Introduction
Apoptosis is a conserved and irreversible process,
which allows cells to undergo a tightly controlled form of death
and is essential for normal development and homeostasis (1,2).
Apoptosis is a process, which involves the body removing unused or
harmful cells in order to protect itself, and the deregulation of
apoptosis is the predominant obstacle in the successful treatment
of cancer. Inhibiting the expression of anti-apoptotic factors or
inducing pro-apoptotic factors contributes to the apoptosis of
cancer cells (3). The B-cell
lymphoma-2 (Bcl-2) apoptotic protein family, one of the key
apoptotic gene families, contains pro-apoptotic and anti-apoptotic
members, all of which act as critical regulators of apoptosis
(4,5). Members of the Bcl-2 protein family
include three subgroups of proteins, which either promote cell
survival, including Bcl-2 and B-cell lymphoma-extra large (Bcl-xl),
initiate cell killing, including Bcl-2-like protein 11 and BH3
interacting-domain death agonist or activate the effector pathways
of apoptosis, including B-cell-associated X protein (Bax) and Bcl-2
homologous antagonist/killer (6).
Previous studies have demonstrated that small
non-protein-coding molecules, termed microRNAs (miR) serve key
roles in homeostatic processes. These processes include cell
development, proliferation and apoptosis (7–9), in
addition to the regulation of gene expression at the translational
level (10) by recognizing and
binding to the 3′ untranslated region (3′UTR) of the target mRNA
with its seed region (11).
However, the role of miR in apoptosis remains to be fully
elucidated. Previous studies have demonstrated that miRs are
important in this process, particularly in regulating the Bcl-2
gene family (12–14). miR-29 has been reported to promote
apoptosis through a mitochondrial pathway, which involves myeloid
leukemia cell (Mcl)-1 and Bcl-2 (15), while the downregulation of
miR-125b, has been reported to inhibit the expression of Bcl-2 and
promote apoptosis in hepatocellular carcinoma (HCC) (16). It has been reported that miR-125b
is able to reduce the cellular proliferation and cell cycle
progression of HCC cells by targeting Mcl-1 and the interleukin 6
receptor (17). Bcl-w
downregulation by miR-122 has been reported to result in apoptosis
in the Hep3B and HepG2 HCC cell lines (18). miRs are also able to modulate
cancer cell sensitivity to anticancer therapy via the Bcl-2 gene
family (19). Qiu et al
(20) reported that miR-503
regulates cell apoptosis by targeting Bcl-2, and modulates the
resistance of non-small cell lung cancer cells to cisplatin (DDP).
miRNA-195 is downregulated in BEL-7402/5-fluorouracil (5-FU) cells,
and overexpression of miRNA-195 sensitizes BEL-7402/5-FU cells to
5-FU by targeting Bcl-w to increase cell apoptosis (21).
Bcl-xl, an anti-apoptotic Bcl-2 family member, is
regulated by miRs (22,23). However, the mechanisms underlying
the association between miRNAs and Bcl-xl remain to be elucidated.
The present study aimed to investigate whether miR-133a and miR-326
contribute to the regulation of chemotherapy resistance, mediated
by Bcl-xl, by examining cell apoptosis in the HepG2 HCC cell
line.
Materials and methods
Materials
The human HepG2 HCC cell line was purchased from the
Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum
(FBS) were obtained from GE Healthcare Life Sciences (Beijing,
China). 5-FU and DDP were purchased from Sigma-Aldrich (St. Louis,
MO, USA). The miR mimics were purchased from Shanghai GenePharma
Co., Ltd. (Shanghai, China).
Identification of miR target sites
The miRanda (http://www.microrna.org/), TargetScan (http://www.targetscan.org/vert_61/), Pictar
(http://pictar.mdc-berlin.de/cgi-bin/PicTar_vertebrate.cgi)
and miRBase (http://www.mirbase.org/search.shtml) algorithms were
used to predict the putative target genes of miR-133a and
miR-326.
Cell culture
The human HepG2 HCC cell line was maintained in
DMEM, supplemented with 4.5 g/l glucose, 10% FBS and 1%
penicillin/streptomycin (GE Healthcare Life Sciences) in a
humidified atmosphere containing 5% CO2 at 37°C.
Construction of 3′UTR-luciferase reporter
plasmids
The full-length Bcl-xl cDNA construct containing the
entire 3′UTR was synthesized and cloned into a pcDNA3.1 plasmid
(Landbiology, Guangzhou, China). Site directed mutagenesis using
the QuikChange Site-Directed Mutagenesis kit (Agilent Technologies
GmbH, Waldbronn, Germany) using the following primers (Shanghai
Novland Co., Ltd., Shanghai, China): Forward,
5′-GCTCCCATGACCATACTGAGCCTGGTTCTGGGCCCAAGACAGATGCC-3′ and reverse
5′-GGCATCTGTTCTTGGGCCCAGAACCAGGCTCAGTATGGTCATGGGAGC-3′. The 3′UTR
(wild-type and mutant) of Bcl-xl was synthesized and was inserted
downstream of the Renilla luciferase gene of the
dual-luciferase. The primer for Bclxl was designed, and targeted
fragments were obtained by PCR amplification (Stratagene, Santa
Clara, CA, USA). Subsequently, the specificity of the PCR primers
was analyzed through digesting PCR products with restriction
endonuclease. The target gene was then linked with the vector and
recombinant plasmids were amplified by transformation into E.
coli (Landbiology). Positive clones were selected randomly and
sequenced for verification.
Luciferase assay
The wild-type and mutant Bcl-xl 3′UTR-luciferase
reporter plasmids were co-transfected with a miR-133a or miR-326
mimic (Shanghai GenePharma, Co., Ltd.) into the HEK923 cells
(American Type Culture Collection, Manassas, VA, USA) using
Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA,
USA). At 48 h post-transfection, the cells were assayed for
luciferase activity using a Dual-Luciferase Assay system (Promega
Corporation, Madison, WI, USA), according to the manufacturer's
instructions. The Renilla luciferase activities were
normalized to the corresponding activities of firefly luciferase.
For each transfection, the average luciferase activity was
calculated from three replicates.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RT-qPCR was performed, as previously reported
(24). Total RNA was extracted
from the cells using TRIzol reagent (Tiangen Biotech Co., Ltd.,
Beijing, China), according to the manufacturer's instructions. The
primer sequences for Bcl-xl, Bax, GAPDH, miR-133a, miR-326 and U6
were as follows: Bcl-xl, forward 5′-GCTGGTGGTTGACTTTCTCTCCTAC-3′
and reverse 5′-CCTCAGTCCTGTTCTCTTCCACATC-3′; Bax, forward
5′-CCCTTTTCTACTTTGCCAGCA-3′ and reverse 5′-GGAGTCTCACCCAACCACCC-3′;
GAPDH, forward 5′-CATGAGAAGTATGACAACAGCCT-3′ and reverse
5′-AGTCCTTCCACGATACCAAAGT-3′; miR-133a, forward
5′-TCATATTTGGTCCCCTTCAACC-3′ and reverse
5′-TATCGTTGTTCTCCACTCCTTCAC-3′; miR-326, forward
5′-ACTGTCCTTCCCTCTGGGC-3′ and reverse
5′-AATGGTTGTTCTCCACTCTCTCTC-3′; and U6 small nuclear RNA, forward
5′-ATTGGAACGATACAGAGAAGATT-3′ and reverse
5′-GGAACGCTTCACGAATTTG-3′. The U6 small nuclear RNA was used as an
internal control for miR quantification. To determine the mRNA
expression levels of Bcl-xl and Bax, the total RNA were reverse
transcribed into cDNA using a Custom Reverse Transcription kit. The
qPCR analyses of Bcl-xl and Bax were performed using the
above-mentioned primers, and normalized to GADPH. SYBR green qPCR
was performed on a MX3000P PCR System (Agilent Technologies, Inc.,
Santa Clara, CA, USA). The cycling programme consisted of 40
cycles, with each cycle consisting of 3 min at 95°C, 12 sec at 95°C
and 40 sec at 62°C. The relative expression ratios of Bcl-xl, Bax,
miR-133a, miR-326 were calculated using the 2−ΔΔCt
method (25).
Cell transfection
The cells were transfected using Lipofectamine 2000
transfection reagent according to the manufacturer's instructions
(Invitrogen Life Technologies) The sequences for the two miRNA
mimics were as follows: miR-133a: 5′-uuugguccccuucaaccagcug-3′;
miR-326: 5′-ccucugggcccuuccuccag-3′. At 1 day prior to
transfection, the cells were plated in the appropriate quantity of
growth medium without antibiotics in order to reach 80–90%
confluency at the time of transfection. For each transfection
sample, miRNA mimics- Lipofectamine 2000 complexes were prepared as
follows: miRNA mimics (Shanghai GenePharma, Co., Ltd.) were diluted
in the appropriate quantity of Opti-minimum essential medium (MEM;
Gibco-BRL, Grand Island, NY, USA) without serum and mixed gently.
Lipofectamine 2000 was diluted in the appropriate quantity of
Opti-MEM without serum then mixed gently. The samples were
incubated for 5 min at room temperature. The diluted miRNA mimics
were then combined with the diluted Lipofectamine 2000 and
incubated for 20 min at room temperature to allow complex formation
to occur. The miRNA mimics-Lipofectamine 2000 complexes were added
to each well containing cells and medium. The cells were incubated
at 37°C in a CO2 incubator until harvesting of cells and
the assay for the target gene.
Western blot analysis
At 48 h post-transfection, the cell lysates were
prepared by lysis in radioimmunoprecipitation assay buffer (CWBIO,
Beijing, China) with protease inhibitors. Centrifugation was then
performed at 10,000–14,000 × g for 15 min at 4°C, and the protein
concentration was determined using a bicinchoninic acid assay
(CWBIO). The proteins (30–50 µg) were separated on 10%
SDS-polyacrylamide gels (CWBIO) and were transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). A PageRuler prestained protein ladder (CWBIO) was used as a
molecular marker. A single membrane was cut into three parts at the
43 kD, 30 kD and 20 kD bands, and incubated with anti-β-actin
(1:1,000; cat. no. 12620), anti-Bcl-xl (1:1,000; cat. no. 2764) and
anti-Bax (1:1,000; cat. no. 5023) monoclonal rabbit primary
antibodies (Cell Signaling Technology, Inc., Danvers, MA, USA),
respectively. The proteins were detected using a horseradish
peroxidase-conjugated goat anti-rabbit IgG secondary antibody
(1:4,000; cat. no. CW0103; CWBIO) and a ChemiLucent ECL Detection
system (EMD Millipore, Billerica, MA, USA). Quantification of the
protein bands was performed using AlphaImager 2200 software
(National Institutes of Health, Bethesda, MD, USA).
MTT analysis
The HepG2 cells in the exponential growth phase were
seeded into 96-well plates at a density of 4×103
cells/well and were incubated for 12 h at 37°C. The cells were
transfected with either the miR-133a mimic, miR-326 mimic or the
negative control (NC). At 24 h post-transfection, various
concentrations of 5-FU (0, 5, 50, 500 and 5,000 µM), or DDP
(0, 6.25, 12.5, 25, 50 and 100 µM) were added. Subsequent to
incubation for 24 h, 20 µl MTT (Amresco LLC, Solon, OH, USA)
solution, at a concentration of 5 mg/ml, was added to each well for
4 h at 37°C. The culture medium was then removed, and the insoluble
formazan crystals were dissolved in 150 µl dimethyl
sulf-oxide (Beijing Dingguo Changsheng Biotechnology Co., Ltd.,
Beijing, China). Following agitation for 10 min, the absorbance at
490 nm (A490) was optically monitored using a 318-microplate reader
(Shanghai Sanco Instrument Co., Ltd., Shanghai, China).
Statistical analysis
SPSS software, version 18.0 (SPSS, Inc., Chicago,
IL, USA) was used for statistical analysis. All values are
expressed as the mean ± standard deviation. One-way analysis of
variance was performed to evaluate the significance of the
differences between samples. P<0.05 was considered to indicate a
statistically significant difference.
Results
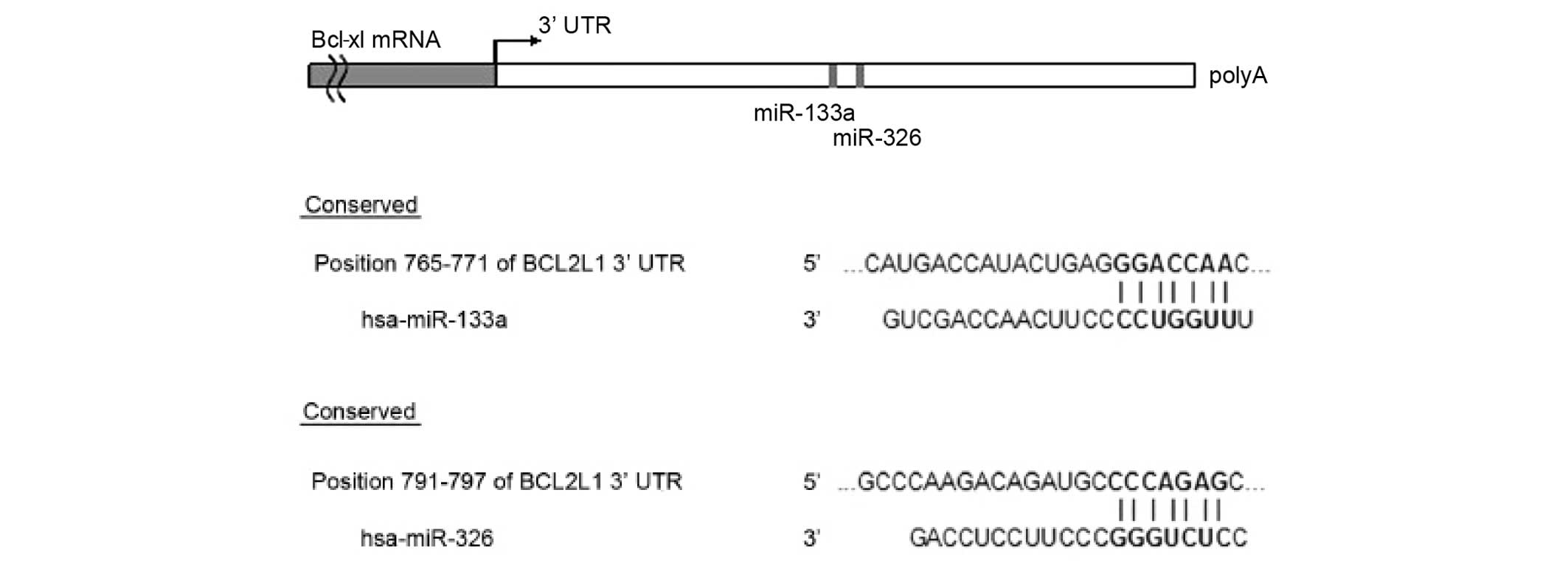
miR-133a and miR-326 share a common
target gene, Bcl-xl
To examine the mechanism of miR-133a and miR-326,
the TargetScan, Pictar, miRanda and miRBase computational programs
were used to predict putative terget genes for the two miRs. The
programs predicted the common target gene for miR-133a and miR-326,
as the human Bcl-xl gene (Fig.
1).
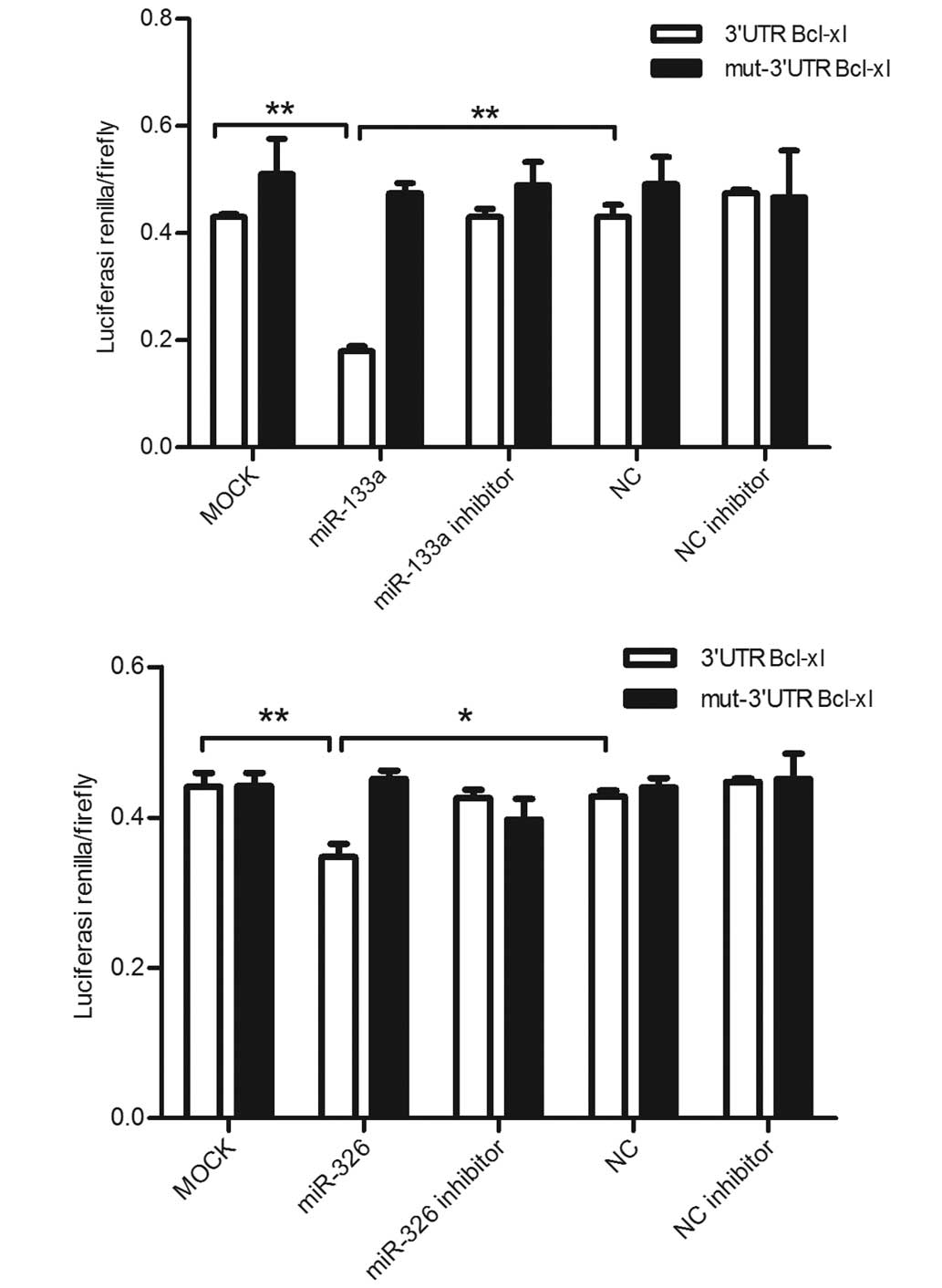
Confirmation of putative miR binding
sites for miR-133a and miR-326 using luciferase reporter
assays
The present study confirmed the predicted binding
sites recognized by miR-133a and miR-326 using the pmirGLO
dual-luciferase vector. The wild-type, containing binding sited
with the Bcl-xl3′UTR or mutant, containing binding sites with
deletion of the Bcl-xl were cloned downstream of firefly luciferase
using the pmirGLO vector and were co-transfected with miR-133a or
miR-326 in the HEK293 cells. As demonstrated in Fig. 2, in the cells transfected with the
vector containing the Bcl-xl 3′UTR fragment with the binding site
for Bcl-xl, the luciferase activity was significantly inhibited
following transfection of miR-133a compared with the cells that
were transfected with the NC miR mimics and untreated cells.
Similar results were observed in the cells transfected with
miR-326.
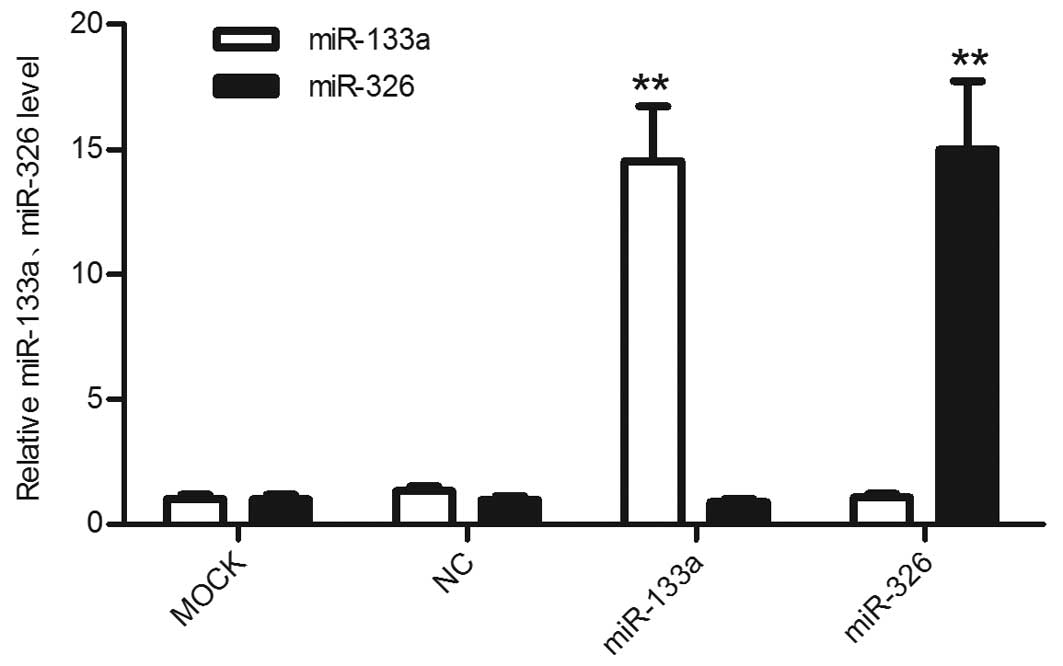
Expression levels of miR-133a and miR-326
are significantly upregulated subsequent to transfection
Using RT-qPCR, the differences in the expression
levels of miR-133a and miR-326 between the mimic-transfected cells
and untreated cells were assessed. The miR expression levels in the
HepG2 cells transfected with the miR-133a or miR-326 mimics were
significantly upregulated compared with the control cells and the
untreated cells (Fig. 3).
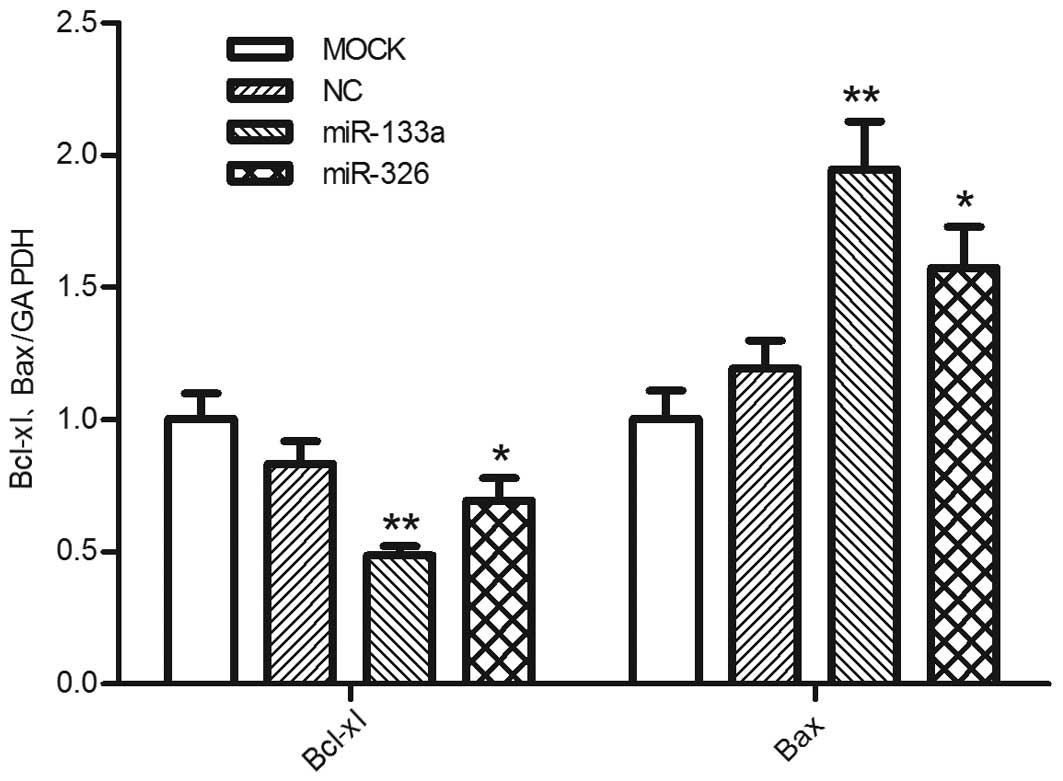
miR-133a and miR-326 downregulate the
mRNA expression of Bcl-xl
RT-qPCR was performed to assess whether miR-133a and
miR-326 affected the mRNA expression of Bcl-xl. The data
demonstrated that transfection with either miR-133a or miR-326
reduced the mRNA expression levels of Bcl-xl compared with the
negative control group, whereas the expression levels of Bax were
increased, as expected (Fig. 4).
This suggested that mRNA degradation may be an important mechanism
underlying the miR-133a and miR-326-mediated post-transcriptional
regulation of Bcl-xl.
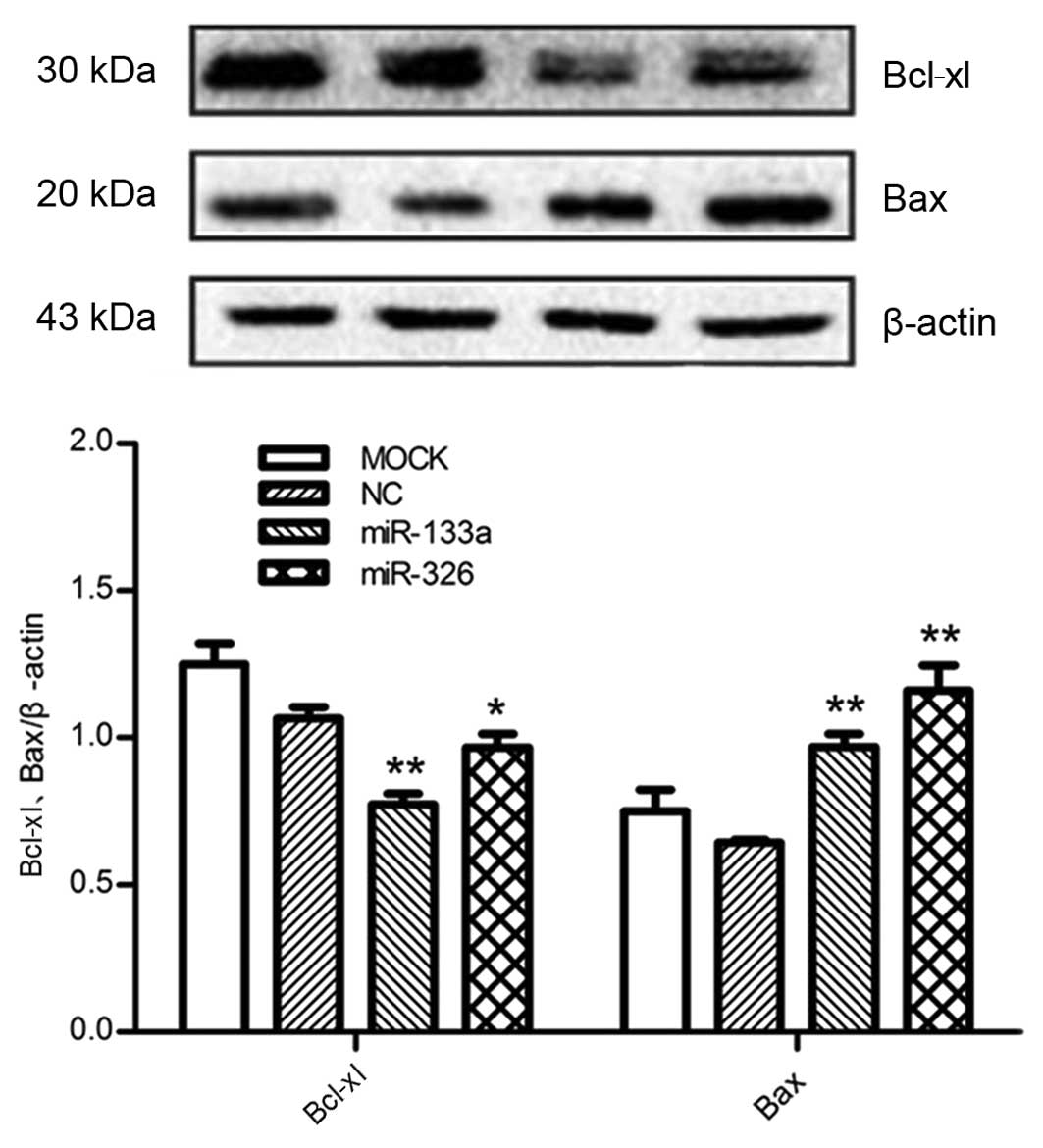
miR-133a and miR-326 significantly alter
the protein expression of Bcl-xl
In order to examine the effect of miR-133a and
miR-326 on the endogenous expression of Bcl-xl, the miR-133a,
miR-326 or negative control mimics were transfected into HepG2
cells. The results demonstrated that enhanced expression of
miR-133a or miR-326 significantly reduced the protein expression
levels of Bcl-xl compared with negative control and
mock-transfected cells, whereas the protein levels of Bax exhibited
the opposite effects (Fig. 5),
consistent with the changes in mRNA levels.
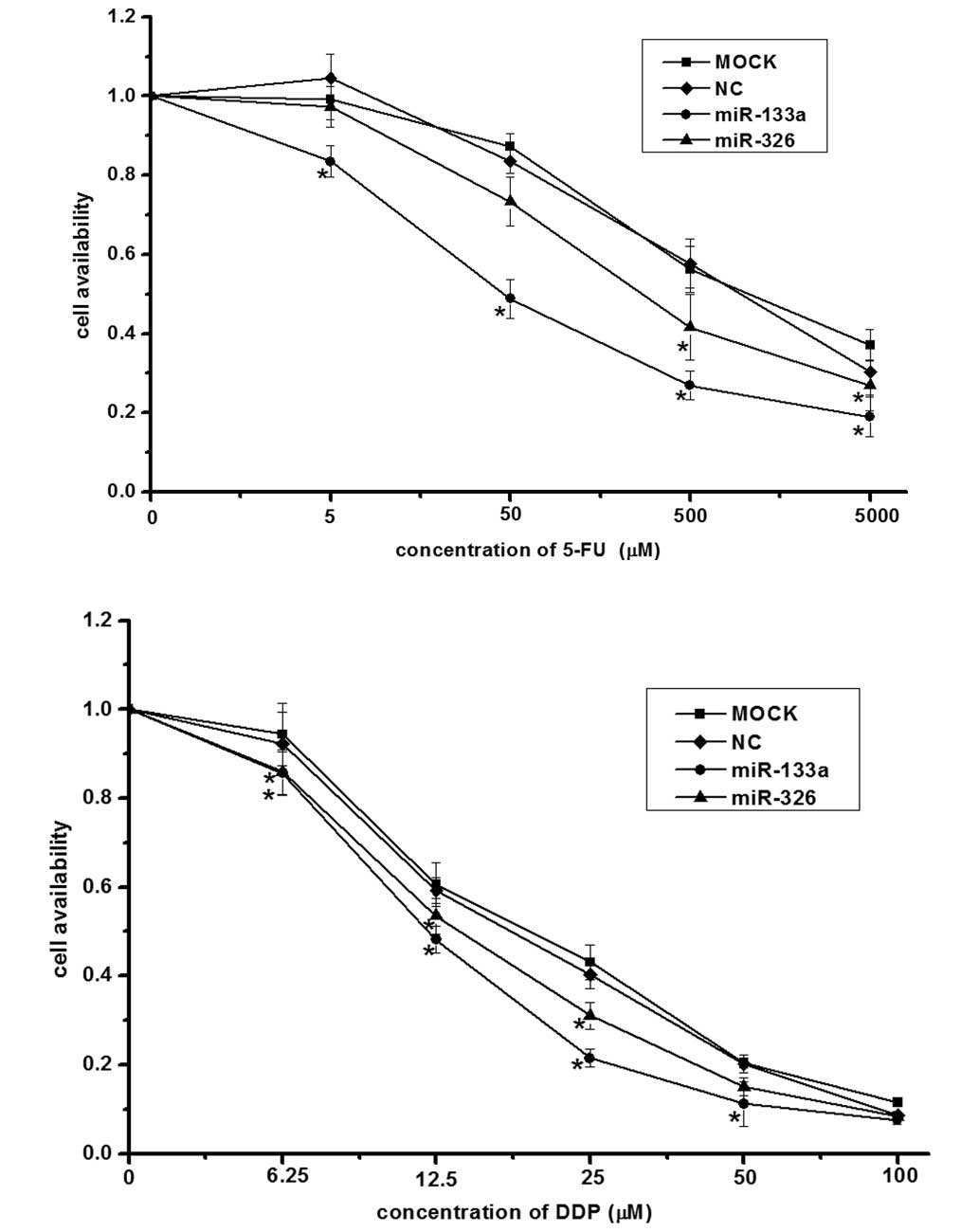
miR-133a and miR-326 sensitize HepG2
cells to 5-FU and DDP
To investigate whether miR-133a and miR-326
sensitize HepG2 cells to 5-FU and DDP, the miR-133a mimic, miR-326
mimic or negative control plasmids were transfected into the HepG2
cells. An MTT cell proliferation assay was performed to determine
the rate of cell survival. The results indicated that transfection
with the miR-133a or miR-326 mimics significantly reduced cell
viability compared with negative control cells and the
mock-transfected cells (Fig.
6).
Discussion
Thousands of human genes have been implicated as
potential miR targets (26), and
previous studies have suggested that the downregulation of miRs is
involved in cancer progression (27,28).
miRs have also been reported to function as oncogenes or tumor
suppressors by targeting specific cancer-associated genes (29,30).
Although the involvement of miRs has been observed in various types
of cancer, the molecular mechanisms by which they modulate cancer
progression and the behavior of cancer cells, including cell
apoptosis and drug resistance, remain to be fully elucidated. In
the present study, miR-133a and miR-326 sensitized the HepG2 HCC
cell line to 5-FU and DDP. Furthermore, transfection of the cells
with miR-133a and miR-326 led to the marked induction of HepG2 cell
apoptosis. In the present study, Bcl-xl was further characterized
as the common functional target of miR-133a and miR-326, using
computational prediction programs, and the specific binding
associations were confirmed using a dual-luciferase reporter assay.
Transfection of the HepG2 cells with the miR-133a and miR-326
mimics significantly repressed the mRNA and protein expression
levels of Bcl-xl in the HepG2 cells. These findings, together with
those of the previous studies, indicate the importance of miR-133a
and miR-326 in HepG2 drug resistance and implicate the potential
application of miR-133a and miR-326 in cancer therapy.
Apoptosis is a process, which requires circumvention
for tumor progression to occur. The human body removes unused or
harmful cells to protect itself, however, cancer cells evolve to
resist cell death, avoiding the surveillance system and surviving
in the tumor environment (31). A
previous study demonstrated that miR-133a is downregulated in
osteosarcoma cell lines, and is able to reduce cell proliferation
and promote cell apoptosis by targeting and repressing the
expression of Bcl-xl and Mcl-1 (32). It has also been reported that the
overexpression of miR-326 causes cell cycle arrest at the
G1 phase, delays cell proliferation and enhances
apoptosis by targeting the nin one binding protein in human glioma
cell lines (33). An additional
report demonstrated that miR-326 is involved in chemotherapy
resistance in breast cancer by directly modulating the expression
of multidrug resistance-associated protein 1 (34). In the present study, the HepG2 HCC
cell line was used as a novel model to investigate the
apoptosis-associated genes by which miR-133a and miR-326 exert
their functions. Bcl-xl was confirmed as a common target of
miR-133a and miR-326, and miR-133a and miR-326 were confirmed as
being important in the promotion of apoptosis and cancer cell drug
resistance. The findings of the present study, and the previously
mentioned studies, demonstrated that miR-133a and miR-326 may
target multiple proteins, which function in or cooperate in
different cellular processes, including cell apoptosis.
In conclusion, the present study demonstrated the
functions of miR-133a and miR-326 in the regulation of Bcl-xl
expression, and in Bcl-xl-mediated cell apoptosis and drug
resistance. These results contribute to a further understanding of
apoptosis-associated multiple drug resistance in tumor cells.
Additionally, as the introduction of a single miR can modulate
complex genes, miRs may be used as potential prognostic markers and
therapeutic targets for the treatment of HCC.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81372579 and
30900625); the Science and Technology Department Projects of Hunan
Province (grant no. 2012FJ2016); the Science and Technology
Development Project of Hengyang (grant no. 2012KS13); the Hunan and
Innovation Fund Project of the Hunan Higher Education Institution
(grant no. 13K084) and the Construction Projects of Provincial Key
Disciplines.
References
|
1
|
Witko-Sarsat V: Apoptosis, cell death and
inflammation. J Innate Immun. 2:201–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ghatage DD, Gosavi SR, Ganvir SM and
Hazarey VK: Apoptosis: Molecular mechanism. J Orofac Sci.
4:103–107. 2012. View Article : Google Scholar
|
|
3
|
Schultz DR and Harrington WJ Jr:
Apoptosis: Programmed cell death at a molecular level. Semin
Arthritis Rheum. 32:345–369. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zinkel S, Gross A and Yang E: BCL2 family
in DNA damage and cell cycle control. Cell Death Differ.
13:1351–1359. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Petros AM, Olejniczak ET and Fesik SW:
Structural biology of the Bcl-2 family of proteins. Biochim Biophys
Acta. 1644:83–94. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lynam-Lennon N, Maher SG and Reynolds JV:
The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos
Soc. 84:55–71. 2009. View Article : Google Scholar
|
|
8
|
Chen G, Umelo IA, Lv S, et al: miR-146a
inhibits cell growth, cell migration and induces apoptosis in
non-small cell lung cancer cells. PLoS One. 8:e603172013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Di Leva G, Garofalo M and Croce CM:
MicroRNAs in Cancer. Annu Rev Pathol. 9:287–314. 2014. View Article : Google Scholar :
|
|
10
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Farazi TA, Hoell JI, Morozov P and Tuschl
T: MicroRNAs in human cancer. Adv Exp Med Biol. 774:1–20. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu J, Zhu X, Wu L, et al: MicroRNA-122
suppresses cell proliferation and induces cell apoptosis in
hepatocellular carcinoma by directly targeting Wnt/β-catenin
pathway. Liver Int. 32:752–760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang X, Huang F, Yang D, et al:
Expression of microRNA-122 contributes to apoptosis in H9C2
myocytes. J Cell Mol Med. 16:2637–2646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang Y, Zheng J, Sun Y, et al: MicroRNA-1
regulates cardiomyocyte apoptosis by targeting Bcl-2. Int Heart J.
50:377–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiong Y, Fang JH, Yun JP, et al: Effects
of microRNA-29 on apoptosis, tumorigenicity and prognosis of
hepatocellular carcinoma. Hepatology. 51:836–845. 2010.
|
|
16
|
Zhao A, Zeng Q, Xie X, et al:
MicroRNA-125b induces cancer cell apoptosis through suppression of
Bcl-2 expression. J Genet Genomics. 39:29–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jia HY, Wang YX, Yan WT, et al:
MicroRNA-125b Functions as a Tumor Suppressor in Hepatocellular
Carcinoma Cells. Int J Mol Sci. 13:8762–8774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin CJ, Gong HY, Tseng HC, et al: miR-122
targets an anti-apoptotic gene, Bcl-w, in human hepatocellular
carcinoma cell lines. Biochem Biophys Res Commun. 375:315–320.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kontos CK, Christodoulou MI and Scorilas
A: Apoptosis-related BCL2-Family members: Key players in
chemotherapy. Anticancer Agents Med Chem. 14:353–374. 2014.
View Article : Google Scholar
|
|
20
|
Qiu T, Zhou L, Wang T, et al: miR-503
regulates the resistance of non-small cell lung cancer cells to
cisplatin by targeting Bcl-2. Int J Mol Med. 32:593–598.
2013.PubMed/NCBI
|
|
21
|
Yang X, Yin J, Yu J, et al: miRNA-195
sensitizes human hepatocellular carcinoma cells to 5-FU by
targeting BCL-w. Oncol Rep. 27:250–257. 2012.
|
|
22
|
Qin B, Xiao B, Liang D, et al: MicroRNA
let-7c inhibits Bcl-xl expression and regulates ox-LDL-induced
endothelial apoptosis. BMB Rep. 45:464–469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo R, Wang Y, Shi WY, et al: MicroRNA
miR-491–5p targeting both TP53 and Bcl-XL induces cell apoptosis in
SW1990 pancreatic cancer cells through mitochondria-mediated
pathway. Molecules. 17:14733–14747. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ling HY, Ou HS, Feng SD, et al: CHANGES IN
microRNA (miR) profile and effects of miR-320 in insulin-resistant
3T3-L1 adipocytes. Clin Exp Pharmacol Physiol. 36:e32–39. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (−ΔΔCT) Method. Methods. 25:402–408.
2001. View Article : Google Scholar
|
|
26
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Khatri R and Subramanian S: MicroRNA-135b
and Its circuitry networks as potential therapeutic targets in
colon cancer. Front Oncol. 3:2682013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nian W, Ao X, Wu Y, et al: miR-223
functions as a potent tumor suppressor of the Lewis lung carcinoma
cell line by targeting insulin-like growth factor-1 receptor and
cyclin-dependent kinase 2. Oncol Lett. 6:359–366. 2013.PubMed/NCBI
|
|
29
|
Georgantas RW, Streicher K, Zhu W, et al:
MicroRNA oncogenes and tumor suppressors controlling malignant
melanoma cell growth, apoptosis, migration, and invasion. J Clin
Oncol. 29:85492011.
|
|
30
|
Wang XF, Shi ZM, Wang XR, et al: MiR-181d
acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2.
J Cancer Res Clin Oncol. 138:573–584. 2012. View Article : Google Scholar
|
|
31
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ji F, Zhang H, Wang Y, et al:
MicroRNA-133a, downregulated in osteosarcoma, suppresses
proliferation and promotes apoptosis by targeting Bcl-xl and Mcl-1.
Bone. 56:220–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou J, Xu T, Yan Y, et al: MicroRNA-326
functions as a tumor suppressor in glioma by targeting the Nin one
binding protein (NOB1). PLoS One. 8:e684692013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liang Z, Wu H, Xia J, et al: Involvement
of miR-326 in chemotherapy resistance of breast cancer through
modulating expression of multidrug resistance-associated protein 1.
Biochem Pharmacol. 79:817–824. 2010. View Article : Google Scholar :
|