Introduction
Urotensin II (U II) is an effective vasoconstrictor
present in different types of mammals, including humans, which has
been demonstrated to be associated with a variety of cardiovascular
diseases in previous years (1). U
II is the most powerful vasoconstrictor known in the human body
(2). The isolated human
G-protein-coupled receptor 14 (UT) is the specific receptor for U
II (2), and it is predominantly
expressed in the brain, heart, kidney, adrenal gland and placenta
(3,4). The binding of U II to UT results in a
range of biological effects (4,5),
specifically various cardiovascular effects, including promoting
the growth of endothelial cells and vascular smooth muscle cells,
cardiac fibrocyte proliferation, the constriction of vessels and
lowering of the heart rate.
Previous studies demonstrated that in the aortic
coarctation-induced pressure-overload rat model, plasma U II levels
were markedly increased, and this was involved in the pressure
overload-induced myocardial remodeling process (6). In addition, U II not only directly
contributed to cardiac hypertrophy (7), but also promoted myocardial fibrosis
by stimulating the proliferation of cardiac fibroblasts (8) and collagen synthesis (9). These results suggested that, in
addition to affecting hemodynamics, U II may also be involved in
myocardial fibrosis by increasing collagen synthesis (10). The administration of the UT
antagonist, SB-611812, markedly improves the cardiac function of
the experimental animals and markedly reduces myocardial remodeling
(11). However, whether or not U
II is involved in the occurrence and progression of myocardial
fibrosis in the pressure-overload rat model, and the potential
mechanism of action, remains to be elucidated. The present study
aimed to investigate the association between myocardial fibrosis
and systematic changes in the expression levels of U II and UT in a
chronic pressure-overload rat model induced by coarctation of the
abdominal aorta (CAA). The effect of changes in the expression
levels of collagen (Col) I and Col III was also investigated.
Simultaneously, a primary neonatal rat cardiac fibroblast
cultivation model was established in order to investigate the
effect of U II receptor antagonists and protein kinase A
(PKA)-specific inhibitors on myocardial fibrosis. The effect of
systematic changes in the expression levels of U II, UT, Col I and
Col III was also investigated to assess the role of U II in
myocardial fibrosis, and its association with the cyclic adenosine
monophosphate (cAMP)-PKA signal transduction pathway in the chronic
pressure-overload rat model.
Materials and methods
Materials
U II (rat) and KT-5720 were purchased from Tocris
Bioscience (Bristol, UK); goat anti-rat U II polyclonal antibody
(cat no. sc-21098) was purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA); rabbit anti-rat anti-Col I (cat no.
ab34710) and III (cat no. ab7778) polyclonal antibodies were
purchased from Abcam (Cambridge, UK); goat anti-rabbit
immunoglobulin G (IgG) PV-6000 working fluid was purchased from
Xingsheng Biotech Co., Ltd. (Nanjing, China); Dulbecco's modified
Eagle's medium, fetal bovine serum, collagenase and trypsin were
purchased from Invitrogen Life Technologies (Carlsbad, CA,
USA).
Animals
A total of 45 Sprague-Dawley rats weighing 200–220 g
were supplied by the Experimental Animal Center of Shanxi Medical
University (Shanxi, China) and were fed a regular diet. Animals
were kept at 20 ± 2°C and a relative humidity of 40–55% with a 12-h
light/dark cycle. The experimental animal license number was
SYXK:2006–0015. The animal experiments were performed according to
the guidelines on experimental animal care and use, issued by the
American National Institutes of Health (NIH Publication, 8th
Edition, 2011) (12). The present
study was performed strictly in accordance with the recommendations
in the Guide for the Care and Use of Laboratory Animals of the
National Institutes of Health. The animal use instructions were
reviewed and approved by the Institutional Animal Care and Use
Committee of Shanxi Medical University.
Establishment of the CAA model
Following skin preparation, the rats were
anesthetized by intraperitoneal injection of 10% chloral hydrate (3
ml/kg; Chengdu Kelong Chemical Co., Ltd., Chengdu, China). An
incision was made in the middle of sub-xiphisternal abdomen, and
the abdominal cavity was opened layer by layer. The vagina vasorum
of the abdominal aorta was stripped at the top of the left and
right renal arteries, and the abdominal aorta was isolated. A
tip-blunt no. 7 needle (Northwest Medical Devices Co., Ltd.,
Chengdu, China) was placed inside the aorta along the direction of
the blood vessel, and a no. 4 surgical thread was used to ligate
the abdominal aorta around the needle. The needle was slowly
withdrawn to achieve partial stenosis of the abdominal aorta and
the abdomen was subsequently closed layer by layer. In the sham
group of animals, the surgical thread was only wound around the
abdominal aorta following the opening of the abdomen; it was not
used to ligate the abdominal aorta (13). On postoperative day 3, 100,000
units of penicillin (North China Pharmaceutical Co., Ltd.,
Shijiazhuang, China) were intraperitoneally injected to prevent
infection. All animals were placed in the feeding room at a
temperature of 24±3°C with artificial lighting (light and dark
cycles of 12 h each). The animals were fed a standard rat diet and
had free access to water. Group I consisted of control,
sham-operated animals (n=15); Group II consisted of 4 week
post-surgery CAA rats (n=10); Group III consisted of 8 week
Post-surgery CAA rats (n=10); Group IV consisted of 12 week
post-surgery CAA rats (n=10).
Throughout the course of the experiment, all 15
sham-operated rats survived and the survival rate was 100%. In the
CAA group animals, 9 rats were alive 4 weeks following surgery
(mortality rate 10%), at 8 weeks post-surgery, 8 rats survived
(mortality rate of 20%) and at 12 weeks post-surgery, 6 rats
survived (mortality rate of 40%).
Echocardiographic examination
A General Electric Vivid 7 echocardiographic machine
(GE Healthcare, Pittsburgh, PA, USA) was used to perform the
echocardiography 4, 8 and 12 weeks following surgery. The rats were
anesthetized using 10% chloral hydrate (3 ml/kg), their chest hair
was removed and echocardiography was subsequently performed using a
10S probe (the probe frequency was 11.0 MHz). Motion-mode
ultrasound was used to record the curves of the left ventricle, the
interventricular septum and the left ventricular (LV) posterior
wall, using the parasternal long axis view. The end-diastolic
interventricular septal thickness (IVSTd), end-diastolic LV
posterior wall thickness (LVPWTd), end-diastolic LV diameter (LVDd,
all measured in mm) and LV ejection fraction (EF, recorded as a
percentage), were measured. Each parameter was measured three times
and the average was calculated.
Detection of the hemodynamic
response
Following the induction of anesthesia, the rats were
fixed on the operating table. A longitudinal incision (1.5–2 cm)
was made in the neck skin and the right common carotid artery was
separated. The common carotid artery at the distal end of heart was
ligated and the proximal end was gripped with an artery clamp. A
small opening was made in the common carotid artery and a 1 mm
polyethylene catheter (American Health & Medical Supply
International Corp, Scarsdale, NY, USA), pre-filled with heparin
saline, was inserted. A RM6240B-type biological-function
experimental system (Chengdu Instrument Company, Chengdu, China)
was used to record the LV systolic pressure (LVSP), the LV
end-diastolic pressure (LVEDP) and the maximum increases and
decreases in the LV pressures (±dP/dtmax).
Myocardial histological examination
Following the completion of the observation period
(12 weeks), the rats were weighed and anesthetized, and a sample of
~3 ml abdominal aorta blood was obtained. The blood sample was
subsequently centrifuged at 574 ×g for 15 min to separate the
serum. The serum was subsequently cryopreserved at −20°C for future
experiments. The heart was rapidly removed and lavaged with
pre-chilled saline. The surrounding tissues were dissected and the
liquid on the surface, and inside the cardiac chambers was removed
using filter paper (Sangon Biotech, Shanghai, China). The partial
LV free-wall was subsequently cut and placed into 4%
paraformaldehyde fixative prior to examination under a light
microscope (CME; Leica Microsystems, Wetzlar, Germany). The
myocardial tissue was paraffin-embedded prior to morphological
examination of the myocardial cells, including hematoxylin and
eosin (HE), and Masson staining.
Immunohistochemistry
Immunohistochemical staining was performed to
determine the expression levels of U II, UT, Col I and Col III in
the cardiac tissues. Conventional paraffin sections were cut 2.0
µm thick and washed with phosphate buffered saline (PBS)
three times for 5 min. Antigen retrieval was performed for 2 min in
high-pressure hot citrate solution (Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China). The tissue sections were
subsequently cooled to room temperature and washed three times for
2 min with PBS. The primary antibodies, anti-U II (1:150), anti-UT
(1:200), anti-Col I (1:150) and anti-Col III (1:150) were applied
dropwise, and incubated for 1 h at 37°C. The tissue sections were
cooled to room temperature for 60 min prior to washing three times
for 2 min with PBS. The secondary antibody (goat anti-rabbit IgG)
PV-6000 working solution (Zhongshan Golden Bridge Biotechnology
Co., Ltd.) was subsequently added dropwise, and the tissue sections
were incubated at 37°C for 20 min, prior to washing three times for
2 min with PBS. The proteins were detected using diaminobenzidine
staining, and were subsequently counterstained with hematoxylin
prior to dehydration and mounting with neutral gum. The Aperio
scanscope scanning system (Aperio, Buffalo Grove, IL, USA) was used
for digital pathological scanning.
Determination of the cAMP content
The double-antibody sandwich ABC-ELISA kit (Senxiong
Biotechnology Co., Ltd., Shanghai, China) was used to measure the
plasma cAMP levels in the CAA model rats at 4, 8 and 12 weeks,
according to the manufacturer's instructions.
Cultivation of neonatal rat
fibroblasts
A thoracotomy was performed on 1 to 3-day-old
Sprague Dawley rats under aseptic conditions to obtain the hearts.
Following digestion with collagenase (0.04%) and trypsin (0.08%)
(Invitrogen Life Technologies), 10% serum-containing medium was
added to form a cell suspension. In accordance with the different
wall-adherence durations of fibroblasts and cardiomyocytes,
differential adhesion was performed for 1.5 h in order to obtain
the cardiac fibroblasts. The passaged fibroblasts were subsequently
confirmed using an anti-vimentin monoclonal antibody (1:100; cat
no. ab8978; Abcam, Cambridge, UK) and immunocytochemistry; the
purity was 95%.
Grouping
Generations 3–5 of the cells were used in the
present study. The cells were grouped as follows: i) Control group
with no stimulation; ii) U II group, where the cells were
stimulated with 10−8 mol/l U II; iii) U II+KT-5720
group, where the cells were stimulated with 10−8 mol/l U
II + 1 mol/l KT-5720; and iv) U II+SB-611812 group, where the cells
were stimulated with 10−8 mol/l U II + 1 mol/l
SB-611812. The changes in corresponding indexes of each group were
observed at the specified time points.
The protein concentrations of Col I and Col III in
the cell supernatants were determined using an ELISA [Rat Collagen
Type I,III (Col I,III) ELISA kit; Tong Wei Biological Technology
Co., Ltd., Shanghai, China] according to the manufacturer's
instructions. The cells were seeded into 48-well plates using the
above-mentioned method and were incubated with serum-free medium
for 24 h prior to the application of the various stimuli for 48 h.
The supernatant was subsequently collected and the protein
concentrations of Col I and Col III were determined by ELISA. The
ELISA procedure was performed, according to the manufacturer's
instructions, and each experiment was performed three times.
Western blotting
Following stimulation, the cell lysates were
prepared using radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology, Shanghai, China). The samples were
mixed with 5X sample buffer, boiled for 5 min and centrifuged
(13,225 ×g, 15 min, 4°C). The protein concentration in the
supernatant was determined using a bicinchoninic acid (BCA) protein
quantitative kit (Wuhan Boster Biological Technology Co., Ltd.)
according to the manufacturer's instructions. 10% SDS-PAGE (Sangon
Biotech) was performed using 50 µg total protein from the
cell lysates. Following electrophoresis, the proteins were
transferred onto nitrocellulose membranes (Wuhan Boster Biological
Technology, Co., Ltd.) and blocked with bovine serum albumin (Wuhan
Boster Biological Technology Co., Ltd., Hubei, China) for 1 h at
room temperature. The membranes were subsequently incubated with
primary antibodies against Col I (1:5,000) and Col III (1:7,500)
overnight at 4°C. Following the washing of the membranes, the
secondary antibodies were applied and the membranes were incubated
for 1 h, followed by enhanced chemiluminescence (ECL) detection
using an ECL kit (Applygen Technologies Inc, Beijing, China). Band
Scan Imaging software (Glyko, Novato, CA, USA) was used to analyze
the intensity of the bands.
Statistical analysis
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
was used for statistical analysis. The quantitative data are
expressed as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Echocardiography results
As shown in Table
1, at 4 weeks following surgery, the IVSTd of the model group
had increased to a greater extent compared with the sham group, and
the LVPWTd exhibited a non-significant thickening trend
(P>0.05). The LVDd and EF were not significantly altered. The
IVSTd, LVPWTd and LVDd of the model group were significantly
increased relative to the sham group 8 weeks following surgery
(P<0.05), and the EF was decreased (P<0.05). This trend
continued for 12 weeks following surgery: The IVSTd, LVPWTd and
LVDd of the model group were significantly increased relative to
the sham group (P<0.01), and the EF was decreased
(P<0.01).
 | Table IComparison of IVSTd, LVPWTd, LVDd and
EF between the two groups. |
Table I
Comparison of IVSTd, LVPWTd, LVDd and
EF between the two groups.
| Group | N | Time (weeks) | IVSTd (mm) | LVPWTd (mm) | LVDd (mm) | EF (%) |
|---|
| Sham | 5 | 4 | 1.72±0.14 | 1.74±0.12 | 4.81±0.25 | 72.32±5.68 |
| 5 | 8 | 1.85±0.11 | 1.75±0.45 | 4.85±0.16 | 73.12±5.11 |
| 5 | 12 | 1.75±0.12 | 1.76±0.13 | 4.86±0.13 | 74.15±5.16 |
| Model | 6 | 4 | 2.34±0.11a | 1.84±0.25 | 4.91±0.48 | 70.70±5.94 |
| 6 | 8 | 2.40±0.03a | 2.52±0.24a | 6.83±0.28a | 63.15±5.23a |
| 6 | 12 | 1.72±0.17b | 2.87±0.35b | 7.05±0.14b | 45.16±4.21b |
Changes in the hemodynamic
parameters
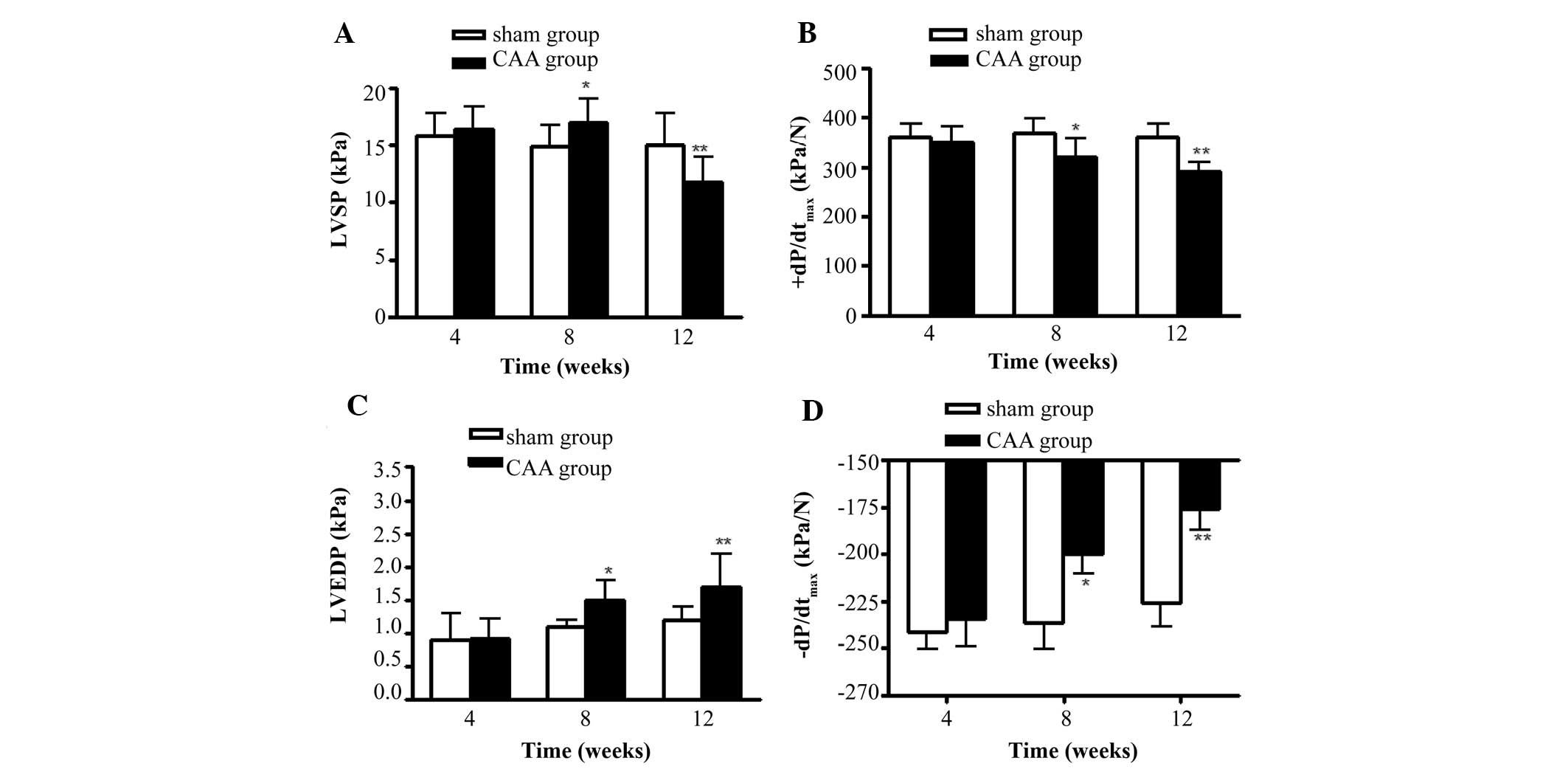
As shown in Fig. 1,
compared with the sham group, the levels of LVSP, +dP/dtmax,
−dP/dtmax and LVEDP were not significantly altered in the model
group 4 weeks following surgery. The +dP/dtmax and −dP/dtmax levels
were significantly decreased at 8 weeks following surgery
(P<0.05), whereas the LVEDP was significantly increased
(P<0.05). These changes were more pronounced at 12 weeks
following surgery (P<0.01). The LVSP level temporarily increased
8 weeks following surgery (P<0.05) and significantly decreased
12 weeks following surgery (P<0.01).
Morphological examination of the
myocardial tissue biopsies
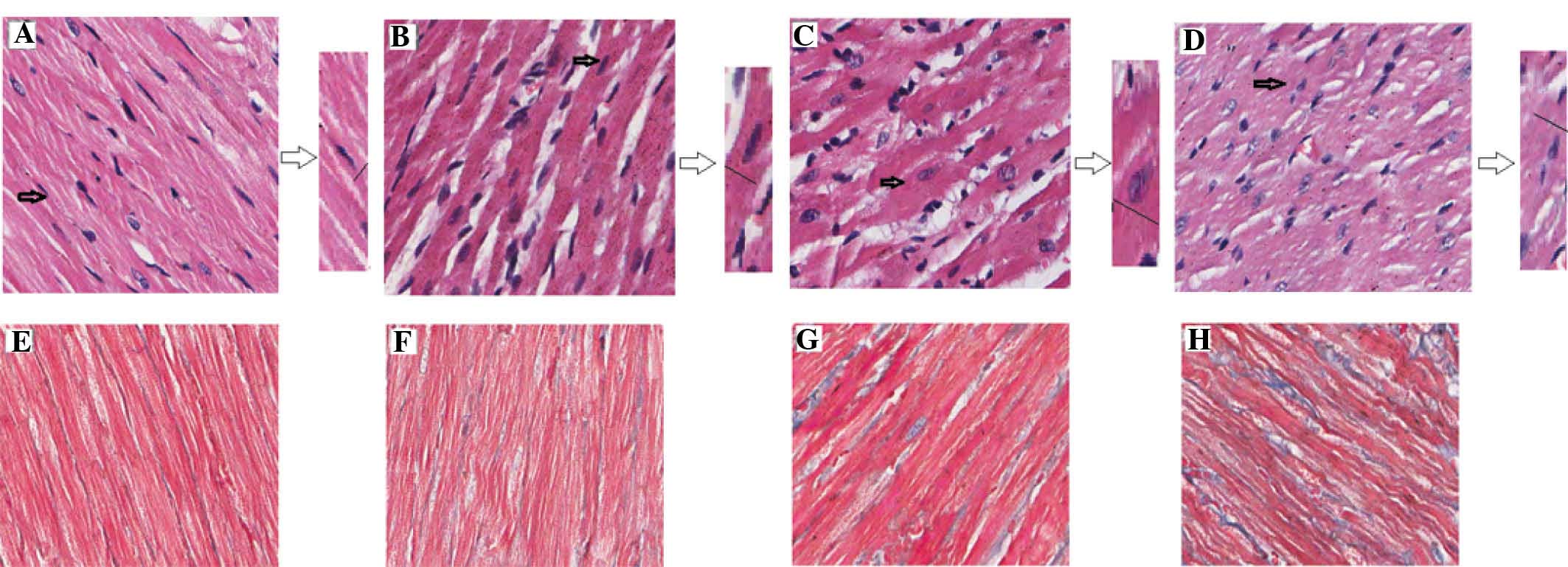
HE staining revealed that, compared with the sham
group, the diameter of the myocardial cells in the model group was
increased. The cells in the model group were arranged irregularly
with disorganized muscle fibers; the muscular fibers were loose and
exhibited edema, with increased gaps between them, and the fibrous
connective tissues around the partial muscle bundles exhibited
hyperplasia (Fig. 2). Masson
staining highlighted the collagen fibers with a blue coloration,
whereas the muscle fibers and cellulose were colored red. Compared
with the sham group, the expression of collagen fibers in the model
group cardiac tissues gradually increased, particularly by the week
12 (Fig. 2).
Immunohistochemical staining results
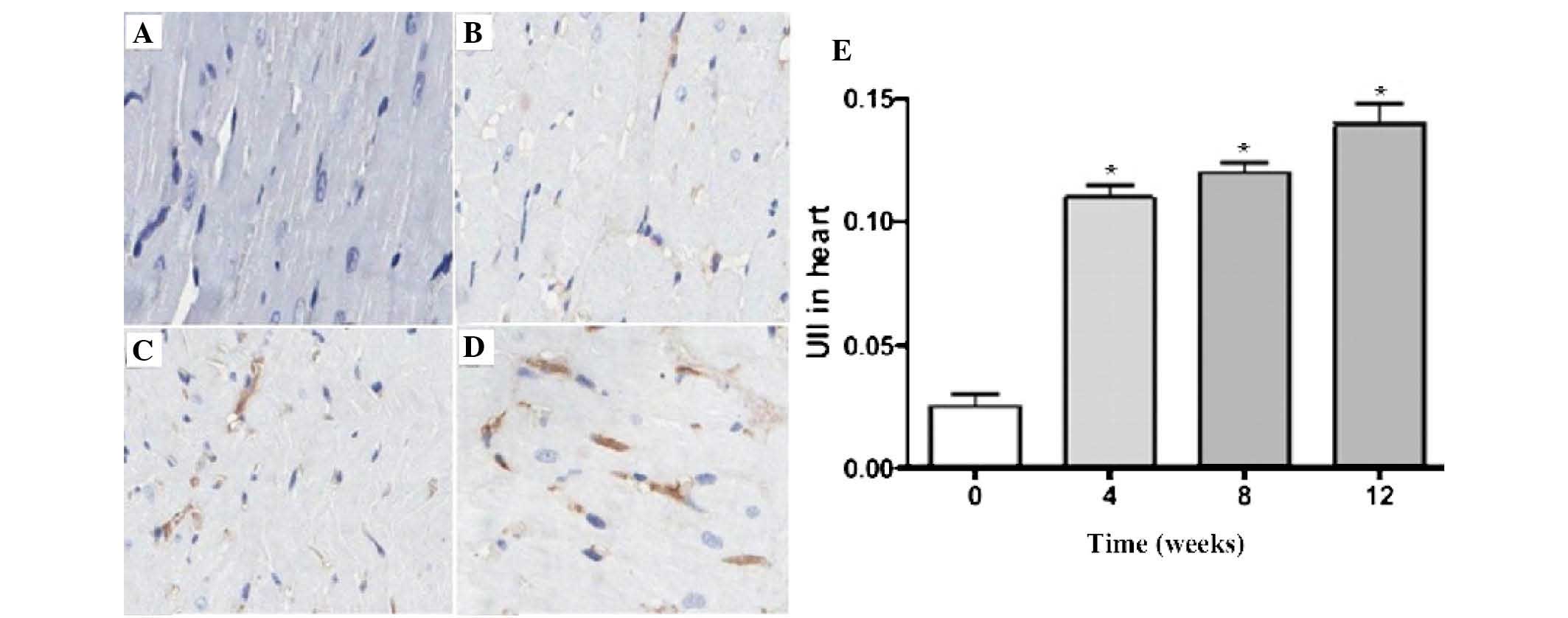
Fig. 3 demonstrated
the expression of U II in the rat myocardial tissues. The darker
areas indicated U II expression inside the myocardial interstitium.
Compared with the sham group, U II expression in the model group
gradually increased over time. The quantitative analysis revealed
that the increased U II expression in the model group was
time-dependent (P<0.05).
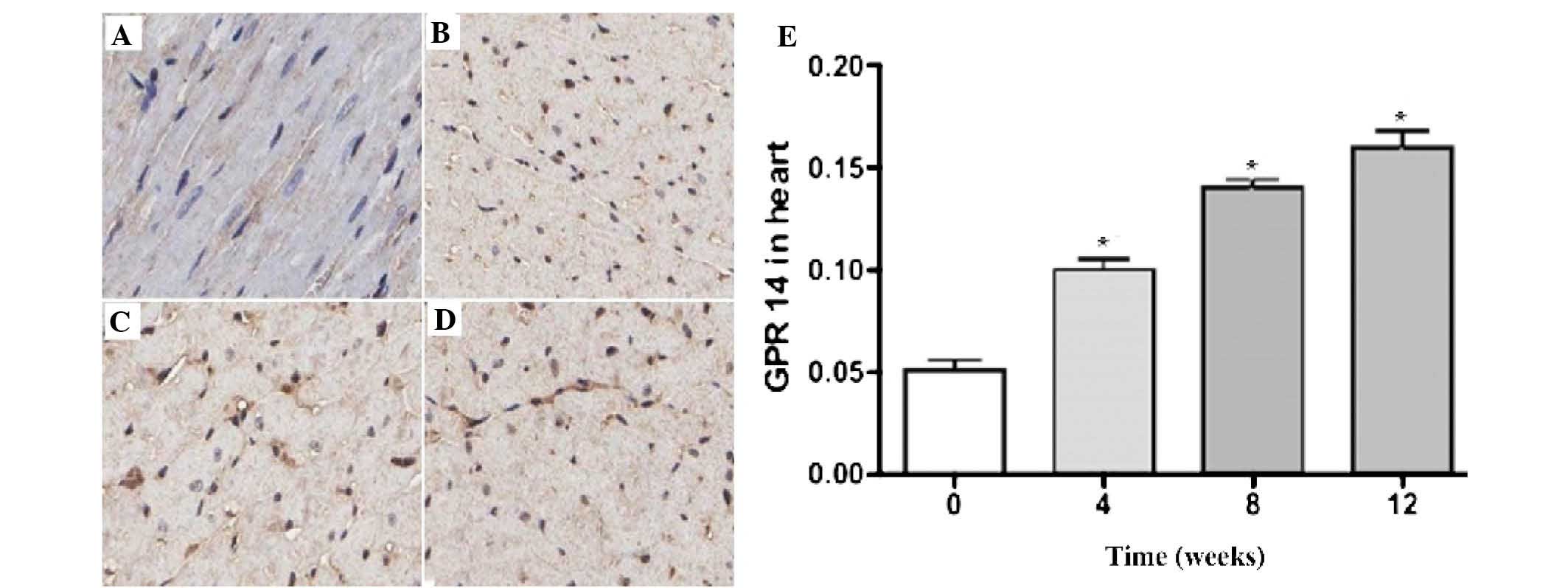
Fig. 4 demonstrated
the expression of UT in the rat myocardial tissues; the darker
areas of staining indicate UT expression in the cytoplasm of the
myocardial cells. There was a low expression level of UT in the
cytoplasm of the sham group myocardial cells. Compared with the
sham group, the expression of UT in the model group gradually
increased over time. The quantitative analysis revealed that the
increased expression of UT in the model group was time-dependent
(P<0.05).
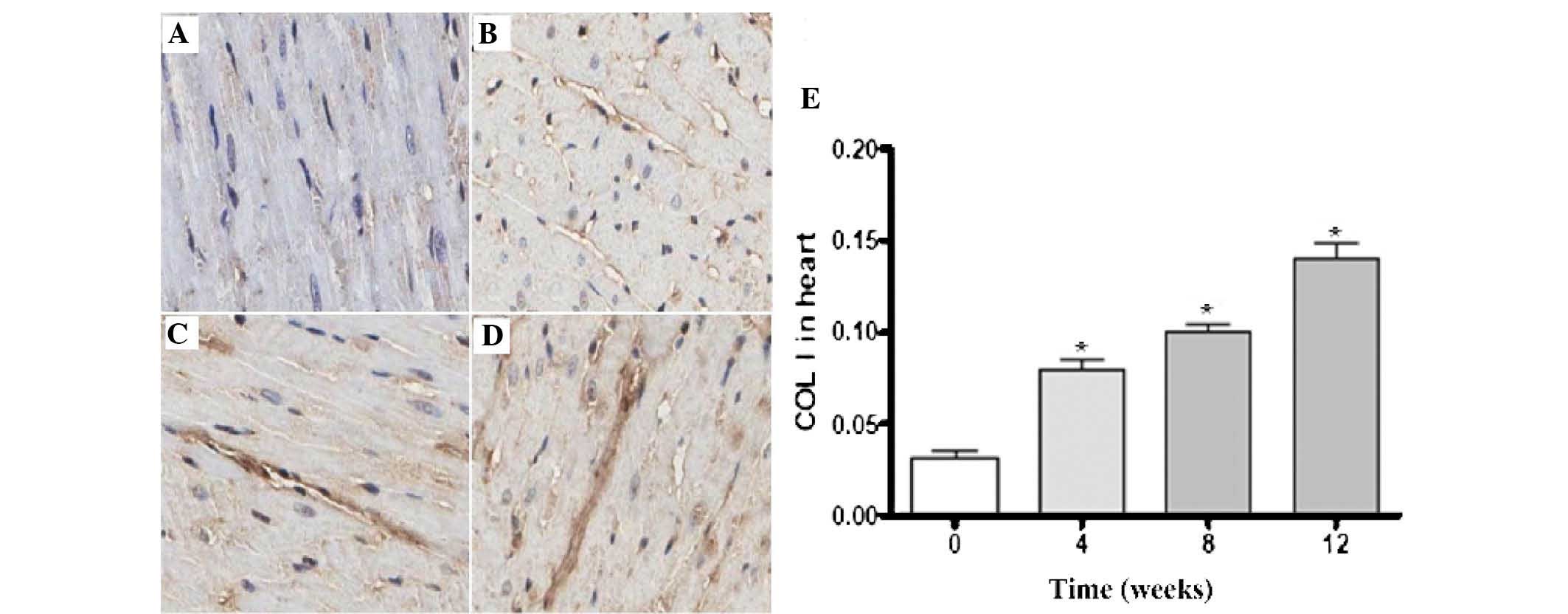
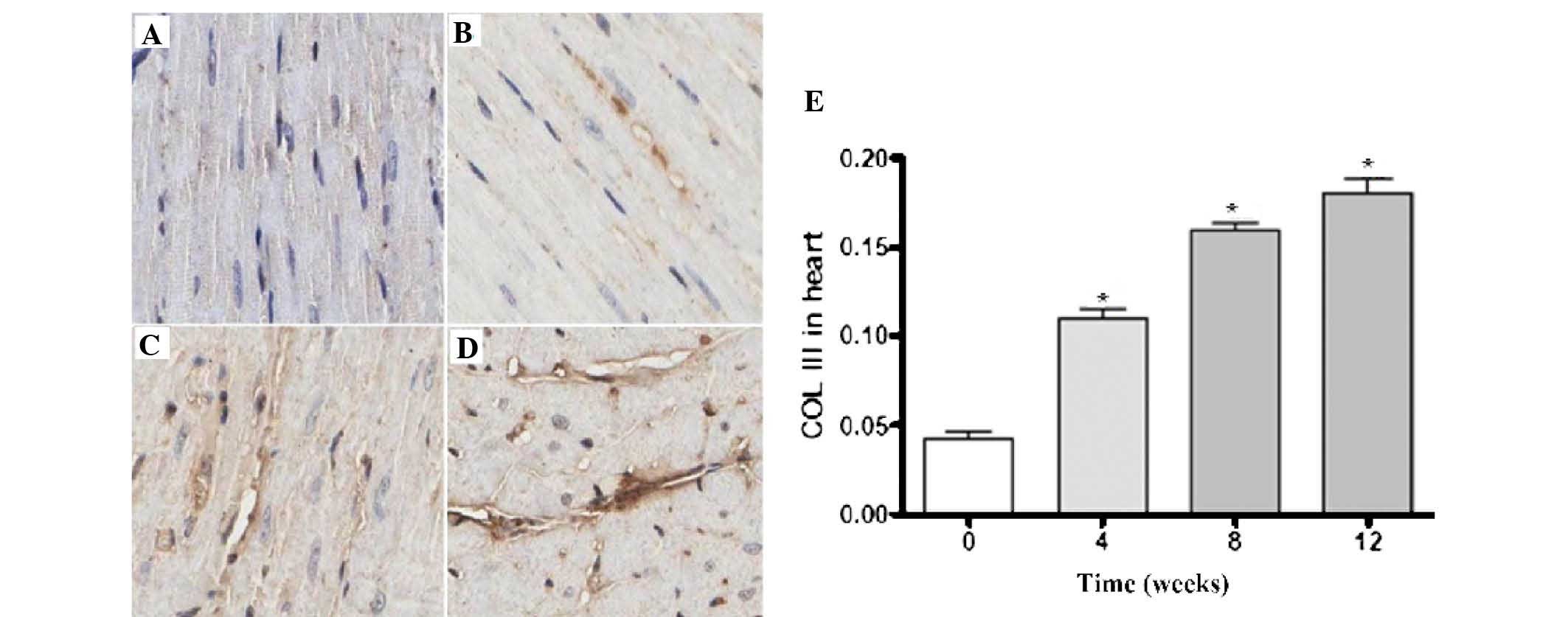
Figs. 5 and
6 revealed the expression levels
of Col I and Col III in the rat myocardial tissues; the darker
areas of staining indicated the expression of Col I and Col III in
the myocardial interstitial tissues. There was a small quantity of
Col I and Col III expression in the sham group. Compared with the
sham group, the expression levels of Col I and Col III in the model
group gradually increased over time. The quantitative analysis
revealed that the increased expression of Col I and Col III in the
model group was time-dependent (P<0.05).
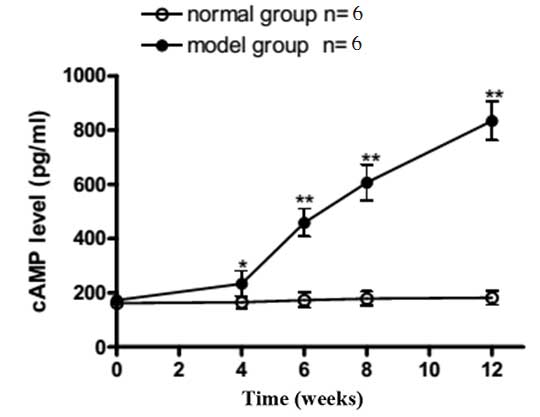
cAMP concentration determination using
the ABC-ELISA method
As shown in Fig. 7,
the plasma cAMP concentration in the model group was higher
compared with the sham group at week 4 (P<0.05). At weeks 8 and
12, the plasma cAMP concentrations exhibited a time-dependent
increase, which was more significant compared with that observed in
week 4 (P<0.01).
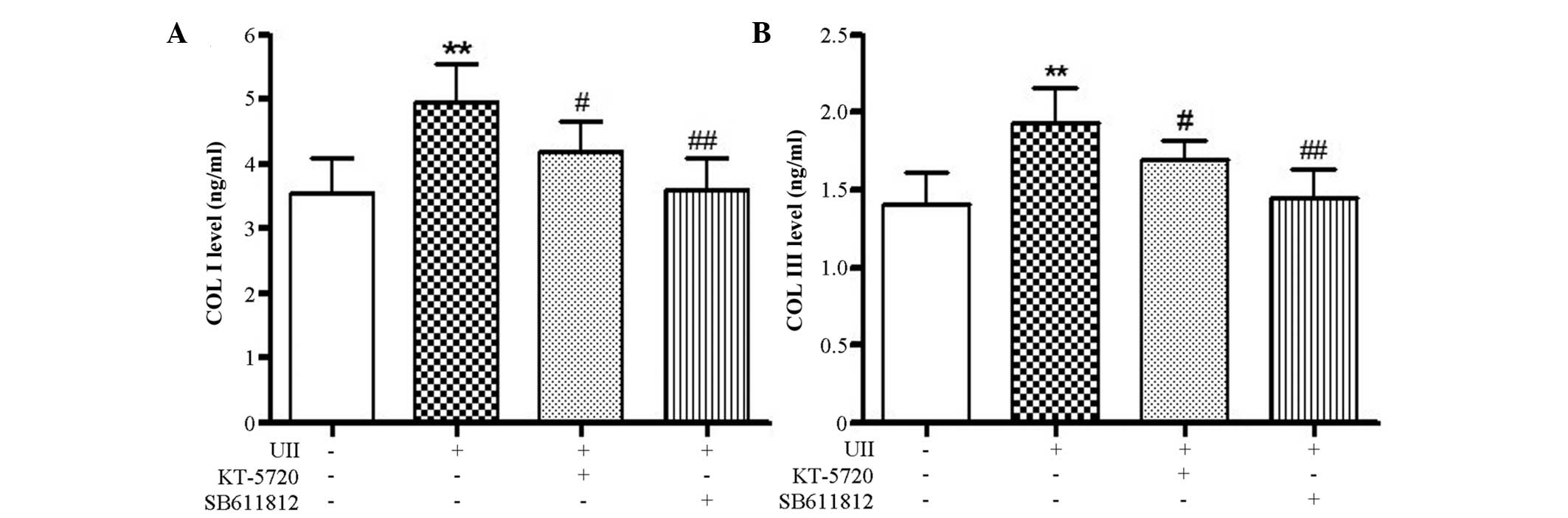
Determination of Col I and Col III
concentration using the ELISA method
As shown in Fig. 8,
stimulation with 10−8 mol/l U II significantly
stimulated the accumulation of Col I and Col III protein in the
fibroblasts. This difference was statistically significant when
compared with the control group (P<0.01). The administration of
10−8 mol/l U II and either the U II receptor antagonist,
SB-611812, or the PKA-specific inhibitor, KT-5720, resulted in a
significant decrease in Col I and Col III levels compared with the
U II treatment group (P<0.05). These results indicated that the
U II-stimulated synthesis of Col I and Col III in fibroblasts may
be achieved through the PKA pathway.
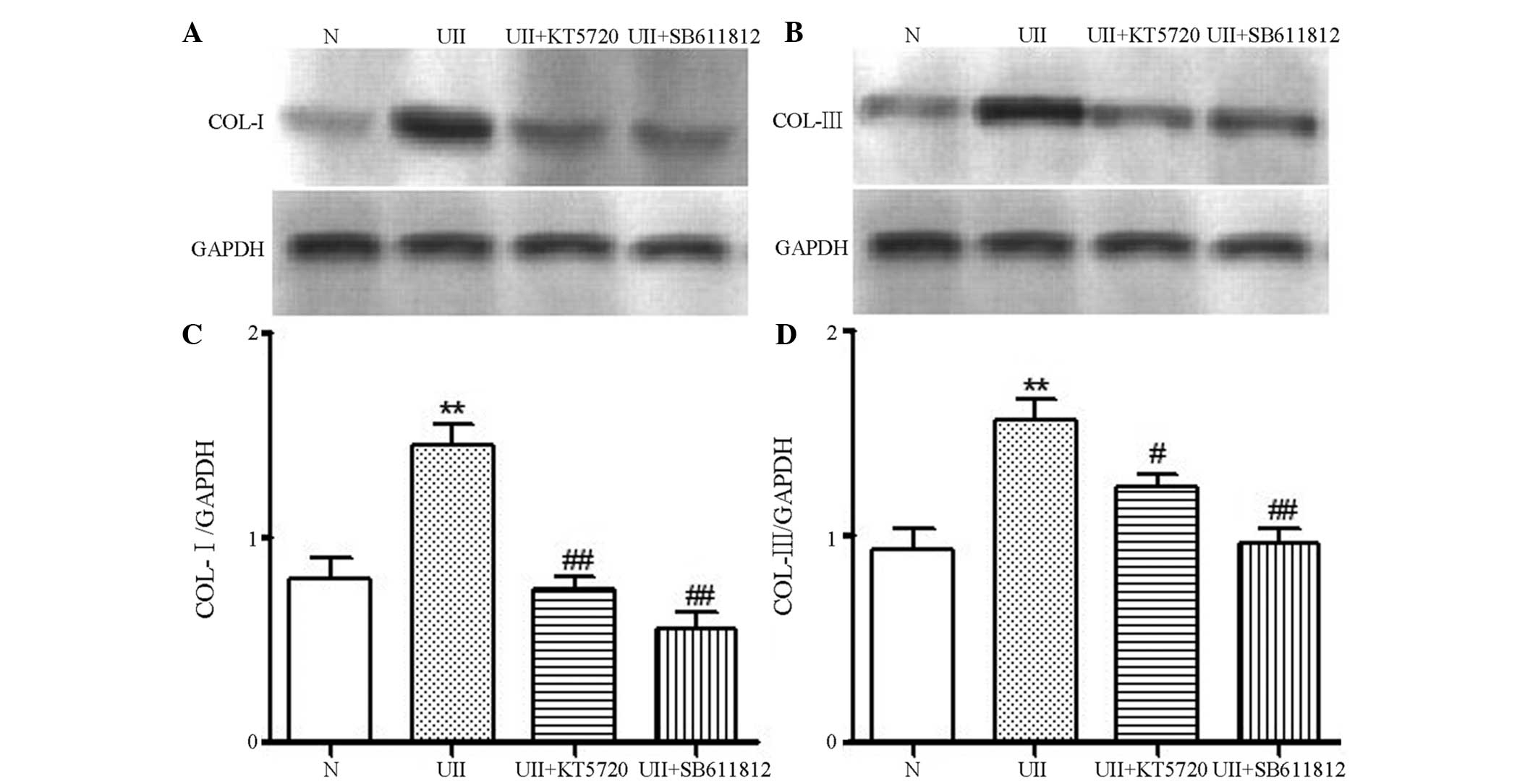
Protein expression levels of Col I and
Col III in fibroblasts
As shown in Fig. 9,
there was a small quantity of Col I and Col III expression in the
fibroblasts from the control group. Following stimulation with
10−8mol/l U II, the expression of Col I and Col III
increased significantly (P<0.01). The administration of
10−8 mol/l U II and either the U II receptor antagonist,
SB-611812, or the PKA-specific inhibitor, KT5720, resulted in a
significant decrease in Col I and Col III levels compared with the
U II treatment group (P<0.05). These results indicated that the
PKA pathway may be involved in the U II-stimulated synthesis of Col
I and Col III in the fibroblasts.
Discussion
Heart failure is an important condition, which
endangers human lives and chronic pressure-overload is an important
cause of heart failure. LV pressure overload can induce cardiac
hypertrophy, fibrosis and systolic dysfunction (14–16).
As the duration of LV pressure overload increases, LV hypertrophy
can gradually develop, eventually leading to heart failure
(14,15). CAA has been demonstrated to
successfully replicate chronic pressure-overload heart failure and
provides an appropriate experimental model (17).
U II was identified as a type of vasoactive peptide
in mammals. Previous studies revealed that U II serum levels were
markedly higher in patients with clinical heart failure (18,19),
and were also higher in the cardiac tissues of patients with
end-stage heart failure (2).
Similarly, animal experiments revealed that, in the coronary artery
ligation-induced myocardial infarction heart failure rat model, the
expression of ventricular U II and its receptor UT revealed a
gradual time-dependent increase in concentration (7). The chronic perfusion of U II into
rats can induce diastolic dysfunction and stimulate collagen
synthesis (20). U II can also
induce the mRNA expression levels of Col I and Col III in neonatal
cardiac fibroblasts through the transforming growth factor β1
signaling pathway (21,22). These previous studies indicated
that U II exerts an important role in heart failure and myocardial
fibrosis. A study performed previously on the role and signal
transduction mechanisms of U II have principally concentrated on
blood vessel regulation (23).
However, whether U II is involved in the development of chronic
pressure in overload-induced rat myocardial fibrosis, and the
possible mechanisms of this involvement, remain to be elucidated.
In the present study, CAA was used to establish a rat model, and
echocardiography and hemodynamic detection indicated that, with a
prolonged modeling time, the systolic and diastolic functions of
the model group rats decreased, confirming the use of the chronic
pressure-overload heart failure model.
Furthermore, HE and Masson staining confirmed that
the CAA-established chronic pressure-overload heart failure rat
model exhibited clear myocardial fibrosis. Additionally, the degree
of fibrosis gradually increased as the modeling time increased.
Immunohistochemical staining revealed that the protein expression
levels of U II, UT, Col I and Col III in the myocardial tissues of
the model group markedly increased. As myocardial fibrosis
progressed, the levels of these proteins exhibited a time-dependent
increase, suggesting that U II and its receptor, UT, may be
involved in the occurrence and development of myocardial fibrosis
in the pressure-overload rats.
Contemporary studies have demonstrated that, in
addition to its effects on hemodynamics, U II may be involved in
myocardial fibrosis by increasing collagen synthesis (10). The UT antagonist, SB-611812,
markedly reduced cardiac remodeling (11). The mechanism of U II-induced
collagen synthesis may involve the PKC, mitogen-activated protein
kinase or calcineurin (24)
pathways, although the precise mode of U II action in myocardial
cells remains to be fully elucidated (25–27).
The Camp-PKA pathway is a major pathway of cellular signal
transduction, which regulates multiple protein activities, the
expression of numerous genes and diverse cellular functions. The
specific action of cAMP in cells is considered to be regulated via
the activation of the cAMP-dependent PKA signaling pathway
(28). Our previous study used the
Langendorff rat isolated-heart model and observed that different
concentrations of U II (EC50 10−8 mol/l)
produced a dose-dependent inhibition of cardiac function; a more
marked inhibitory effect was observed in the CAA-induced heart
failure rat model (29). The
PKA-specific inhibitor, KT5720, inhibits the U II-mediated heart
function inhibition in normal and heart failure model rats, and at
the cellular level, KT5720 was observed to inhibit the U
II-stimulated inhibition of L-type Ca2+ currents in rat
cardiomyocytes. Therefore, U II may inhibit cardiac L-type
Ca2+ currents through the PKA pathway and this may
account for the inhibitory effect of U II on cardiac functions
(30). In order to further
investigate the mechanism(s) of U II in myocardial fibrosis and the
association with the cAMP-PKA pathway, an in vitro neonatal
rat fibroblast experiment was performed. The administration of U II
and the antagonists, KT5720 and SB-611812, indicated that
10−8 mol/l U II significantly stimulated the synthesis
of Col I and Col III in fibroblasts, and that KT5720 and SB-611812
significantly reduced the U II-stimulated synthesis and expression
of Col I and Col III. These results indicated that KT5720 and
SB-611812 significantly inhibited U II-induced Col synthesis in
cardiac fibroblasts. Taken together with the in vivo
experiments, which deomonstrated that plasma cAMP concentrations in
the model group gradually increased with the severity of myocardial
fibrosis, the results suggested that the cAMP-PKA signaling pathway
may regulate U II-promoted collagen synthesis in cardiac
fibroblasts, and therefore is involved in the process of pressure
overload-induced myocardial fibrosis in rats.
In conclusion, in the CAA-induced chronic
pressure-overload rat model, the extent of heart failure and
myocardial fibrosis gradually increased with time. Similarly, the
expression levels of U II, UT, Col I and Col III in myocardial
tissues significantly increased with time, suggesting that U II may
exert an important role in the myocardial fibrosis process in the
pressure-overload rat model. The in vitro experiments
revealed that the cAMP-PKA signaling pathway regulated the effects
of U II on Col synthesis in cardiac fibroblasts, and that this
effect was mediated by UT and antagonized by UT inhibition.
Therefore a novel signaling pathway associating U II and myocardial
fibrosis was putatively been identified, although further studies
are required in animal models.
Acknowledgments
This study was supported by the Natural Science
Foundation of Shanxi Province (no. 2012011036-1), the Shanxi
Provincial Scientific Research Projects Foundation of
Abroad-Studying Personnel (no. 2012-7), the Shanxi Provincial
University Scientific Research Projects Foundation of
Abroad-Studying and Returning Personnel (no. 2011-63), the Selected
Scientific Research Projects Foundation of Abroad-Studying
Personnel, Office of Human Resources, Shanxi Province (no.
2013-68), the Selected Scientific Research Projects Foundation of
Abroad-Studying and Returning Personnel, the Shanxi Province (no.
2010-97), Technology Innovation Foundation of Shanxi Medical
University (no. 2010-7) and the Shanxi Provincial Scientific
Research Projects Foundation of Abroad-Studying and Returning
Personnel (no. 2009–9).
References
|
1
|
Ross B, McKendy K and Giaid A: Role of
urotensin II in health and disease. Am J Physiol Regul Integr Comp
Physiol. 298:R1156–R1172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ames RS, Sarau HM, Chambers JK, Willette
RN, Aiyar NV, Romanic AM, Louden CS, Foley JJ, Sauermelch CF,
Coatney RW, et al: Human urotensin-II is a potent vasoconstrictor
and agonist for the orphan receptor GPR14. Nature. 401:282–286.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Totsune K, Takahashi K, Arihara Z, Sone M,
Satoh F, Ito S, Kimura Y, Sasano H and Murakami O: Role of
urotensin II in patients on dialysis. Lancet. 358:810–811. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsushita M, Shichiri M, Imai T, Iwashina
M, Tanaka H, Takasu N and Hirata Y: Co-expression of urotensin II
and its receptor (GPR14) in human cardiovascular and renal tissues.
J Hypertens. 19:2185–2190. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ban Y, Watanabe T, Suguro T, Matsuyama TA,
Iso Y, Sakai T, Sato R, Idei T, Nakano Y, Ota H, et al: Increased
plasma urotensin-II and carotid atherosclerosis are associated with
vascular dementia. J Atheroscler Thromb. 16:179–187. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen Z, Xu J, Ye Y, Li Y, Gong H, Zhang G,
Wu J, Jia J, Liu M, Chen Y, et al: Urotensin II inhibited the
proliferation of cardiac side population cells in mice during
pressure overload by JNK-LRP6 signalling. J Cell Mol Med.
18:852–862. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tzanidis A, Hannan RD, Thomas WG, Onan D,
Autelitano DJ, See F, Kelly DJ, Gilbert RE and Krum H: Direct
actions of urotensin II on the heart: Implications for cardiac
fibrosis and hypertrophy. Circ Res. 93:246–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi L, Ding W, Li D, Wang Z, Jiang H,
Zhang J and Tang C: Proliferation and anti-apoptotic effects of
human urotensin II on human endothelial cells. Atherosclerosis.
188:260–264. 2006. View Article : Google Scholar
|
|
9
|
Zhang YG, Li J, Li YG and Wei RH:
Urotensin II induces phenotypic differentiation, migration, and
collagen synthesis of adventitial fibroblasts from rat aorta. J
Hypertens. 26:1119–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao J, Ding W, Song N, Dong X, Di B, Peng
F and Tang C: Urotensin II-induced collagen synthesis in cultured
smooth muscle cells from rat aortic media and a possible
involvement of transforming growth factor-β1/Smad2/3 signaling
pathway. Regul Pept. 182:53–58. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bousette N, Pottinger J, Ramli W, Ohlstein
EH, Dhanak D, Douglas SA and Giaid A: Urotensin-II receptor
blockade with SB-611812 attenuates cardiac remodeling in
experimental ischemic heart disease. Peptides. 27:2919–2926. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington (DC): 2011
|
|
13
|
Ma L, Liu J, Chu N, et al: The
relationship between cardiac hypertrophy index and cardiac function
in chronic heart failure rats. J Capit Med Univ (Chin). 31:596–599.
2010.
|
|
14
|
Kehat I and Molkentin JD: Molecular
pathways underlying cardiac remodeling during pathophysiological
stimulation. Circulation. 122:2727–2735. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Berenji K, Drazner MH, Rothermel BA and
Hill JA: Does load-induced ventricular hypertrophy progress to
systolic heart failure? Am J Physiol Heart Circ Physiol.
289:H8–H16. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Creemers EE and Pinto YM: Molecular
mechanisms that control interstitial fibrosis in the
pressure-overloaded heart. Cardiovasc Res. 89:265–272. 2011.
View Article : Google Scholar
|
|
17
|
Bayer AL, Heidkamp MC, Patel N, Porter MJ,
Engman SJ and Samarel AM: PYK2 expression and phosphorylation
increases in pressure overload-induced left ventricular
hypertrophy. Am J Physiol Heart Circ Physiol. 283:H695–H706. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lapp H, Boerrigter G, Costello-Boerrigter
LC, Jaekel K, Scheffold T, Krakau I, Schramm M, Guelker H and
Stasch JP: Elevated plasma human urotensin-II-1ike immunoreactivity
in ischemic cardiomyopathy. Int J Cardiol. 94:93–97. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Russell FD, Meyers D, Galbraith AJ, Bett
N, Toth I, Kearns P and Molenaar P: Elevated plasma levels of human
urotensin-II immunoreactivity in congestive heart failure. Am J
Physiol Heart Circ Physiol. 285:H1576–H1581. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tran L, Kompa AR, Kemp W, Phrommintikul A,
Wang BH and Krum H: Chronic urotensin-II infusion induces diastolic
dysfunction and enhances collagen production in rats. Am J Physiol
Heart Circ Physiol. 298:H608–H613. 2010. View Article : Google Scholar
|
|
21
|
Dai HY, Kang WQ, Wang X, Yu XJ, Li ZH,
Tang MX, Xu DL, Li CW, Zhang Y and Ge ZM: The involvement of
transforming growth factor-beta1 secretion in urotensin II-induced
collagen synthesis in neonatal cardiac fibroblasts. Regul Pept.
140:88–93. 2007. View Article : Google Scholar
|
|
22
|
Dai HY, He T, Li XL, Xu Wl and Ge ZM:
Urotensin-2 promotes collagen synthesis via ERK1/2-dependent and
ERK1/2-independent TGF-β1 in neonatal cardiac fibroblasts. Cell
Biol Int. 35:93–98. 2011. View Article : Google Scholar
|
|
23
|
Domínguez-Rodríguez A, Díaz I,
Rodríguez-Moyano M, Calderón-Sánchez E, Rosado JA, Ordóñez A and
Smani T: Urotensin-II signaling: Mechanism in rat coronary artery:
Role of STIM1 and Orai1-dependent store operated calcium influx in
vasoconstriction. Arterioscler Thromb Vasc Biol. 32:1325–1332.
2012. View Article : Google Scholar
|
|
24
|
Song QG, Lü JZ and Yang JX: Effect of
urotensin II on the proliferation of cardiac Myofibroblasts and its
intracellular signaling mechanism in new-born SD rats. J Xi'an
Jiaotong Univ. 28:157–160. 2007.
|
|
25
|
Kemp W, Kompa A, Phrommintikul A, Herath
C, Zhiyuan J, Angus P, McLean C, Roberts S and Krum H: Urotensin II
modulates hepatic fibrosis and portal hemodynamic alterations in
rats. Am J Physiol Gastrointest Liver Physiol. 297:G762–G767. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rossowski WJ, Cheng BL, Taylor JE, Datta R
and Coy DH: Human urotensin II-induced aorta ring contractions are
mediated by protein kinase C, tyrosine kinases and Rho-kinase:
Inhibition by somatostatin receptor antagonists. Eur J Pharmacol.
438:159–170. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang YG, Li YG, Liu BG, Wei RH, Wang DM,
Tan XR, Bu DF, Pang YZ and Tang CS: Urotensin II accelerates
cardiac fibrosis and hypertrophy of rats induced by isoproterenol.
Acta Pharmacol Sin. 28:36–43. 2007. View Article : Google Scholar
|
|
28
|
Staudt Y, Mobini R, Fu M, Felix SB, Kühn
JP and Staudt A: Beta1-adrenoceptor antibodies induce apoptosis in
adult isolated cardiomyocytes. Eur J Pharmacol. 466:1–6. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li CJ and Han QH: Effect of Urotensin II
on cardiac function in heart failure rats and its mechanism of
action. Chinese J Integr Med Cardio Cerebrovasc Dis. 6:38–40.
2008.
|
|
30
|
Han QH, Liu WY, Li CJ, Wang R and Shi HT:
The effect of urotensin II on cardiac function of rats and its
electrophysiological mechanism. Chinese J Integr Med Cardio
Cerebrovasc Dis. 9:1479–1481. 2011.
|