zheng Z, Chen H, Li J, Li T, Zheng B,
Zheng Y, Jin H, He Y, Gu Q and Xu X: Sirtuin 1-mediated cellular
metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and
therapeutic effects of metformin. Diabetes. 61:217–228. 2012.
View Article : Google Scholar :
|
|
6
|
Kowluru RA and Chan PS: Metabolic memory
in diabetes - from in vitro oddity to in vivo problem: Role of
apoptosis. Brain Res Bull. 81:297–302. 2010. View Article : Google Scholar
|
|
7
|
Kowluru RA, Chakrabarti S and Chen S:
Re-institution of good metabolic control in diabetic rats and
activation of caspase-3 and nuclear transcriptional factor
(NF-kappaB) in the retina. Acta Diabetol. 41:194–199. 2004.
View Article : Google Scholar
|
|
8
|
Kowluru RA, Zhong Q and Kanwar M:
Metabolic memory and diabetic retinopathy: Role of inflammatory
mediators in retinal pericytes. Exp Eye Res. 90:617–623. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sen R and Baltimore D: Inducibility of
kappa immunoglobulin enhancer-binding protein Nf-kappa B by a
posttranslational mechanism. Cell. 47:921–928. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar
|
|
11
|
DiDonato JA, Mercurio F and Karin M:
NF-kappaB and the link between inflammation and cancer. Immunol
Rev. 246:379–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kowluru RA, Koppolu P, Chakrabarti S and
Chen S: Diabetes-induced activation of nuclear transcriptional
factor in the retina and its inhibition by antioxidants. Free Radic
Res. 37:1169–1180. 2003. View Article : Google Scholar
|
|
13
|
Yu J and Auwerx J: Protein deacetylation
by SIRT1: An emerging key post-translational modification in
metabolic regulation. Pharmacol Res. 62:35–41. 2010. View Article : Google Scholar
|
|
14
|
Haigis MC and Sinclair DA: Mammalian
sirtuins: Biological insights and disease relevance. Annu Rev
Pathol. 5:253–295. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baur JA: Resveratrol, sirtuins and the
promise of a DR mimetic. Mech Ageing Dev. 131:261–269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai W, Ramdas M, Zhu L, Chen X, Striker GE
and Vlassara H: Oral advanced glycation endproducts (AGEs) promote
insulin resistance and diabetes by depleting the antioxidant
defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci USA.
109:15888–15893. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsai KL, Huang PH, Kao CL, Leu HB, Cheng
YH, Liao YW, Yang YP, Chien Y, Wang CY, Hsiao CY, et al: Aspirin
attenuates vinorelbine-induced endothelial inflammation via
modulating SIRT1/AMPK axis. Biochem Pharmacol. 88:189–200. 2014.
View Article : Google Scholar
|
|
18
|
Kim ES, Isoda F, Kurland I and Mobbs CV:
Glucose-induced metabolic memory in Schwann cells: prevention by
PPAR agonists. Endocrinology. 154:3054–3066. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang W, Bai L, Qiao H, Lu Y, Yang L, Zhang
J, Lin R, Ren F, Zhang J and Ji M: The protective effect of
fenofibrate against TNF-α-induced CD40 expression through
SIRT1-mediated deacetylation of NF-κB in endothelial cells.
Inflammation. 37:177–185. 2014. View Article : Google Scholar
|
|
20
|
Wang W, Lin Q, Lin R, Zhang J, Ren F,
Zhang J, Ji M and Li Y: PPARalpha agonist fenofibrate attenuates
TNF-α-induced CD40 expression in 3T3-L1 adipocytes via the
SIRT1-dependent signaling pathway. Exp Cell Res. 319:1523–1533.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
No authors listed. The effect of intensive
diabetes therapy on the development and progression of neuropathy.
The Diabetes Control and Complications Trial Research Group. Ann
Intern Med. 122:561–568. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Writing Team for the Diabetes Control and
Complications Trial/Epidemiology of Diabetes Interventions and
Complications Research Group: Sustained effect of intensive
treatment of type 1 diabetes mellitus on development and
progression of diabetic nephropathy: the Epidemiology of Diabetes
Interventions and Complications (EDIC) study. JAMA. 290:2159–2167.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ihnat MA, Thorpe JE and Ceriello A:
Hypothesis: The 'metabolic memory', the new challenge of diabetes.
Diabet Med. 24:582–586. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
LeRoith D, Fonseca V and Vinik A:
Metabolic memory in diabetes-focus on insulin. Diabetes Metab Res
Rev. 21:85–90. 2005. View
Article : Google Scholar
|
|
26
|
Baker RG, Hayden MS and Ghosh S: NF-κB,
inflammation and metabolic disease. Cell Metab. 13:11–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Salminen A, Kauppinen A, Suuronen T and
Kaarniranta K: SIRT1 longevity factor suppresses NF-kappaB-driven
immune responses: Regulation of aging via NF-kappaB acetylation?
Bioessays. 30:939–942. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yao H and Rahman I: Perspectives on
translational and therapeutic aspects of SIRT1 in inflammaging and
senescence. Biochem Pharmacol. 84:1332–1339. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie J, Zhang X and Zhang L: Negative
regulation of inflammation by SIRT1. Pharmacol Res. 67:60–67. 2013.
View Article : Google Scholar
|
|
30
|
Keech AC, Mitchell P, Summanen PA, O'Day
J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson
E, et al: Effect of fenofibrate on the need for laser treatment for
diabetic retinopathy (FIELD study): A randomised controlled trial.
Lancet. 370:1687–1697. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ginsberg HN, Elam MB, Lovato LC III,
Crouse JR III, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein
HC, Probstfield J, et al: Effects of combination lipid therapy in
type 2 diabetes mellitus. N Engl J Med. 362:1563–1574. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Planavila A, Iglesias R, Giralt M and
Villarroya F: Sirt1 acts in association with PPARalpha to protect
the heart from hypertrophy, metabolic dysregulation and
inflammation. Cardiovasc Res. 90:276–284. 2011. View Article : Google Scholar
|
|
33
|
Biscetti F, Gaetani E, Flex A, Aprahamian
T, Hopkins T, Straface G, Pecorini G, Stigliano E, Smith RC,
Angelini F, et al: Selective activation of peroxisome
proliferator-activated receptor (PPAR)alpha and PPAR gamma induces
neoangiogenesis through a vascular endothelial growth
factor-dependent mechanism. Diabetes. 57:1394–1404. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Okayasu T, Tomizawa A, Suzuki K, Manaka K
and Hattori Y: PPARalpha activators upregulate eNOS activity and
inhibit cytokine-induced NF-kappaB activation through AMP-activated
protein kinase activation. Life Sci. 82:884–891. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shen W, Gao Y, Lu B, Zhang Q, Hu Y and
Chen Y: Negatively regulating TLR4/NF-kappaB signaling via PPARα in
endotoxin-induced uveitis. Biochim Biophys Acta. 1842:1109–1120.
2014. View Article : Google Scholar : PubMed/NCBI
|