Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an
autosomal dominant disease characterized by combined occurrence of
tumors/hyperplasia in the parathyroid gland, gastrointestinal
endocrine tissue, anterior pituitary and other tissues. The
responsible gene, MEN1, has been mapped to chromosomal
region 11q13 (1,2). The gene is composed of 10 exons and
encodes a 610-amino acid protein (MENIN) (1,2). It
is hypothesized to be involved in cell growth regulation, cell
cycle, genome stability and synapse plasticity. A two-hit mutation
hypothesis in this gene proposes that the first hit or mutation is
inherited as a germline mutation and the second hit occurs as a
somatic mutation in the predisposed endocrine cell (3). This second mutation may occur by
chromosome loss (e.g. loss of heterozygosity), chromosome loss with
duplication, mitotic recombination or another localized event, such
as a point mutation. As a consequence of these two mutations, both
alleles of the MEN1 gene are inactivated, allowing tumor
growth (3,4). Germline mutations of the MEN1
gene were detected to be inherited or sporadic in patients affected
by MEN1-like tumors (5). Although
MEN1 is a rare disease with an estimated prevalence of 30–200 per
million, the penetration rate is high among MEN1 gene
carriers. Preexisting evidence demonstrated that all carriers were
completely exposed by 50 years of age (6).
Although >1,000 families presenting with MEN1
have been described since the cloning of the gene in 1997 (7), the clinical treatment and long term
follow-up is rarely reported in the literature (8). This case describes a Chinese family
presenting with MEN1, with a mutation in intron 5 of the
MEN1 gene, which has not been described previously. After
four surgical procedures and full follow up over 20 years, the
proband showed a relatively benign prognosis.
Case report
Ethical approval
Written informed consent was obtained from the
proband, her relatives and the 75 healthy controls prior to genetic
testing. This study was approved by the ethics committee of Peking
Union Medical College Hospital (Beijing, China). Written informed
consent was obtained from study participants or the caretakers or
guardians of the minors involved in this study. All clinical
investigations were conducted according to the principles expressed
in the Declaration of Helsinki.
Proband (III-3) patient history
The proband (III-3), a female, was born in a family
native to Benxi, Liaoning Province of China in July 1953. The
patient complained of diarrhea and upper abdominal pain since the
age of 30. At the age of 32, the patient presented with
galactorrhea, secondary amenorrhea, extremity and back pain, and
gross hematuria. She first consulted the Peking Union Medical
College Hospital in March 1988 when she was 35 years old.
Gastroscopic investigation revealed multiple ulcers of the duodenal
bulb and post bulbar area. Endocrine investigation revealed a serum
gastrin level in the fasting state of 550–950 pg/ml (reference
range, 20–160 pg/ml). The ratio of basic acid output (reference
range 3.9±1.98 mmol/h) and maximal acid output was 25.2 (reference
range 3–23 mmol/l), with a ratio of 63.5%. Serum prolactin was 125
ng/ml (reference range, 1.5–11.5 ng/ml). Computer tomography scans
showed pituitary macroadenomas that were less enhanced by the
contrast agent, 3×3×2.5 cm in size, in the pituitary region. The
serum calcium level was 9.6–10.7 mg/dl (reference range 8.4–10.4
mg/dl), and phosphate level was 3.0–4.0 mg/dl (reference range
3.0–5.0 mg/dl). Urine calcium excretion (24 h) was 310 mg
(reference range, <300 mg) and serum parathyroid hormone was
elevated to 37.2 ng/dl (radioimmunoassay normal range, <27
ng/dl). X-ray and ultrasonography did not reveal the presence of
kidney stones. The patient was was diagnosed with MEN1 gastrinoma,
a pituitary tumor and primary hyperparathyroidism. Surgical
treatment was refused by the patient and 20 mg/day of omeprazole
was administered, which relieved diarrhea and upper abdominal
pain.
In the following 2 years, the patient complained of
headaches and her visual field was restricted in her left eye. She
accepted a trans-sphenoidal adenomectomy in May 1992 (at 38 years
of age) in the Peking Union Medical College Hospital. The tumor was
3×3×3 cm in size and sella tursica basement invasion was
identified; the tumor was histological diagnosed as sparsely
granulated cell nonfunctional adenoma. Radiotherapy was also
administered post surgery at a dose of 5,000 rads/month. Following
treatment, the patient's menstrual periods resumed, headaches were
relieved and thyroid function test results were normal. In
addition, the level of adrenocortico-tropic hormone was <13
pg/ml (reference range 25–50 pg/ml); 24 h urine free cortisol level
was 25.8 µg (reference range 60±3.5 µg/24h); serum
gastrin was 180 pg/ml (reference range 50–150 pg/ml); parathyroid
hormone was 36.8 ng/dl (radioimmunoassay normal range <27
ng/dl); serum free calcium ion was 1.31 mmol/l (normal range,
1.1–1.2 mmol/l) and total serum calcium was 10.7 mg/dl (reference
range 2.25–2.75 mmol/l). 131I-MIBI revealed a
radioactivity-dense area in the upper and lower thyroid area.
In September 1993, at 39 years old, the patient
underwent a second surgical procedure because of elevated serum
calcemia and hyperparathyroidism. All four parathyroid glands were
found to be enlarged. All the right side and half of each of the
two left side parathyroid glands (2+1/2+1/2) were resected, which
resulted in normalization of serum calcium and phosphate
levels.
At 40 years old, the patient underwent the advised
pancreatoduodenectomy (whipple procedure) to treat the gastrinoma.
The pancreatic head and neck, duodenal, gallbladder, bile duct,
subtotal gastric (4/5) and pyloric region lymph nodes were removed.
Pathological investigation showed infra-duodenal mucosa and
multiple pancreatic neuroendocrine neoplasias, immunohistochemical
analysis revealed positive cell nuclear staining of gastrin,
chromogranin A and neuron-specific enolase, and negative staining
of 5 hydroxytryptamine, pancreatic polypeptide and glucagon; lymph
node metastases were located in 7/21.
At the age of 45, the patient was diagnosed with
diabetes and insulin treatment was initiated. Thymoma and lung
masses in her left lung were identified at the age of 46; thus, a
fourth surgical procedure was conducted in December 2002. The whole
thymus and one lobe of the left lung were removed from the patient.
Pathological investigation revealed a malignant carcinoid tumor in
the thymus and left lung with lymph nodes metastasis.
Immunohistological analysis revealed that these masses were
positively stained for cytokeratin AE1/AE3 and negatively stained
for chromogranin A and Syn.
The proband, who had poorly-controlled diabetes,
died of cerebral hemorrhage after severe accidental trauma in
December 2007 at the age of 54.
Family history
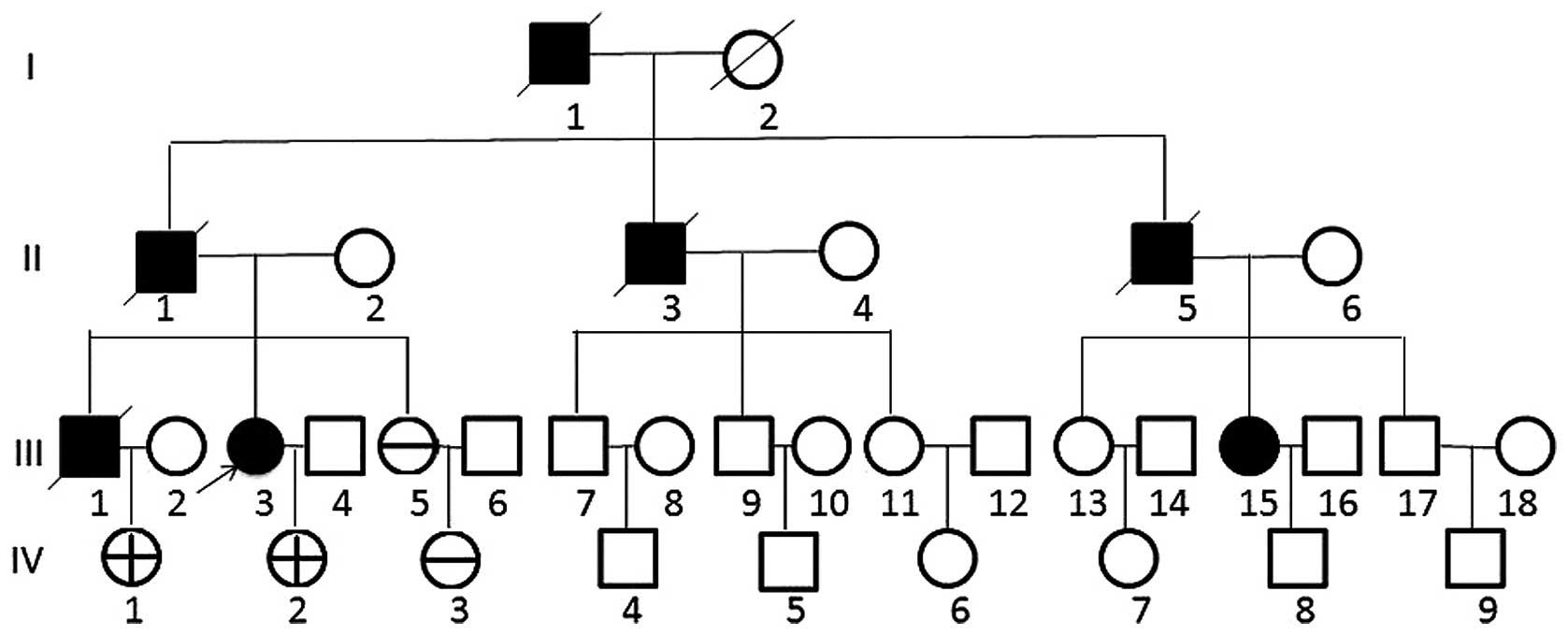
The pedigree of this family is shown in Fig. 1. Five members (III-3, III-5, IV-1,
IV-2 and IV-3) of the family were screened. In order to confirm the
presence of the mutation in the family, 75 healthy controls from a
health checkup population were also enrolled in the present study.
A detailed illness and family history were taken; pituitary,
primary parathyroiddism, hypertension, peptic ulcer history and
family history were negative. Detailed family history and venous
blood samples were first studied in May 2003. The patient's
grandfather (I-1) succumbed to severe stomach disease at 84 years
old. Her father (II-1) was diagnosed with gastrinoma and underwent
surgery at 56 years old, he succumbed to malignant thymoma at 70
years old. An uncle (II-3) succumbed to stomach disease and kidney
stones. An aunt (II-5) had kidney stones, and her daughter (III-15)
had nonfunctional pituitary adenoma and underwent surgery at the
Peking Union Medical College Hospital at the age of 36 years old in
1986. The brother of the proband (III-1) succumbed to gastrinoma of
the pancreas, malignant thymoma with lymph node, brain and bone
metastasis at the age of 55. The proband's sister (III-5) was 47
years old in 2003, her two nieces (IV-1 and IV-3) were 23 and 20
years old, respectively and her daughter (IV-2) was 20 years old;
all four were in good health.
MEN1 gene mutation analysis
Genomic DNA was extracted by standard methods from
peripheral leukocytes. The coding sequence, including 9 coding
exons and 16 splice junctions of the MEN1 gene of leukocyte
DNA, was determined. DNA fragments ranging from 240 to 393 bp in
length were amplified from leukocyte DNA, genomic DNA was amplified
in the presence of 75 ng of each oligonucleotide primer and 1.5
units of pfu Taq polymerase (Tianwei, Beijing, China) in a
final volume of 50 ml of the following solution: 0.5 mmol/l
deoxynucleotide triphosphates, 1 or 2 mmol/l MgCl2, 67
mmol/l Tris (pH 8.8), 16.6 mmol/l ammonium sulfate, 6.7 mmol/l EDTA
and 10 mmol/l 2-mercaptoethanol. The primer sequences used were as
previously described (9,10), derived from the DNA sequence
(Genbank accession no. U93237) and synthesized by Shanghai Bioasia
(Shanghai,China). Amplification proceeded by a denaturation step of
5 min at 95°C, followed by 33 cycles of 1 min at 95°C, 2 min at
55°C and 3 min at 72°C, and a final extension step of 8 min at 72°C
using a PTC-150 Thermocycler (MJ Research, Inc., St. Bruno, QC,
Canada). PCR products were purified with 2% agarose gel
electrophoresis and sequenced with Sanger dideoxy chain termination
(Shanghai Bioasia;), The sequences obtained were matched with the
published MEN1 gene (Genbank accession no. U93237). The
entire coding region of the MEN1 gene, including the
exonintron boundaries, was sequenced. The direct DNA sequencing
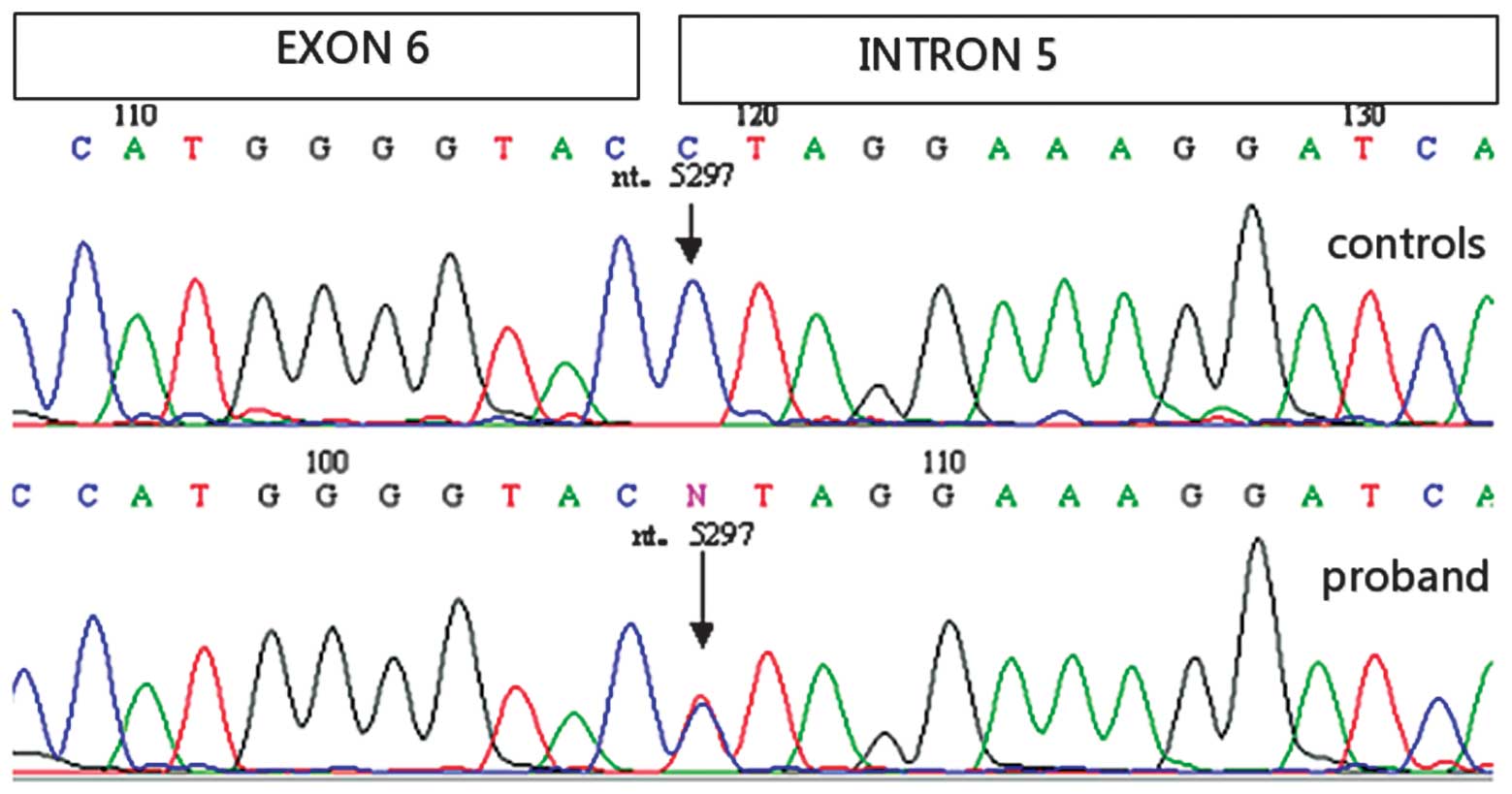
results of the proband (III-3) revealed heterozygous G to A
variation at the nucleotide position-1 of intron 5 (c.825-1G>A
or IVS5-1G>A. DNA mutations in the MEN1 gene were not
observed in the 75 healthy controls (Fig. 2). No other mutations were found in
the coding regions and the exonintron boundaries regions. The
heterozygous germline mutation was also identified in the patient's
daughter (IV-2) and one niece (IV-1), but not in another niece
(IV-3).
Follow up
Familial history in 2003 revealed that the sister
(III-5), nieces (IV-1 and IV-3) and daughter (IV-2) of the proband
were clinically normal, with negative history of stomach disease,
kidney stones, menstrual disorders, hypertension, lipoma and other
endocrine disorders. In addition, serum calcium, phosphate and
alkaline phosphatase levels were all normal.
For private reasons, one carrier (IV-1), who was 32
years old and fathered one child in 2012, was not accepted for
further investigation.
The final follow up occurred in March 2012, the
family history was provided by the proband's sister (III5) and her
daughter (IV-2). The proband's daughter (IV-2) had 2 sons (7 years
old and 7 months old in 2012). She was in good health except from
mild anemia, with no complaints of stomach pain, hypoglycemia or
bladder stones. The laboratory findings are summarized in Table I. The patient was observed to have
an elevated serum parathyroid hormone level and normal serum
calcium level; however, after 8 years of follow up, parathyroid
ultrasound did not detect enlarged parathyroid glands.
 | Table ILaboratory findings of IV-2 tested in
March 2003 and March 2012. |
Table I
Laboratory findings of IV-2 tested in
March 2003 and March 2012.
| Clinical
observation | Reference range | March 2003 | March 2012 |
|---|
| Hemoglobin (g/l) | 110–150 | 135 | 101 |
| Red blood cells
(×1012/l) | 3.5–4.5 | 4.2 | 3.4 |
| ACTH
(pg/µl) | 25.0–65.0 | 28.2 | 32.0 |
| Plasma Glucose
(mmol/l) | 3.1–6.4 | 3.5 | 4.5 |
| Insulin
(µU/ml) | 2.6–24.9 | 5.1 | 6.1 |
| Serum calcium
(mmol/l) | 2.15–2.55 | 2.35 | 2.45 |
| Serum phosphorus
(mmol/l) | 0.87–1.45 | 1.12 | 1.01 |
| Alkaline phosphatase
(IU/l) | 40–150 | 68 | 108 |
| Parathyroid hormone
(pg/ml) | 15.0–65 | 92 | 167 |
| PRL
(ng/µl) | 3.4–24.1 | 15 | 23.0 |
| TSH
(µIU/µl) | 0.30–3.9 | 0.39 | 0.45 |
| Gastrin (pg/ml) | 50–150 | 80 | 235 |
| GH
(ng/µl) | 0–2 | 0.7 | 0.8 |
Discussion
A Chinese MEN1 pedigree was investigated in this
study. MEN1 gene sequence analysis revealed the presence of
a heterozygous mutation c.825-1G>A (IVS 5-1 G>A). Two
carriers were identified and followed in the pedigree. The family
was followed up for over 20 years.
Since the cloning of the gene in 1997, 1,336
mutations and 24 normal allelic variants had been described by
2008. Intron 5 mutations were reported in two different studies
(935-1G>C) (11,12). A Japanese 66-year-old male
presenting with prolactinoma, a gastrin-secreting carcinoid tumor
and multiple parathyroid lessons. The mutation caused transcription
skipping of exon 6 (88 bp), resulting in a frame shift mutation and
premature termination codon. Raghavan et al (12) presented another case of a
35-year-old with multiple gastrinomas, pituitary microadenoma,
hyperparathyroidism, a serum gastrin level of 2,000 pg/ml (normal,
<100 pg/ml), and markedly elevated serum calcium and PTH. The
patient underwent 3.5/4 parathyroid gland excision was performed
and pituitary microadenoma was treated with bromocriptine. The
clinical outcome and follow up of the patients was not mentioned in
these two case reports. The proband in the present study presented
with a mutation that was different but in the same site as the
mutations of these two cases. The proband had a clear family
history and presented with gastrinoma, pituitary tumor, parathyroid
hyperplasia, thymoma and a lung malignant carcinoid tumor. Thus,
this suggests that mutations in this site cause a similar
phenotype.
As reported by the National Cancer Institute summit
meeting in 2007 on GEP-NETs (13),
standardized clinical management is often limited by different
aspects of the disease. For example, the relative rarity, the
limited understanding of tumor biology and behavior, heterogeneous
clinical presentation, and the lack of prospectively evaluated risk
stratification systems, has resulted in incomplete implementation
of staging systems. Evidence for proper follow up and controlled
study of treatment efficacy are poor (14,15).
Patients with MEN1 have a decreased life expectancy, and the
outcomes of current treatments, which are generally similar to
those for the respective tumors occurring in non-MEN1 patients, are
not as successful. This is due to the presence of multiple tumors,
which may be larger, more aggressive, and resistant to treatment,
as well as the presence of metastases (16). These characteristics of MEN1 tumors
thereby render it difficult to achieve a successful cure. For
example, patients with MEN1 often develop multiple submucosal
duodenal gastrinomas, thereby reducing surgical cure rates compared
with similar sporadic solitary tumors, (17). Patients with MEN1 also develop
multiple parathyroid tumors, and subtotal parathyroidectomy has
been shown to result in persistent or recurrent hypercalcemia
within 10 years in 20–60% of patients with MEN1, as opposed to ~4%
in patients without MEN1 (18).
The proband underwent four surgical procedures and was followed up
for 20 years. She died of accidental trauma and had a relatively
stable illness course. This indicates that although multiple organs
and systems were involved in MEN1, MEN1 tumors should be considered
surgically curable diseases if the patients are properly cared for
by multidisciplinary teams comprising relevant specialists with
experience in the diagnosis and treatment of patients with
endocrine tumors. Recent studies support this hypothesis. For
example, duodenal gastrinoma in patients with MEN1 could be
considered a surgically curable disease by pancreaticoduodenectomy
with a high cure rate (19).
In conclusion, an MEN1 pedigree was reported in the
present study. The clinical course of the proband was followed up
for 20 years. The clinical course of the patient indicates that
although MEN1 is a complex disease involving multiple organs and
systems, patients with MEN1 may have a relatively benign prognosis
after proper treatment. MEN1 tumors should thus be considered as
surgically curable.
Acknowledgments
This study was supported by Professor Xunwu Meng.
The MEN1 gene analysis was conducted in Peking Union Medical
College Hospital. The follow up in April 2012 was conducted in
Beijing Chaoyang Hospital.
References
|
1
|
Chandrasekharappa SC, Guru SC, Manickam P,
Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z,
Lubensky IA, Liotta LA, et al: Positional cloning of the gene for
multiple endocrine neoplasia-type 1. Science. 276:404–407. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lemmens I, Van de Ven WJ, Kas K, Zhang CX,
Giraud S, Wautot V, Buisson N, De Witte K, Salandre J, Lenoir G, et
al: Identification of the multiple endocrine neoplasia type 1
(MEN1) gene. The European Consortium on MEN1. Hum Mol Genet.
6:1177–1183. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pannett AA and Thakker RV: Somatic
mutations in MEN type 1 tumors, consistent with the Knudson
'two-hit' hypothesis. J Clin Endocrinol Metab. 86:4371–4374.
2001.PubMed/NCBI
|
|
4
|
Carling T: Multiple endocrine neoplasia
syndrome: Genetic basis for clinical management. Curr Opin Oncol.
17:7–12. 2005. View Article : Google Scholar
|
|
5
|
Leotlela PD, Jauch A, Holtgreve-Grez H and
Thakker RV: Genetics of neuroendocrine and carcinoid tumours.
Endocr Relat Cancer. 10:437–450. 2003. View Article : Google Scholar
|
|
6
|
Brandi ML, Gagel RF, Angeli A, Bilezikian
JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG,
Libroia A, et al: Guidelines for diagnosis and therapy of MEN type
1 and type 2. J Clin Endocrinol Metab. 86:5658–5671. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lemos MC and Thakker RV: Multiple
endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations
reported in the first decade following identification of the gene.
Hum Mutat. 29:22–32. 2008. View Article : Google Scholar
|
|
8
|
Eckel F and Jelic S; ESMO Guidelines
Working Group: Billiary cancer: ESMO clinical recommendation for
diagnosis, treatment and follow-up. Ann Oncol. 20(Suppl 4): 46–48.
2009. View Article : Google Scholar
|
|
9
|
Mailman MD, Muscarella P, Schirmer WJ,
Ellison EC, O'Dorisio TM and Prior TW: Identification of MEN1
mutations in sporadic enteropancreatic neuroendocrine tumors by
analysis of paraffin-embedded tissue. Clin Chem. 45:29–34.
1999.PubMed/NCBI
|
|
10
|
Nagamura Y, Yamazaki M, Shimazu S, Sano K,
Tsukada T and Sakurai A: A novel splice site mutation of the MEN1
gene identified in a patient with primary hyperparathyroidism.
Endocr J. 59:523–530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hai N, Aoki N, Shimatsu A¸ Mori T and
Kosugi S: Clinical features of multiple endocrine neoplasia type 1
(MEN1) phenocopy without germline MEN1 gene mutations: Analysis of
20 Japanese sporadic cases with MEN1. Clin Endocrinol (Oxf).
52:509–518. 2000. View Article : Google Scholar
|
|
12
|
Raghavan R, Shah S, Kondkar AA, Dherai AJ,
Desai D, Chauhan P, Lala M and Ashavaid TF: MEN1 935-1G>C
splicing mutation in an Indian patient with multiple endocrine
neoplasia type 1. Mol Diagn Ther. 11:129–131. 2007. View Article : Google Scholar
|
|
13
|
Modlin IM, Moss SF, Chung DC, Jensen RT
and Snyderwine E: Priorities for improving the management of
gastroen-teropancreatic neuroendocrine tumors. J Natl Cancer Inst.
100:1282–1289. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Machens A, Schaaf L, Karges W, Frank-Raue
K, Bartsch DK, Rothmund M, Schneyer U, Goretzki P, Raue F and
Dralle H: Age related penetrance of endocrine tumours in multiple
endocrine neoplasia type 1 (MEN1): A multicentre study of 258 gene
carriers. Clin Endocrinol (Oxf). 67:613–622. 2007.
|
|
15
|
Ehehalt F, Saeger HD, Schmidt CM and
Grützmann R: Neuroendocrine tumors of the pancreas. Oncologist.
14:456–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thakker RV, Newey PJ, Walls GV, Bilezikian
J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML,
et al: Clinical practice guidelines for multiple endocrine
neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 97:2990–3011.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Imamura M, Komoto I, Ota S, Hiratsuka T,
Kosugi S, Doi R, Awane M and Inoue N: Biochemically curative
surgery for gastrinoma in multiple endocrine neoplasia type 1
patients. World J Gastroenterol. 17:1343–1353. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schreinemakers JM, Pieterman CR, Scholten
A, Vriens MR, Valk GD and Rinkes IH: The optimal surgical treatment
for primary hyperparathyroidism in MEN1 patients: A systematic
review. World J Surg. 35:1993–2005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lopez CL, Falconi M, Waldmann J,
Boninsegna L, Fendrich V, Goretzki PK, Langer P, Kann PH, Partelli
S and Bartsch DK: Partial pancreaticoduodenectomy can provide cure
for duodenal gastrinoma associated with multiple endocrine
neoplasia type 1. Ann Surg. 257:308–314. 2013. View Article : Google Scholar
|