Introduction
Neonatal late-onset septicemia (LOS) is a common
complication of infants under prolonged hospitalization in the
neonatal intensive care unit (1).
LOS occurs in ~10% of all neonates in neonatal intensive care units
and up to 21% of very low birth weight infants experience an
episode of LOS (2,3). Fast and accurate diagnosis is
important for reduction of the mortality of LOS. Although blood
culture remains to be the gold standard in the diagnosis of
bacterial bloodstream infections, this method has certain
limitations, such as a long waiting time for results (at least 48
h), poor sensitivity (10–20%) in detecting fastidious microbes, and
the use of antibiotics before blood specimens are drawn, which may
affect the results obtained from the blood culture (4,5). In
addition, the blood culture method is flawed in detection of
polymicrobial infection (5,6).
The 16S rDNA polymerase chain reaction (PCR), based
on the amplification of 16S rDNA in bacteria, is fast with high
sensitivity and can fully detect the whole bacterial spectrum in
the experimental samples (7). In
combination with denaturing gradient gel electrophoresis (DGGE) and
sequencing, 16S rDNA PCR has been used to detect the pathogens in
the blood of patients suffering from fever or neutropenia (8). However, the use of 16S rDNA PCR in
the diagnosis of neonatal LOS has not yet been reported. The aim of
the present study was to investigate the efficiency of 16S rDNA
PCR-DGGE and sequencing in the detection of bacteria in neonatal
LOS.
Materials and methods
The study protocol was approved by the Institutional
Review Board of the Children's Hospital, Chongqing Medical
University (Chongqing, China). Informed written consent was
obtained from the guardians of the enrolled neonates.
Diagnostic criteria
Signs and symptoms suggestive of clinical sepsis
were: Unstable temperature, lethargy, irritability,
gastrointestinal dysfunction with milk intolerance, vomiting,
abdominal distension or bloody stool, respiratory dysfunction,
sudden increase in respiratory rate or persistent tachypnoea, and
tachycardia or bradycardia. These signs and symptoms were described
in detail in a previous study (9).
Sample collection
From January to May 2012, 60 neonates who were
suspected of neonatal septicemia according to the above diagnostic
criteria in the Department of Neonatology, Children's Hospital of
Chongqing Medical University were enrolled in the present study.
Paired blood samples were collected after careful skin disinfection
and sent for blood culture and molecular analysis. Blood samples
from 10 neonates diagnosed with jaundice (caused by ABO hemolytic
disease, without any evidence of infection or antibiotic treatment)
were collected and served as negative controls. Venous blood (3 ml)
was collected, of which 1 ml was inoculated in the corresponding
culture bottles for aerobic blood culture in the BACTEC 9120 system
(BD Diagnostics, Bergen, NJ, USA), and another 1 ml was used for
anaerobic blood culture in the BacT/Alert system (BioMérieux,
Marcy-l'Etoile, France). The remaining 1 ml venous blood was
collected in a sterile blood collection tube containing EDTA
(Shanghai Kehua Bio-engineering, Shanghai, China) and stored at
−70°C until molecular processing.
DNA extraction
DNA extraction was performed according to the
manufacturer's instructions using the QIAamp DNA Blood Mini kit
(Qiagen, Hilden, Germany) with sterile water as a negative control.
The blood samples were frozen, thawed for 3 cycles, and incubated
at 37°C for 1 h with mixing every 20 min followed by the addition
of 180 μl (40 mg/ml) lysozyme (Sigma-Aldrich, St. Louis, MO,
USA). All DNA extraction reagents except the lysozyme solution were
filtered through a 0.22-μm filter prior to bacterial DNA
extraction. Whole genomic DNA (100 ml), including bacterial genomic
DNA, was dissolved in buffer AE (10 mM Tris·Cl, 0.5 mM EDTA, pH
9.0) and stored at −20°C. PCR amplification was performed using a
PCR amplifier (Eppendorf, Humburg, Germany), with previously
described thermocycling conditions (8).
16S rDNA nested PCR amplification
Primers for amplification of the variable region of
16S rDNA for reaction 1 were as follows: B5,
5′-TCAGATTGAACGCTGGCGGC-3′ and B4, 5′-TATTACCGCGGCTGCTGGCA-3′
(8). The amplified 493-bp products
from reaction 1 were then amplified in reaction 2 with primers P2
(5′-CCTACGGGAGGCAGCAG-3′) and P3 (5′-ATTACCGCGGCTGCTGG-3′) starting
from nucleotide 341 and 534 of the 16S rDNA, respectively (8). A 40-bp GC clamp was attached to the
5′ end of the P2 primer to prevent complete separation of PCR
amplicons during DGGE analysis. The sterile water was filtered
through a 0.22-μm filter to avoid possible contamination.
The PCR mixture of the first amplification was adjusted to a final
volume of 25 μl with sterile water after 12.5 μl of
Premix Taq (Takara Bio Inc., Otsu, Japan), 0.5 μl each
primer (10 μM), and 5 μl DNA template were added. The
PCR mixture of the second amplification was adjusted to a final
volume of 50 μl with sterile water after 1 μl of PCR
product from the first amplification, 25 μl Premix Taq
(Takara Bio Inc.), and 1 μl each primer (10 μM) were
added, with sterile water used as a negative control.
DGGE analysis
DGGE analysis was performed using the Dcode
Universal Mutation system (Bio-Rad, Hercules, CA, USA) following
the manufacturer's instructions. The products from PCR
amplification were first electrophoresed on a 2% (wt/vol) agarose
gel and stained with 4S nucleic acid (Sangon Biotech Co. Ltd.,
Shanghai, China). Only samples with visible target bands were
adopted for further DGGE analysis. Electrophoresis was performed on
8% polyacrylamide gels with a denaturing gradient ranging from 35
to 65% in 1X TAE buffer (0.04 M Tris-acetate, 0.00l M EDTA; pH 8.0)
at 85 V and 60°C for 16 h. Then the gels were incubated in 1X TAE
buffer containing SYBR Green I (Biotech, Beijing, China) for 30 min
and scanned using a Benchtop 3UV Transilluminator (UVP, Inc.,
Upland, CA, USA). Each visible band was excised from the DGGE
polyacrylamide gels, placed in 30 μl of sterile water, and
incubated at 4°C overnight. PCR-DGGE analysis of each sample was
repeated twice.
TA cloning
DNA recovered from excised DGGE bands was amplified
by PCR as described previously (10), and the PCR products were
electrophoresed on a 2% agar gel and purified using the Gel DNA
Extraction kit (Takara Bio Inc.). The purified PCR products (2 ml)
were used to perform TA cloning using the pMD 18-T Vector kit
(Takara Bio Inc.) according to the manufacturer's instructions.
Then the pMD 18-T plasmids containing the PCR amplicons were
transformed to Escherichia coli DH5α-competent cells
(TIANGEN Biotech (Beijing), Co., Ltd., Beijing, China) and cultured
on ampi-cillin-resistant Luria-Bertani (LB) broth at 37°C
overnight.
Confirmation of the cloned DNAs by colony
PCR
Clones on ampicillin-resistant LB media were
collected and blended in 10 μl sterile water. Then colony
PCR was performed to confirm the cloned DNAs using primers M13-RV
(5′-CAGGAAACAGCTATGAC-3′) and M13-M3 (5′-GTAAAACGACGGCCAGT-3′). The
PCR mixture containing 1 μl template (the monoclone blended
in sterile water), 1 μl of each primer (10 μM), and
12.5 μl Premix Taq (Takara Bio Inc.) was adjusted to a final
volume of 25 μl with sterile water. The PCR mixture was
first incubated at 94°C for 7 min followed by a total of 30 cycles
of 30 sec at 94°C, 30 sec at 60°C, and 30 sec at 72°C, with a final
step at 72°C for 5 min. The length of each amplicon was confirmed
by agarose gel electrophoresis.
Sequencing of PCR products
The sequencing of the PCR products was performed on
an ABI 3730×l DNA Analyzer with M13+(−47) primers and BigDye
terminator v3.1 (Applied Biosystems, USA) at Sangon Biotech
(Shanghai, China), and the sequences were analyzed and blasted on
NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastHome).
Confirmation of the efficiency of the DNA
extraction method in spiked samples using the 16S rDNA PCR-DGGE and
sequencing method
E. coli and Staphylococcus epidermidis
cultures at 1×105 CFU/ml (10 ml) each were blended in
blood samples (200 μl) from neonates diagnosed with jaundice
to prepare spiked, 'infected' blood samples, in order to imitate
the bacterial load in septicemia (4). The bacterial DNA was then extracted
following the procedures described above and analyzed by the 16S
rDNA PCR-DGGE and sequencing method.
Pathogen-specific PCR of blood
culture-proven samples
Pathogen-specific PCR was conducted to detect the
DNA of the bacteria confirmed by blood culture but not by the 16S
rDNA PCR-DGGE and sequencing method. Two patients who were
diagnosed with Klebsiella pneumoniae septicemia were
selected. The primers for amplification of K. pneumoniae
were as follows: Forward: 5′-GCGTGGCGGTAGATCTAAGTCATA-3′ and
reverse: 5′-TTCAGCTCCGCCACAAAGGTA-3′. The PCR conditions and method
used to confirm the length of the PCR products were the same as
described above.
16S rDNA PCR-DGGE and sequencing of
spiked samples
Enterococcus faecalis, S. epidermidis and
Pseudomonas aeruginosa, often observed in septicemia, were used
to imitate the situation present in infected blood by a
pyrosequencing method. Genomic DNA of each bacterium was extracted
as described above. The concentration of the DNA was quantified
using NanoDrop 1000 (Nanodrop, Wilmignton, DE, USA) and diluted to
65 ng/μl. The DNA of E. faecalis was 5-fold serially
diluted (served as less dominant bacteria) and blended with a
constant concentration of DNA from the other two bacteria (both
served as dominant bacteria). The set of the mixed DNA served as
the template and was subjected to a series of 16S rDNA PCR
amplifications as described previously (10). In addition, the set of diluted DNA
of E. faecalis alone served as the template and was
subjected to 16S rDNA PCR amplification.
The PCR mixture containing 25 μl Premix Taq
(Takara Bio Inc.), 1 μl of each primer (10 μM), 1
μl diluted DNA of E. faecalis, or 3 μl (mixed
DNA with 1 μl of each bacterial DNA) of bacterial DNA was
adjusted to a final volume of 50 μl with sterile water. The
PCR products were then analyzed by the DGGE and sequencing method
as described above.
Results
Blood culture
From January to May 2012, 60 neonates who were
suspected of neonatal septicemia were enrolled in the present
study. Positive blood culture results occurring >72 h after
birth were reported for 14 of these 60 neonates (Table I). Of these 14 neonates with
positive blood culture, 12 neonates were diagnosed with
mono-bacterial infection and 2 neonates were confirmed to be
infected by two species of bacteria.
 | Table IComparison of sequencing results of
blood culture. |
Table I
Comparison of sequencing results of
blood culture.
| Identified bacterial
species
|
|---|
| Patient | Blood culture | 16S rDNA
PCR-denaturing gradient gel electrophoresis and sequencing
method |
|---|
| 1 | Klebsiella
pneumoniae | Neisseria sp.,
Moraxella sp., |
| Enterobacter
sp., Micrococcus sp. |
| 2 | Klebsiella
pneumoniae | Moraxella sp.,
Acinetobacter sp., |
| Bacillus sp.,
Aeromonas sp. |
| 3 | Escherichia
coli | Escherichia
colia, Vibrio
sp. |
| 4 | Group B
Streptococcus |
Stenotrophomonas sp., |
| Acinetobacter
sp., sphingobacterium |
| 5 | Enterococcus
faecalis | Micrococcus
sp., Klebsiella sp., |
| Enterobacter
sp., Acinetobacter sp. |
| 6 | Serratia
marcescens | Acinetobacter
sp., Enterobacter sp. |
| 7 | Staphylococcus
epidermidis | Staphylococcus
epidermidis, |
| Bacillus sp.,
Halomonas sp., |
|
Propionibacterium sp. |
| 8 | Staphylococcus
aureus | Acinetobacter
sp., Klebsiella sp., |
| Enterobacter
sp., Micrococcus sp. |
| 9 | Enterobacter
cloacae, Klebsiella pneumoniae |
Klebsiella sp.,
Enterobacter sp., |
|
Acinetobacter sp., |
|
Corynebacterium sp. |
| 10 | Enterococcus
faecium, Staphylococcus haemolyticus | Dietzia sp.,
Enterobacter sp., |
| Klebsiella
sp., Proteobacterium, |
| Bacillus
sp. |
| 11 | Enterobacter
aerogenes |
Enterobacter sp., unknown
bacteria |
| 12 | Klebsiella
pneumoniae |
Klebsiella sp. |
| 13 | Group B
streptococcus | Vibrio sp.,
Escherichia coli, |
| Burkholderia
sp. |
| 14 | Streptococcus
agalactiae |
Acinetobacter sp.,
Klebsiella sp., |
| Enterobacter
sp. |
Bacterial spectrum screened by molecular
methods
The sequencing results showed diverse bacterial
species in the blood samples, the majority of which were not
detected by blood culture (Table
I). Only in five cases did the sequencing results match partly
or wholly with the blood culture results. One of these five
patients was diagnosed with K. pneumoniae bloodstream
infection, which was confirmed by molecular methods and blood
culture. In the other four of these five cases, the molecular
method detected a more complex bacterial spectrum, which contained
the blood culture proven bacteria (Table I). In the other 9 cases, blood
culture failed to detect bacteria, such as Neisseria sp.,
Moraxella sp., that were detected by the 16S rDNA PCR-DGGE
and sequencing method. In addition, the 16S rDNA PCR-DGGE and
sequencing method also failed to detect the blood culture-proven
bacteria, such as Klebsiella pneumonia and Enterococcus
faecalis.
Investigation of the reasons leading to
poor correlation between results of blood culture and the molecular
method
The three steps investigating the possible reason
for failure of the molecular method in detecting culture-proven
bacteria were as follows. Firstly, efficiency of the DNA extraction
method was tested. Then pathogen-specific PCR was applied to
confirm the existence of culture-proven bacterial DNA in the sample
DNA solutions. Thirdly, spiked samples were made to imitate the
infection revealed by 16S rDNA PCR-DGGE and sequencing (presence of
'culture-proven pathogen' bacteria with other bacteria detected by
molecular method). The hypothesis that the molecular method-proven
bacterial DNA could interfere with the detection of blood
culture-proven bacteria was tested in these spiked blood samples by
the 16S rDNA PCR-DGGE and sequencing method as described in the
materials and methods.
Efficiency of the DNA extraction
method
The results of the PCR-DGGE and sequencing method
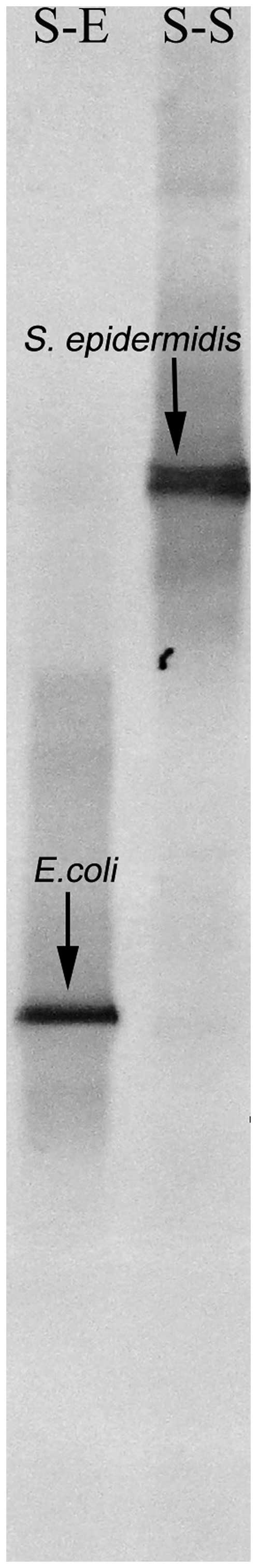
for the spiked 'infected' blood samples demonstrated that the DNA
of the corresponding bacteria in blood was successfully isolated.
The bands on the polyacrylamide gel of the DGGE were identical to
those of E. coli and S. epidermidis, which were
confirmed by subsequent sequencing (Fig. 1).
Detection of blood culture-proven
pathogens by pathogen-specific PCR
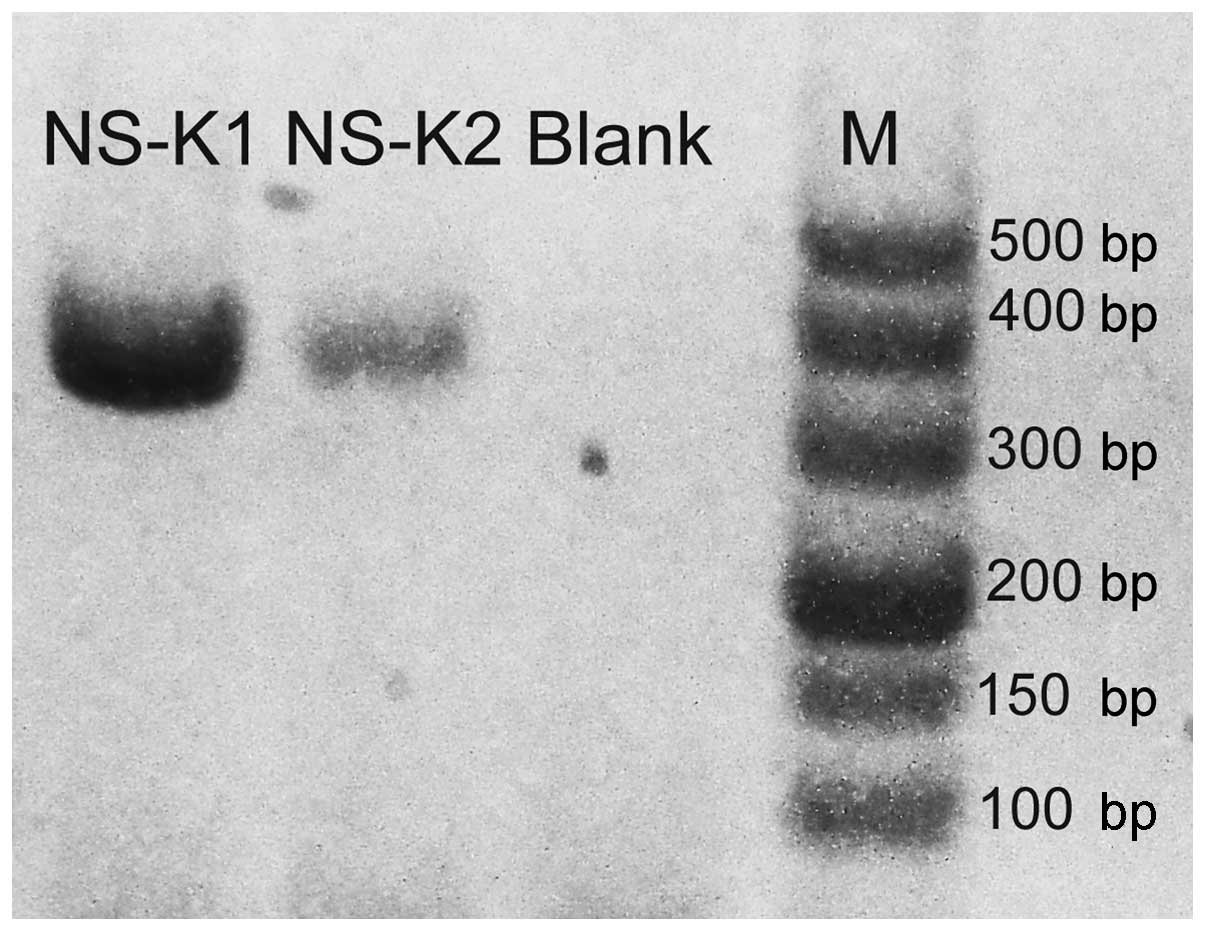
No visible band was found on the gels of two
culture-proven cases after PCR-agarose gel electrophoresis. Hence,
a second PCR amplification of the products from the first
amplification was performed. After the second amplification, the
target gene of K. pneumoniae was observed on the agarose gel
(Fig. 2).
Competitive inhibitory effect of 16S rDNA
PCR-DGGE
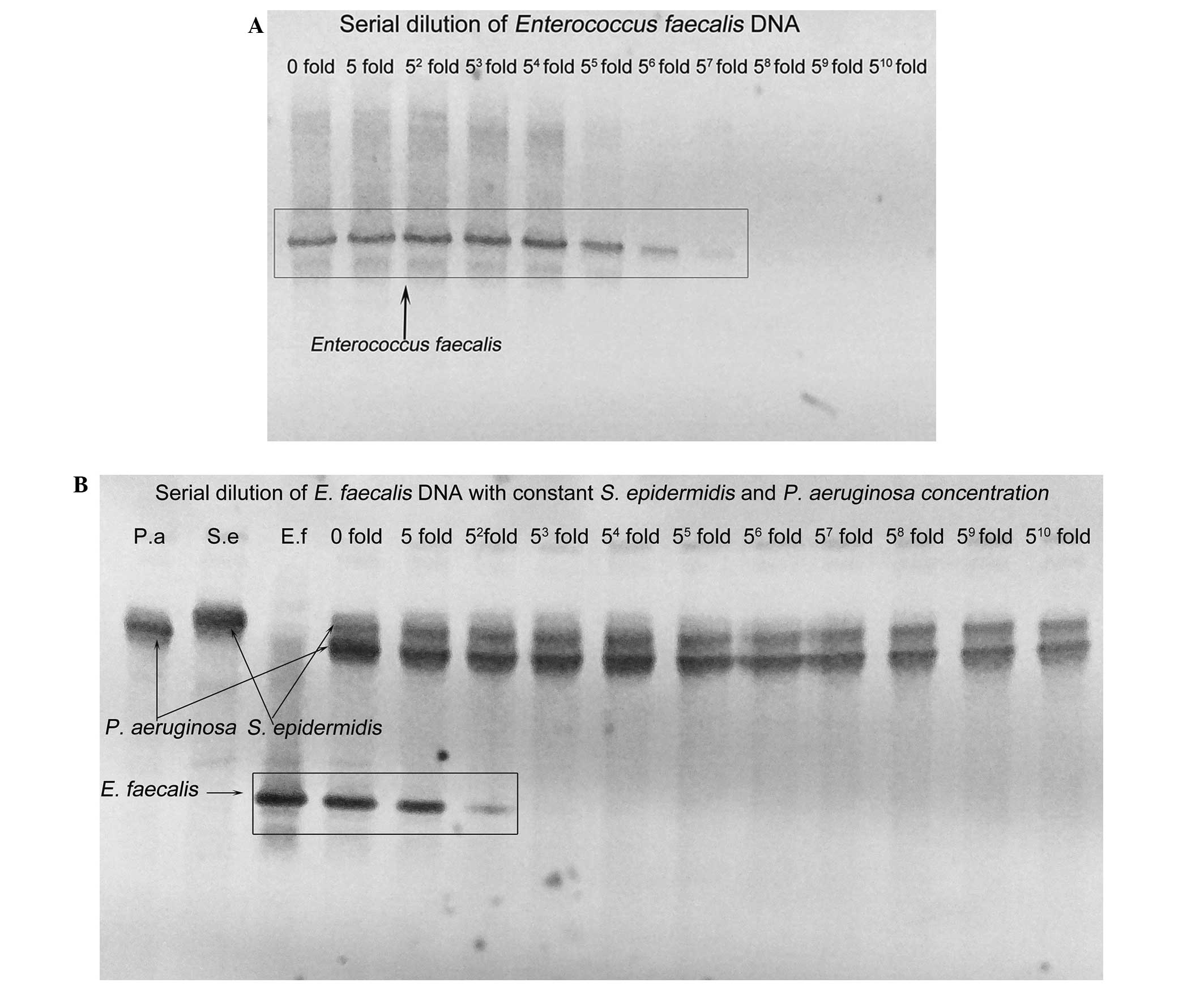
The detection limits of 16S rDNA PCR-DGGE for a
single certain species of bacterial DNA versus mixed species of
bacterial DNA (mixture of other bacterial DNA with this certain
species of bacterial DNA) were completely different with a higher
sensitivity in detecting this certain species of bacterial DNA in
the spiked samples which were free of other bacterial DNA (Fig. 3A and B). When the E.
faecalis DNA was amplified alone even at a 1×57-fold
dilution, the faint band amplified from E. faecalis DNA was
still observed (Fig. 3A). When the
DNAs of the other two species of bacteria were introduced, no band
was observed for E. faecalis DNA even at dilution by
1×53-fold (Fig. 3B).
The detection limit of E. faecalis was different under the
above two situations where the amount of E. faecalis DNA and
16S rDNA were identical throughout, indicating a possible influence
of the introduced bacterial DNA on detecting the E. faecalis
DNA. As the amplifying condition of 16S rDNA PCR was consistent,
the introduced bacterial DNA caused reduced capability in
amplifying E. faecalis DNA. There was lower sensitivity of
16S rDNA PCR in detecting the same copies of E. faecalis DNA
when other bacterial DNA was introduced, compared with detecting
the E. faecalis DNA in the samples containing E.
faecalis DNA alone. This suggests a competitive inhibitory
effect of other bacterial DNA on the E. faecalis DNA during
the 16S rDNA PCR amplification.
Discussion
The recognition of pathogens in the blood is a
crucial aspect in the management of neonates with LOS (6). Although blood culture is regarded as
a gold standard in the clinic, there is a requirement for a more
rapid and sensitive strategy to detect the bacteria in the blood
(5).
In the present study, although a broader bacterial
spectrum was demonstrated by the 16S rDNA PCR-DGGE and sequencing
method as compared with blood culture, it failed to detect
culture-proven bacteria in 9 of the 14 blood culture-positive
cases, indicating its poor correlation with blood culture.
The 16S rDNA PCR technique involves broad-range PCR
amplification of the 16S rDNA regions of different bacterial
species and has been reported by Muyzer et al (11) to be sensitive enough to detect one
species of bacterium in a bacterial community when the bacterium
constitutes >1%. To determine whether the sensitivity is
responsible for the failure of 16S rDNA PCR in the diagnosis of
certain culture-proven cases of septicemia, pathogen-specific PCR
was used in the samples in which blood culture-proven bacteria
failed to be detected by 16S rDNA PCR-DGGE and sequencing method.
Using this procedure the present study aimed to confirm whether the
DNA of culture-proven bacteria was successfully extracted and
existed in the sample DNA solutions. The results of
pathogen-specific PCR demonstrated the presence of a low amount
(only detected after the second amplification) culture-proven
bacterial DNA in the sample DNA solutions. Given the fact that the
16S rDNA PCR used in the present study also went through the second
PCR amplification in nested-PCR, which is similar to the second PCR
amplification in the pathogen-specific PCR, 16S rDNA PCR had the
same capability in detecting these trace amounts of bacterial DNA.
The low load of culture-proven bacterial DNA may not account for
the discrepancy between the 16S rDNA PCR and pathogen-specific PCR
methods. The only difference between these two PCR methods is in
that pathogen-specific PCR primers target a specific species of
bacterial DNA, but 16S rDNA PCR primers target the whole bacterial
spectrum of DNA in the samples. To investigate whether the
detection of blood culture-proven bacterial DNA (such as
Klebsiella pneumonia) interfered with the molecular
method-proven bacterial DNA (such as Neisseria sp.,
Moraxella sp.) in the 16S rDNA PCR amplification among the
patients with poor correlation between the two detection methods,
spiked samples were used to imitate the situation (presence of
other bacterial DNA with blood culture-proven bacterial DNA) and
performed 16S rDNA PCR-DGGE.
The results obtained from the test on spiked samples
in the present study supported the above hypothesis and
demonstrated a competitive inhibitory effect, an unequal
amplification of different bacterial DNAs in the 16S rDNA PCR. The
16S rDNA PCR-DGGE has a higher sensitivity in detecting the
'pathogen' bacterial DNA when the spiked samples were free of other
bacterial 'infection'. However, the introduction of other bacterial
DNAs interferes with the detection of the 'pathogen' bacteria by
the 16S rDNA PCR-DGGE method.
Our findings demonstrate that the 16S rDNA PCR based
molecular methods may cause bias in screening bacteria in LOS due
to the competitive inhibitory effect. A previous study conducted by
Muyzer et al (11) does not
address the exact reason for the limited sensitivity of 16S rDNA
PCR-DGGE. The present study demonstrates that the limited
sensitivity of 16S rDNA PCR-DGGE in detecting bacteria which
constitutes <1% of the whole bacterial community may be
attributed to an unequal PCR amplification among the bacteria in
the bacterial community. For this reason, any practice which leads
to a change in the constitution of the bacterial community should
be noticed and avoided when applying the 16S rDNA PCR based
molecular method. For instance, numerous attempts have been made to
enhance the sensitivity of 16S rDNA PCR, including pre-culture of
blood (incubation before bacterial DNA extraction) to amplify the
amount of the bacteria in the blood samples (12–14).
Although pre-culture is useful for enhancing the detection limit of
16S rDNA PCR, it can induce a competitive inhibitory effect in 16S
rDNA PCR amplification of easy-to-grow bacteria and
fastidious/uncultivable bacteria under certain culture conditions.
Therefore, pre-culture adversely affects the ability to detect
fastidious/uncultivable bacteria with the 16S rDNA PCR based
molecular methods.
Limitations of the present study include a small
sample size and lack of more accurate quantitative methods. The
competitive inhibitory effect was an obstacle to the application of
16S rDNA PCR in the diagnosis of neonatal LOS. Thus, the present
study did not attempt to investigate its use in a large-scale
neonatal population.
Overall, this preliminary investigation focused on
the efficiency of the 16S rDNA PCR-DGGE and sequencing method in
the diagnosis of neonatal LOS. It was demonstrated that a
competitive inhibitory effect caused a bias in 16S rDNA PCR
amplification to screen the bacterial spectrum of neonatal
septicemia. To obtain a higher efficiency of 16S rDNA PCR-DGGE and
sequencing for the diagnosis of LOS, protocols aiming to overcome
the competitive inhibitory effect in 16S rDNA PCR amplification
require development in the future.
Acknowledgments
The authors would like to thank Mr. Yu He and Mr.
Hongdong Li for helpful discussion of the manuscript. This
manuscript has been edited and proofread by Medjaden Bioscience
Limited (Hong Kong, China). This study was supported by the
National Natural Science Foundation of China (grant no. 81370744),
Doctoral Degree Funding from Chinese Ministry of Education (grant
no. 20135503110009), State key clinic discipline project (grant no.
2011–873) and the subproject of National Science & Technology
Pillar Program during the 12th Five-year Plan Period in China
(grant no. 2012BAI04B05).
References
|
1
|
Tsai M-H, Hsu JF, Chu SM, Lien R, Huang
HR, Chiang MC, Fu RH, Lee CW and Huang YC: Incidence, clinical
characteristics and risk factors for adverse outcome in neonates
with late-onset sepsis. Pediatr Infect Dis J. 33:e7–e13. 2014.
View Article : Google Scholar
|
|
2
|
Stoll BJ, Hansen N, Fanaroff AA, Wright
LL, Carlo WA, Ehrenkranz RA, Lemons JA, Donovan EF, Stark AR, Tyson
JE, et al: Late-onset sepsis in very low birth weight neonates: The
experience of the NICHD neonatal research network. Pediatrics.
110:285–291. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rubin LG, Sánchez PJ, Siegel J, Levine G,
Saiman L and Jarvis WR; Pediatric Prevention Network: Evaluation
and treatment of neonates with suspected late-onset sepsis: A
survey of neonatologists' practices. Pediatrics. 110:e422002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paolucci M, Landini MP and Sambri V:
Conventional and molecular techniques for the early diagnosis of
bacteraemia. Int J Antimicrob Agents. 36(Suppl 2): S6–S16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peters RP, van Agtmael MA, Danner SA,
Savelkoul PH and Vandenbroucke-Grauls CM: New developments in the
diagnosis of blood stream infections. Lancet Infect Dis. 4:751–760.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mancini N, Carletti S, Ghidoli N, Cichero
P, Ossi CM, Ieri R, Poli E, Burioni R and Clementi M: Molecular
diagnosis of polymicrobial sepsis. J Clin Microbiol. 47:1274–1275.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sontakke S, Cadenas MB, Maggi RG, Diniz PP
and Breitschwerdt EB: Use of broad range16S rDNA PCR in clinical
microbiology. J Microbiol Methods. 76:217–225. 2009. View Article : Google Scholar
|
|
8
|
Ley BE, Linton CJ, Bennett DM, Jalal H,
Foot AB and Millar MR: Detection of bacteraemia in patients with
fever and neutropenia using 16S rRNA gene amplification by
polymerase chain reaction. Eur J Clin Microbiol Infect Dis.
17:247–253. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ng PC, Cheng SH, Chui KM, Fok TF, Wong MY,
Wong W, Wong RP and Cheung KL: Diagnosis of late onset neonatal
sepsis with cytokines, adhesion molecule and C-reactive protein in
preterm very low birthweight infants. Arch Dis Child Fetal Neonatal
Ed. 77:F221–F227. 1997. View Article : Google Scholar
|
|
10
|
Yu Z and Morrison M: Comparisons of
different hypervariable regions of rrs genes for use in
fingerprinting of microbial communities by PCR-denaturing gradient
gel electrophoresis. Appl Environ Microbiol. 70:4800–4806. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Muyzer G, de Waal EC and Uitterlinden AG:
Profiling of complex microbial populations by denaturing gradient
gel electrophoresis analysis of polymerase chain reaction-amplified
genes coding for 16S rRNA. Appl Environ Microbiol. 59:695–700.
1993.PubMed/NCBI
|
|
12
|
Pammi M, Flores A, Leeflang M and
Versalovic J: Molecular assays in the diagnosis of neonatal sepsis:
A systematic review and meta-analysis. Pediatrics. 128:e973–e985.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jordan JA, Butchko AR and Durso MB: Use of
pyrosequencing of 16S rRNA fragments to differentiate between
bacteria responsible for neonatal sepsis. J Mol Diagn. 7:105–110.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jordan JA and Durso MB: Comparison of 16S
rRNA gene PCR and BACTEC 9240 for detection of neonatal bacteremia.
J Clin Microbiol. 38:2574–2578. 2000.PubMed/NCBI
|