Introduction
Epilepsy is one of the most common neurological
conditions and is a set of chronic brain diseases caused by
abnormal discharge of neurons within a brief time-frame (1). Voltage-dependent calcium channels
(VDCCs) have important effects on neurotransmitter release and
membrane excitability (2,3). CaV1.2 is the most predominant VDCC
located in certain dendrites of hippocampal and cortical neurons
(4–6). Previous studies have demonstrated
that abnormal expression of CaV1.2 is present in different epilepsy
phenotypes (7–9). At early stages during and following
pilocarpine-induced status epilepticus, significant changes of
CaV1.2 have been found in different groups of hippocampal neurons
(7). However, no changes were
observed in the protein expression of CaV1.2 in inferior colliculus
neurons of genetically epilepsy-prone rats, compared with control
Sprague-Dawley rats (3). There are
at least four binding-sites linked to calmodulin (CaM) and several
potential CaM-dependent protein kinase II (CaMKII)-mediated
phosphorylation sites in CaV1.2 (8–11).
Binding of Ca2+ produces a conformational change in CaM,
exposing hydrophobic residues that promote interactions of the
Ca2+/CaM complex with CaV1.2 (12). The Ca2+/CaM complex can
enhance the affinity of CaM and the activity of CaV1.2 (13–16).
Thus, CaM is important as a Ca2+ sensor for
Ca2+-dependent facilitation and inactivation (17,18).
CaMKII is a multifunctional serine/threonine kinase, which can
mediate several Ca2+-dependent neuronal processes, and
it accounts for 0.5–1.0% of total brain protein and up to 2% of
hippocampal protein (19,20). CaMKII is activated by
auto-phosphorylation when it is combined with the
Ca2+/CaM complex. Additionally, phosphorylated-CaMKII
(p-CaMKII) exhibits its biological activity by the phosphorylation
of other target proteins (21,22).
With the involvement of CaMKII, the activity of CaV1.2 is promoted
by CaM (23,24). A previous study demonstrated
immediate inhibition of cortical and hippocampal CaM kinase II
activity in homogenate following the development of status
epilepticus in a rat pilocarpine model (25). However, the roles of CaV1.2, CaM
and CaMKII, and their interactions in epilepsy are controversial
and remain to be fully elucidated. The tremor rat model (TRM;
tm/tm) is an animal model of epilepsy, which is a genetic mutant
first discovered in a Kyoto-Wistar colony (26). TRM is regarded as a useful model of
epilepsy due to its similarity in pathogenesis and treatment to the
human condition (27). Hippocampal
neurons exposed to Mg2+-free solution are well suited to
biochemical and electrophysiological investigations to elucidate
the cellular mechanisms, which lead to epileptogenesis (28), as the spontaneous epileptiform
discharges produced by hippocampal neurons exposed to
Mg2+-free solution is similar to the electrical activity
of epileptic seizures in humans. Thus, the aim of the present study
was to investigate the alterations of the selected
Ca2+/CaV1.2/CaM/CaMKII pathway in the TRM and in
cultured hippocampal neurons exposed to Mg2+-free
solution, in vivo and in vitro, respectively. These
investigations aimed to obtain an insight into the pathways in
producing neuronal excitability.
Materials and methods
Animals
The TRM is an animal model of epilepsy and the tm
genetic mutant was identified in a Kyoto-Wistar colony (29). In the present study, 15 normal
Wistar rats and 15 TRMs aged between 9 and 12 weeks were housed in
individual cages under a controlled environment (12:12 h light/dark
cycle, 50–70% humidity, 24°C) with free access to food and water.
The present study was performed in strict accordance with the
recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health (15,16).
The animal use protocol was reviewed and approved by the
Institutional Animal Care and Use Committee of China Medical
University (Shenyang, China).
Electroencephalographic (EEG)
determination and evaluation
The experimental animals (five animals per group)
were anesthetized with 10% chloral hydrate (intraperitonal, 0.3
ml/100 g) and the animals were implanted with EEG electrodes
(BL-420F; Taimeng Co., Ltd., Chengdu, China). Cortical and
hippocampal electrodes were chronically implanted onto the cortex
(3.0 mM lateral and 3.0 mM rostral to the bregma on the cranium)
and in the left hippocampus (2.0 mM lateral and 4.0 mM caudal to
the bregma and 3.0 mM from the cortical surface), respectively
(30). Over the 7 days following
this procedure, the EEG was recorded using a pen-writing polygraph.
Following habituation of the rats in a soundproof box (40×40×40 cm)
for >20 min, the EEG was recorded for 30 min. The rat was
exposed to puff stimulation on the face by a researcher every 5 min
to ensure the animal remained aroused during the recording period.
Changes in the EEG during absence-like seizures consistently
correlated with the abnormal seizure behavior, as described
previously (29). When 5–7 Hz
spike-wave-like complexes in the cortical and hippocampal EEG
lasted for >1 sec, the response was considered as one
absence-like seizure. When the time interval between two
independent 5–7 Hz spike-wave-like complexes was <1 sec, the two
events were considered as a single seizure.
Primary neuronal cell cultures
Hippocampal neuronal cells, which were obtained from
rats born within 1 day of each other, were dissociated in Hanks'
balanced salt solution containing 0.125% trypsin (Sigma-Aldrich,
St. Louis, MO, USA) for 10 min at 37°C. The cells were then plated
onto dishes at 2×105 cells/cm2 in Dulbecco's
modified Eagle's medium (Gibco-BRL, Invitrogen Life Technologies,
Carlsbad, CA, USA) containing 15% fetal bovine serum and
penicillin-streptomycin, (100 U/ml, Gibco-BRL). Following plating
for 24 h, the medium was replaced with Neurobasal™ medium
supplemented with 2% B27 (Gibco-BRL). The hippocampal neuronal cell
media was refreshed every 3–4 days. Hippocampal neurons at the 12th
day of in vitro culture were used in the following
experiments.
Establishment of the hippocampal neuronal
culture model
The in vitro hippocampal neuronal culture
model was established according to Sombati's method (31). For primary neuronal cell cultures,
maintenance medium was replaced with physiological recording
solution for 3 h (Beyotime Institute of Biotechnology, Haimen,
China) with or without MgCl2 (1 mM). The solution
contained 145 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 2 mM
CaCl2, 10 mM glucose and 0.002 mM glycine (pH 7.3), the
osmolarity of which was adjusted to 290±10 mOsm using sucrose.
Continuous epileptiform high-frequency bursts were induced by
exposing the neuronal cultures to physiological recording solution
without MgCl2 (Mg2+-free) for 3 h, following
which the culture was exposed to physiological recording solution.
For double-labeling immunofluorescence and western blotting, the
cultures were exposed to physiological solution for 8 h following
treatment with Mg2+-free solution for 3 h prior to use
of the hippocampal neurons in these experiments. For measurement of
intracellular calcium concentration, the cultures were exposed to
Mg2+-free solution of 3 h, following which the
hippocampal neurons were used immediately.
Electrophysiological recordings
The electrophysiological recording method used in
the present study was that used in our previous study (32). The extracellular bath contained 135
mM NaCl, 5.4 mM KCl, 1.0 mM MgCl2, 0.33 mM
NaH2PO4, 10 mM HEPES and 5.5 mM glucose (pH
7.4; NaOH). The pipette solution contained 50 mM K-Aspartate, 20 mM
KCl, 20 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 0.2 mM
CaCl2, 13.6 mM NaCl and 3 mM K2ATP3 (pH 7.4; KOH;
Beyotime Institute of Biotechnology). Patch-clamp electrodes were
obtained with capillary tubes pulled with a P-97 puller
(Gibco-BRL). The electrodes had a resistance of ~2.5–4 MΩ. Patch
clamp was performed using an AxoPatch 200B amplifier (Axon
Instruments, Foster City, CA, USA). To record action potential,
cultures, including the normal cultured neurons and the neurons
treated with Mg2+-free solution, were transferred to the
stage of an inverted microscope (TMS-1015, Olympus, Tokyo, Japan).
Patch-clamp was performed utilizing the whole-cell current-clamp
gap-free recording mode.
Western blot analysis
Proteins from the tissues and neurons were extracted
by lysis in radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology) with ultrasonication, followed by
centrifugation and retrieval of the supernatant. The concentrations
of extracted proteins from the tissues and neurons were determined
using a bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). The protein samples (50 µg per lane) were
separated by 10% SDS-PAGE (Beyotime Institute of Biotechnology) and
transferred onto polyvinylidene difluoride membranes (Motimo
Membrane Technology Co., Ltd., Tianjin, China). The membranes were
blocked for 1 h at room temperature with 5% bovine serum albumin
(BSA) in tris-buffered saline with Tween 20 (TTBS; Beyotime
Institute of Biotechnology) containing 50 mM Tris-HCl; 0.1%
Tween-20 and 154 mM NaCl (pH 7.4), followed by 2 h incubation. The
membranes were then incubated overnight at 4°C in TTBS containing
the following primary antibodies: Rabbit anti-CaV1.2 (1:500; cat
no. ab-81095; Abcam, Cambridge, MA, USA), rabbit anti-CaMKII
(1:500; cat no. sc-9035; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), rabbit anti-p-CaMKII (1:500; cat no. sc-3228; Santa Cruz
Biotechnology, Inc.; the epitope corresponding to a short amino
acid sequence containing phosphorylated Thr-286 of CaMKII α), mouse
anti-CaM (1:800; cat no. sc-137079; Santa Cruz Biotechnology, Inc.)
and β-actin (1:1,000; cat no. sc-47778; Santa Cruz Biotechnology,
Inc.). Following several washes in TTBS, the membranes were
incubated for 1 h at room temperature in horseradish
peroxidase-conjugated goat anti-mouse IgG or horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:5,000; cat nos.
sc-2005 and sc-2054, respectively; Santa Cruz Biotechnology, Inc.)
for 2 h at room temperature. Immunoreactive bands were visualized
using an enhanced chemiluminescence kit. The levels of protein were
evaluated by measuring the optical densities of the protein bands
using Scion Image (4.03) for Windows image-analysis software.
β-actin was used as a control to demonstrate that equal quantities
of protein were loaded. Similarly, CaMKII was used as a control to
demonstrate that equal quantities of p-CaMKII were loaded.
Double-labeling immunofluorescence
The TRMs and control rats were anesthetized with 10%
chloral hydrate (i.p., 0.35 ml/100 g). Following intracardial
perfusion with 4% paraformaldehyde, the brains of the rats were
rapidly removed and placed in 4% paraformaldehyde solution for 24 h
at room temperature and 30% sucrose solution for 24 h at 4°C. The
dehydrated-fixed brains were then frozen and 10-µm serial
coronal sections were cut using a cryostat (CM1900 UV; Leica
Microsystems GmbH, Wetzlar, Germany). These sections, and the
Mg2+-free treated and normal hippocampal neurons, were
rinsed in phosphate-buffered saline (PBS; 0.01 M; pH 7.4) for 15
min and incubated in PBS (0.01 M; pH 7.4), which was supplemented
with 0.25% Triton X-100 (Sigma-Aldrich) and 10% BSA (Gibco-BRL) for
1 h for blocking and penetration. The sections and neurons were
then incubated overnight at 4°C in a mixture of the following
primary antibodies: Mouse anti-CaM (1:100; Santa Cruz
Biotechnology, Inc.) and rabbit anti-CaV1.2 (1:80; Abcam) or mouse
anti-CaM (1:100; Santa Cruz Biotechnology, Inc.) and rabbit anti
p-CaMKII (1:100; Santa Cruz Biotechnology, Inc.). PBS without
primary antibodies was used as a negative control. Following
rinsing with 0.01 M PBS three times, for 5 min each, the sections
and neurons were incubated with fluorescein isothiocyanate (FITC)-
and Cy3-conjugated goat anti-mouse or anti-rabbit antibodies
(1:200; ZSGB-BIO, Beijing, China) for 2 h at room temperature in
the dark. Finally, sections from different brain regions, including
the hippocampus (CA1, CA3 and DG), temporal cortex and cerebellum,
were examined. The sections and neurons were examined using a
Confocal laser scanning biological microscope (Fluoview FV500;
Olympus Corporation, Tokyo, Japan). CaV1.2 and p-CaMKII were
labeled with FITC-emitting green light, CaM was labeled with
tetramethylrhodamine-emitting red light, with yellow light
representing the co-localization of CaV1.2 and CaM or p-CaMKII and
CaM. The co-localized cells that were clearly yellow were selected
as positive cells, and the number of positive cells in every 100
cells was determined.
Acute dissociation of neurons
Acute dissociation of neurons was performed using a
previous method with certain modifications (33). The experimental animals were
anesthetized with 10% chloral hydrate (intraperitoneal, 0.3 ml/100
g) and then quickly sacrificed by decapitation. The brains were
rapidly removed and placed in Hank's balanced salt solution (Gibco
Life Technologies). The tissues (hippocampus, temporal cortex and
cerebellum) were separated and cut into 400 µm-thick slices.
These slices were incubated in artificial cerebrospinal fluid
(Beyotime Institute of Biotechnology) containing 126 mM NaCl, 5 mM
KCL, 2 mM CaCl2, 25 mM NaHCO3, 1.5 mM
NaH2PO4, 2 mM MgSO4 and 10 mM
glucose (32°C; 5% CO2; pH adjusted to 7.4 with NaOH) for
at least 1 h. The tissue slices were dissociated using 0.125%
trypsin (Sigma-Aldrich) for 30 min at 37°C, and the dissociated
tissues were then rinsed three times in extracellular fluid
containing 130 mM NaCl; 5.4 mM KCl; 1 mM MgCl2; 1 mM
CaCl2; 10 mM HEPES and 25 mM glucose (32°C; 5%
CO2; pH adjusted to 7.4 with NaOH), and triturated
mechanically with a graded series of fire-polished Pasteur pipettes
of ~500, 300 and 150 µm (Leiqi Experiment Equipment Co.,
Ltd., Hangzhou, China, successively. Following standing for 2 min,
the upper cells in the cell suspension were plated onto culture
dishes (1×106 cells/ml) in extracellular fluid. The
acute isolated neurons were used within 6 h. All experimental
procedures were maintained in a humidified atmosphere under 5%
CO2.
Measurement of intracellular calcium
concentration ([Ca2+]i)
The measurement of [Ca2+] I was
performed, according to that described in our previous study
(34). The acutely dissociated
neurons and Mg2+-free treated hippocampal neurons were
incubated for 20 min with 5 µm fluo-3 acetoxymethylester
(fluo-3/AM; Molecular Probes Life Technologies, Carlsbad, CA, USA;
32°C; 5% CO2; pH adjusted to 7.4 with NaOH). The neurons
of the control group and model group were plated into two cell
culture cover glasses in the same dish. Thus, the control and model
neurons were treated using the same procedure. The labeled neurons
were then rinsed three times with PBS and cultured for 30 min to
exclude non-specific staining of the extracellular fluid (32°C; 5%
CO2). The preparations were observed and quantitatively
analyzed using a confocal laser scanning biological microscope
(Fluoview FV500; Olympus, Corporation). The visual field was
selected where at least five neurons contained fluo-3/AM, which was
excited by the 488 nm line of a 200 mW argon ion laser and captured
at wavelengths >505 nm. The data were expressed as relative
fluorescence intensities. Confocal imaging was performed on three
separate fields of cells for each group. The concentration of
intracellular Ca2+ was expressed as the relative
fluorescent intensity using ImageJ 1.46 software (National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Values are expressed as the mean ± standard
deviation, and statistical analysis were performed using SPSS 13.0
statistical software (SPSS, Inc., Chicago, IL, USA). Statistical
significance was determined using Student's t-test and
one-way analysis of variance was used for comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
Spontaneous discharge activity in the EEG
recording in the TRM and Mg2+-free treatment model
The EEG recording of The control rats and TRMs were
detected in the hippocampus and cerebral cortex, respectively. It
did not reveal any abnormal discharges in The control rat
hippocampus or cerebral cortex (Fig.
1Aa), however, a 6 Hz spike and wave complex was recorded
during absence-like seizure in the TRM hippocampus and cerebral
cortex (Fig. 1Ab). In addition,
the whole-cell current-clamp technique was used to record the
electrical physiological activities of normal neurons (n=6) and
Mg2+-free treated neurons (n=6). The results
demonstrated that representative neurons during 3 h exposure to
physiological recording solution containing 1 mM MgCl2
produced a normal baseline activity, presenting with occasional
action potentials (Fig. 1Ba).
However, exposure of the Mg2+-free treated neurons,
which were exposed for 3 h to Mg2+-free recording
solution, produced continuous high frequency epileptiform
discharges (Fig. 1Bb). These data
indicated that the in vivo and in vitro modelS of
epileptic discharge in the present study had been established
successfully.
Abnormal protein expression of CaV1.2,
CaM, p-CaMKII and CaMKII in the TRM and Mg2+-free
treatment model
Western blot analysis was performed to quantify and
compare selected the protein expression levels in the TRMs (n=6)
and control rats (n=6). The western blot was probed with antibodies
to CaV1.2, CaM, p-CaMKII and CaMKII, and their respective
anticipated bands at 240 kDa, 17 kDa and 50 kDa were detected,
respectively. In the TRMs, hippocampal quantification analysis
revealed that the levels of expression of CaV1.2 (P=0.0017) and CaM
(P=0.0001) were increased significantly, while those of p-CaMKII
(P=0.0001) were decreased significantly, compared with the control
rats. No change was observed in the expression of CaMKII (P=0.08),
compared with control groups (Fig.
2A). Notably, the situation was reversed in the protein
expression patterns of the TRM temporal cortex. The expression
levels of of CaV1.2 (P=0.0001) and CaM (P=0.0130) were
significantly lower than those in the control group. Compared with
the control rats, the expression of p-CaMKII (P=0.0017) was
increased significantly. No change was detected in the expression
of CaMKII (P=0.77), compared with the control group (Fig. 2B). In addition, these findings
verified upregulation of the expression of CaM (P=0.0001) and
downregulation in the expression of CaV1.2 (P=0.0019) at the
protein level in the TRM cerebellum. No changes occurred in the
protein expression of p-CaMKII (P=0.3700) or CaMKII (P=0.72),
compared with the control group (Fig.
2C). In the hippocampal Mg2+-free neuron
epileptiform discharge model, no significant differences were
observed in the expression of CaM (P=0.9799), compared with the
control neurons, while p-CaMKII (P=0.0006) and CaV1.2 (P=0.0001)
were decreased, compared with the control neurons. No change was
observed in the protein expression of CaMKII (P=0.43), compared
with the control group (Fig.
2D).
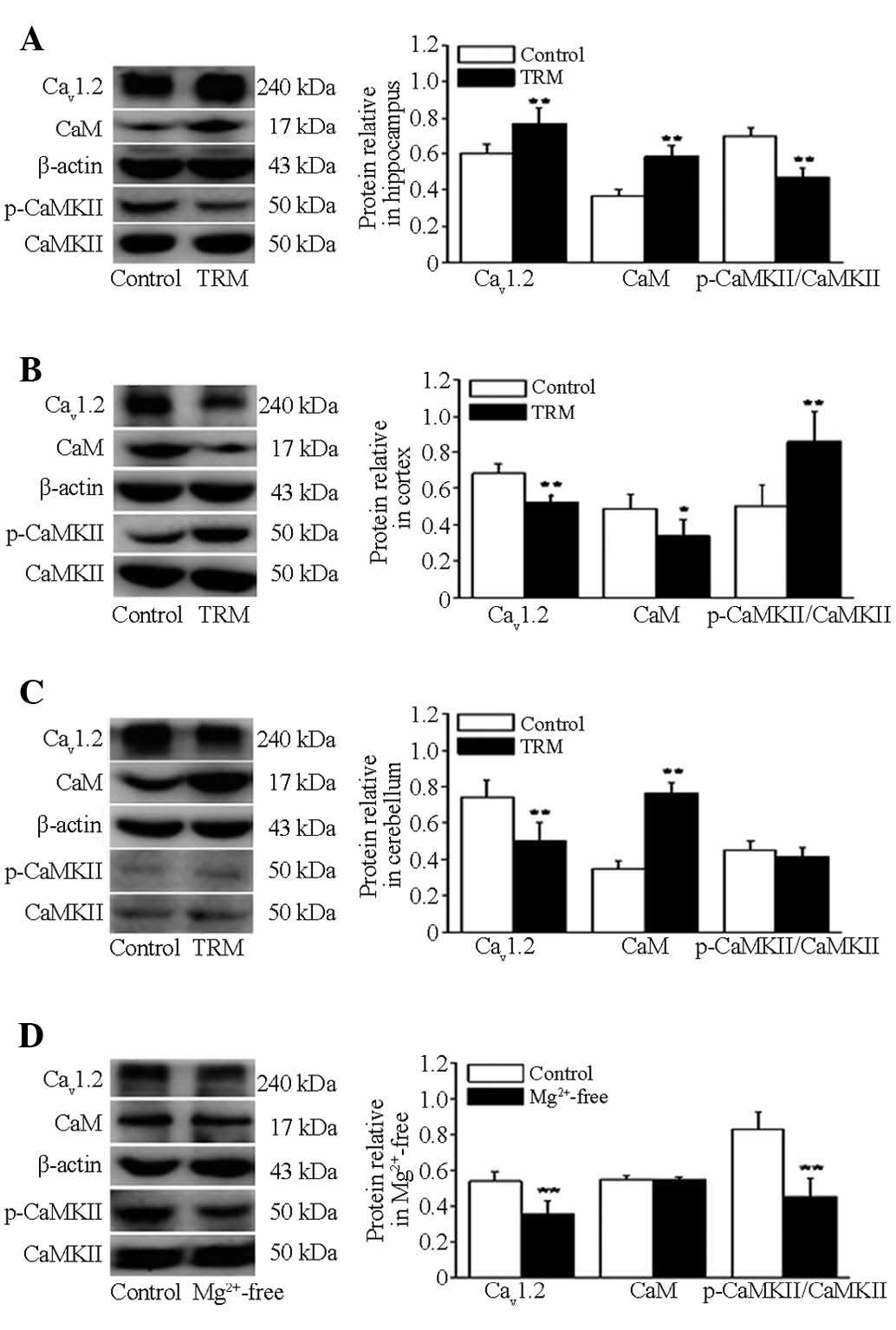 | Figure 2Protein expression levels of CaV1.2,
CaM, p-CaMKII and CaMKII in the TRM and hippocampal cultured
neurons exposed to Mg2+-free by western blotting. (A)
Immunoblots and quantitative analysis of the protein levels of
CaV1.2, CaM, p-CaMKII and CaMKII in the TRM hippocampal and control
groups (n=6). (B) Immunoblots and quantitative analysis of the
protein levels of CaV1.2, CaM, p-CaMKII and CaMKII in the TRM
cortex and control groups (n=6). (C) Immunoblots and quantitative
analysis of the protein levels of CaV1.2, CaM, p-CaMKII and CaMKII
in the TRM cerebellum and control groups (n=6). (D) Immunoblots and
quantitative analysis of the protein levels of CaV1.2, CaM,
p-CaMKII and CaMKII in the in vitro model (n=6) and control
groups (n=6). **P<0.01, vs. control group;
*P<0.05, vs. control group (analysis of variance
followed by Student's t-test). Data are presented as the mean ±
standard deviation. TRM, tremor rat model; CaM, calmodulin; CMKII,
CaM-dependent protein kinase II; p-, phosphorylated. |
Co-localization of CaV1.2 with CaM, and
p-CaMKII with CaM in the TRM and Mg2+-free treatment
model
Immunofluorescence analyses were performed in the
two model (n=5) and control group (n=5) using anti-CaV1.2, anti-CaM
and anti-p-CaMKII to detect the distributions and expression
levels. As shown in Fig. 3A, in
the TRM hippocampus, compared to control group, the CaV1.2 and CaM
immunopositive neurons were stained markedly in CA1 and CA3 areas,
while in DG areas, they were not expressed as highly as they were
in the CA1 or CA3 areas. CaV1.2 was concentrated in the soma and
proximal dendrites of the pyramidal neurons (green), and CaM was
primarily localized to the cytoplasm (red). It was of interest to
note that CaV1.2 co-localized with CaM. Overlapping localization of
these proteins (yellow) were evident in the CA1 (P=0.0044), CA3
(P=0.0050) and DG (P=0.0060) areas when the images of their
individual staining patterns were merged (Fig. 3B). As indicated in Fig. 3C and D, in the TRM cortex, the
number of co-localization cells for CaV1.2-CaM was decreased,
compared with that of the normal rats (P=0.0417). In the
cerebellum, the number of co-localization cells for CaV1.2-CaM was
not changed, compared with the controls (P=0.4771). In the
Mg2+-free treated neuron model, CaV1.2 was localized to
the cell membrane, while CaM was localized to the cell membrane and
the cytoplasm. The number of co-localization cells for CaV1.2-CaM
in the Mg2+-free treated model was decreased compared
with normal neurons (P=0.0075). In addition, p-CaMKII
immunopositive neurons (green) were localized in the cytoplasm of
cells in hippocampus. Compared with the controls, the numbers of
co-localization cells of CaM-p-CaMKII in the CA1 (P=0.0480) and CA3
(P=0.0289) of the TRM hippocampus were increased, respectively,
however, there was no change in the DG (P=0.5675, Fig. 4A and B). It was clear that the
distribution pattern of p-CaMKII was widely localized in the
cytoplasm of cortical cells, which was similar to that observed in
the hippocampus (Fig. 4C). There
was no significant change of overlapping localization of p-CaMKII
and CaM in the temporal cortex (P=0.9631; Fig. 4C and D). In the cerebellum of the
TRM, the number of CaM-p-CaMKII-positive cells was also unchanged
(P=0.7792; Fig. 4C and D).
However, in the in vitro model, there was a decrease in the
number of p-CaMKII and CaM co-localization neurons, compared with
the controls (P=0.0059; Fig. 4C and
D).
 | Figure 3Co-localization of CaV1.2 and CaM in
the TRM and in vitro model, detected using
immunofluorescence. CaV1.2 was labeled with fluorescein
isothiocyanate-emitting green light, CaM was labeled with
tetramethylrhodamine-emitting red light, with yellow light
indicating the co-localization of CaV1.2 and CaM. (A) TRM
hippocampus CA1, CA3 and DG region. (B) Number of positive cells
for CaV1.2-CaM in the TRM hippocampus. **P<0.01, vs.
control group; *P<0.05, vs. control group (ANOVA
followed by Student's t-test; n=5). (C) TRM cortex, cerebellum and
Mg2+-free hippocampal neurons. (D) Number of positive
cells for CaV1.2-CaM in the TRM cortex, cerebellum and in
Mg2+-free hippocampal neurons. **P<0.01,
vs. control group; *P<0.05, vs. control group (ANOVA
followed by Student's t-test, n=5). Scale bar=50 µm. Data
are presented as the mean ± standard deviation. TRM, tremor rat
model; CaM, calmodulin; CMKII, CaM-dependent protein kinase II;
ANOVA, analysis of variance. |
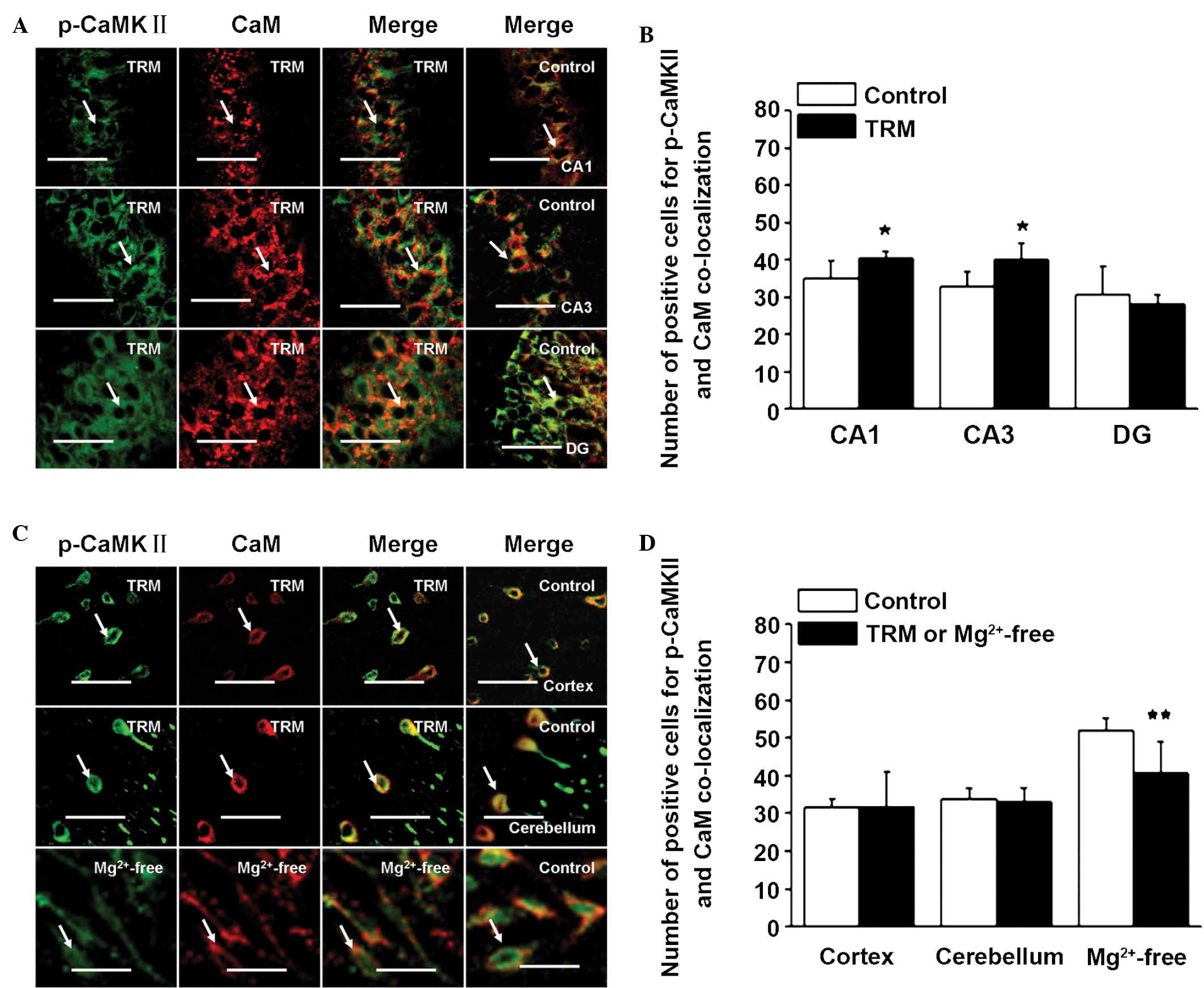 | Figure 4Co-localization of p-CaMKII and CaM
in the TRM and in vitro model, detected using
immunofluorescence. The p-CaMKII was labeled with fluorescein
isothiocyanate-emitting green light and CaM was labeled with
tetramethylrhodamine-emitting red light, with yellow light
indicating the co-localization of p-CaMKII and CaM. (A) TRM
hippocampus CA1, CA3 and DG regions. (B) Number of positive cells
for p-CaMKII-CaM in the TRM hippocampus. **P<0.01,
vs. control group; *P<0.05, vs. control group (ANOVA
followed by Student's t-test; n=5). (C) TRM cortex, cerebellum and
Mg2+-free hippocampal neurons. (D) Number of positive
cells for p-CaMKII-CaM in the TRM cortex, cerebellum and in
Mg2+-free hippocampal neurons. **P<0.01,
vs. control group; *P<0.05, vs. control group (ANOVA
followed by Student's t-test; n=5). Scale bar=50 µm.
Data are presented as the mean ± standard deviation. TRM, tremor
rat model; CaM, calmodulin; CMKII, CaM-dependent protein kinase II;
p-, phosphorylated. |
Measurement of intracellular calcium
concentration in the TRM and Mg2+-free treatment
model
Compared with the controls (n=5), the
[Ca2+] I relative fluorescence intensity of the model
groups (n=5), including the TRMs (hippocampus, cortex and
cerebellum; Fig. 5A-C) and
Mg2+-free hippocampal neurons (Fig. 5D) were increased, as shown in
Fig. 5E and F.
Discussion
The predominant aims of the present study were to
examine the changes in Ca2+/CaV1.2/CaM/CaMKII in the TRM
and in the hippocampal Mg2+-free neuron epileptiform
discharge models; and to demonstrate the possible correlation
between CaV1.2 and the CaM/CaMKII pathway in these models.
Epileptic models are fundamental to the investigation of the
pathogenesis and possible treatments for epilepsy. TRMs were used
in the present study as they are similar to the relevant human
disease (27). TRM is an ideal
model for the investigation of absence-like seizures, and seizures
linked with the tm mutant, which was mapped to rat chromosome 10
(35). Furthermore, the paroxysmal
occurrence of a 5–7 Hz spike-wave complexes can be recorded in the
hippocampal and cortical areas after 8 weeks (29). In addition, the hippocampal
Mg2+-free neuron epileptiform discharge model is a
common model of epileptiform discharge in vitro (36).
In the present study, Mg2+-free treated
hippocampal neurons produced continuous high frequency epileptiform
discharges, which corresponded to observations in our previous
study using whole-cell current-clamp (32). In the present study, the
[Ca2+] I fluorescence intensity was increased in the
TRMs (hippocampus, temporal cortex and cerebellum) and in the in
vitro model, indicating that Ca2+ mayt be a key
element in the epileptogenesis of these two models.
[Ca2+] I led to the depolarization of cells, activating
the inflow of Na+ and further enhancing epilepsy (37,38).
Previous studies demonstrated that status epilepticus causes
sustained elevation of intracellular calcium levels in the
hippocampal neuronal culture model (39,40).
In addition, calcium influx is enhanced in hippocampal CA3 neurons
of spontaneously epileptic rats (41), which is in agreement with the
results of the present study. A prominent finding in the present
study was that the protein expression of CaV1.2 was increased
significantly in the TRM hippocampus. However, the protein
expression levels of CaV1.2 were decreased in the cortex,
cerebellum and in in vitro culture. Previous studies have
demonstrated that VDCC is essential in neuronal excitability
(42). Transient and selective
upregulation of CaV3.2 subunits on the mRNA and protein levels
following status epilepticus causes an increase in cellular T-type
Ca2+ currents and a transitional increase in intrinsic
burst firing (43). Enhancements
of Ca2+ influx into hippocampal CA3 neurons, due to the
easier activation properties of VDCCs, are involved in SER
epileptic seizures (44). In
addition, VDCC currents are enhanced in the hippocampus of patients
with temporal lobe epilepsy (45).
Another study reported that the expression of CaV1.2 was
significantly reduced in Stargazer mouse cerebellar synapses,
compared with their non-ataxic littermates, however, no differences
were detected in hippocampal synapses (5). Accordingly, these findings have
indicated the importance of CaV1.2 in the epileptic brain. The
increased expression of CaV1.2 in TRM may result in an increase in
the number of Ca2+ channels, following which
Ca2+ current may be elevated, and a long-lasting
depolarization shift accompanied by repetitive firing may be
induced, which may contribute to the enhanced neuronal excitability
in epilepsy (41). Thus, the
present study revealed that the upregulated expression of CaV1.2 in
the hippocampus of TRM may be involved in the generation of
epileptiform activity and underlie, at least in part, the observed
seizure phenotype in TRM.
The present data also demonstrated the upregulation
of co-localization of CaM and CaV1.2 in the TRM hippocampus, and
the combination of CaM and CaV1.2 may activate the Ca2+
channel and, ultimately, elevate the level of excitability in
neurons. Notably, the distribution and expression of CaM and CaV1.2
in the cortex was opposite to that observed in the hippocampus in
the TRM, indicating a possible compensatory response aimed at
counteracting hyperexcitability in the cortex of the TRM. Several
previous studies have reported controversy in the change of CaMKII
in different models of epilepsy. Selective suppression of CaMKII
activity results in alterations in Ca2+ homeostasis and
the development of SREDs in hippocampal neurons (46). Additionally, the expression of
CaMKIIα is decreased in pentylenetetrazol-kindled rats (47). Another study demonstrated that the
expression of p-CaMKII is upregulated in dendritic spines during
epileptiform activity in vitro (48). This may be due to factors including
different epilepsy models and brain regions. The present study
demonstrated that the expression of p-CaMKII in the hippocampus was
downregulated. This may be due to Ca2+ being overloaded
in epilepsy, which upregulated the expression of CaM, following
which Ca2+/CaM-dependent enzymes were adjusted and the
activity of CaMKII was restrained. However, the expression of
p-CaMKII in the cortex was upregulated, which disagreed with the
expression in the hippocampus and may be a compensatory response in
the cortex. Notably, the expression levels of p-CaMKII were
particularly low in the cerebellum. Thus, sustained Ca2+
overload may have caused a sharp increase in CaMKII
auto-phosphorylation, which resulted in the reduction in CaMKII and
p-CaMKII. In the in vitro model, the expression levels of
p-CaMKII and CaV1.2 were downregulated, while that of CaM remained
unchanged. Similar to previous studies, the reason for this may be
that SREDs inhibited the activity of CaMKII and restrained its
substrate (23,24).
The limitation of the present study lies in the
in vivo and in vitro models, which may represent
different types of models. To a certain extent, the hippocampal
neuronal culture model can be considered an acute seizures model,
and the neurons treated with Mg2+-free solution for 3 h
may be not sufficient to fully undergo the molecular and cellular
changes associated with the development of epileptogenesis. By
contrast, TRM rats are genetic epileptic animals exhibiting
spontaneous seizures, which can be considered a chronic model of
epilepsy. Therefore, it is not surprising that the discrepancy was
observed in the results of these two models. A noteworthy findings
of the present study was that the CaV1.2-CaM and CaMKII-CaM
complexes were widely co-localized in the TRM hippocampus,
indicating that the association between these proteins may be
involved in TRM seizures. The present study hypothesized that when
[Ca2+] I increases in TRM, Ca2+ and CaM are
combined to form the Ca2+/CaM complex, facilitating the
affinity of CaM and CaV1.2. Additionally, CaMKII was activated by
auto-phosphorylation when it combined with the Ca2+/CaM
complex. Furthermore, increased CaM can be co-localized to the
membrane with CaV1.2, leading to the upregulation and increased
activity of CaV1.2, which contribute to enhanced neuronal
excitability and results in TRM seizures. Collectively, the present
study demonstrated abnormal changes in the
Ca2+/CaV1.2/CaM/CaMKII signaling pathway in TRMs and in
the hippocampal neuronal culture model. Altering the expression of
CaV1.2, CaMKII and CaM may lead these to become potential targets
for therapy in epilepsy or seizures. TRMs and the hippocampal
neuronal culture model can be screened for effective specific VDCC
subtypes for the treatment of epilepsy or seizures.
Acknowledgments
This study was financially supported by Natural
Science Foundation of China (grant nos. 81471323, 81001429 and
31471091). The authors would like to thank Dr Tadao Serikawa (Kyoto
University, Kyoto, Japan) for the provision of the TRM strain.
References
|
1
|
Moseley BD, Wirrell EC, Wong-Kisiel LC and
Nickels K: Early onset epilepsy is associated with increased
mortality: A population-based study. Epilepsy Res. 105:410–414.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakatani Y, Masuko H and Amano T: Effect
of lamotrigine on Na(v)1.4 voltage-gated sodium channels. J
Pharmacol Sci. 123:203–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
N'Gouemo P, Yasuda R and Faingold CL:
Seizure susceptibility is associated with altered protein
expression of voltage-gated calcium channel subunits in inferior
colliculus neurons of the genetically epilepsy-prone rat. Brain
Res. 1308:153–157. 2010. View Article : Google Scholar
|
|
4
|
Arikkath J and Campbell KP: Auxiliary
subunits: Essential components of the voltage-gated calcium channel
complex. Curr Opin Neurobiol. 13:298–307. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leitch B, Shevtsova O, Guevremont D and
Williams J: Loss of calcium channels in the cerebellum of the
ataxic and epileptic stargazer mutant mouse. Brain Res.
1279:156–167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Obermair GJ, Szabo Z, Bourinet E and
Flucher BE: Differential targeting of the L-type Ca2+ channel alpha
1C (CaV1.2) to synaptic and extrasynaptic compartments in
hippocampal neurons. Eur J Neurosci. 19:2109–2122. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu JH, Long L, Tang YC, Hu HT and Tang FR:
Ca(v)1.2, Ca(v)1.3 and Ca(v)2.1 in the mouse hippocampus during and
after pilocarpine-induced status epilepticus. Hippocampus.
17:235–251. 2007. View Article : Google Scholar
|
|
8
|
Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham
AJ, Mohler PJ, Anderson ME and Colbran RJ: L-type Ca2+ channel
facilitation mediated by phosphorylation of the beta subunit by
CaMKII. Mol Cell. 23:641–650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hao LY, Wang WY, Minobe E, Han DY, Xu JJ,
Kameyama A and Kameyama M: The distinct roles of calmodulin and
calmodulin kinase II in the reversal of run-down of L-type Ca(2+)
channels in guinea-pig ventricular myocytes. J Pharmacol Sci.
111:416–425. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Minobe E, Asmara H, Saud ZA and Kameyama
M: Calpastatin domain L is a partial agonist of the
calmodulin-binding site for channel activation in Cav1.2 Ca2+
channels. J Biol Chem. 286:39013–39022. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang WY, Hao LY, Minobe E, Saud ZA, Han DY
and Kameyama M: CaMKII phosphorylates a threonine residue in the
C-terminal tail of Cav1.2 Ca(2+) channel and modulates the
interaction of the channel with calmodulin. J Physiol Sci.
59:283–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dolmetsch RE, Pajvani U, Fife K, Spotts JM
and Greenberg ME: Signaling to the nucleus by an L-type calcium
channel-calmodulin complex through the MAP kinase pathway. Science.
294:333–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Asmara H, Minobe E, Saud ZA and Kameyama
M: Interactions of calmodulin with the multiple binding sites of
Cav1.2 Ca2+ channels. J Pharmacol Sci. 112:397–404. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dick IE, Tadross MR, Liang H, Tay LH, Yang
W and Yue DT: A modular switch for spatial Ca2+ selectivity in the
calmodulin regulation of CaV channels. Nature. 451:830–834. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo F, Minobe E, Yazawa K, Asmara H, Bai
XY, Han DY, Hao LY and Kameyama M: Both N- and C-lobes of
calmodulin are required for Ca2+-dependent regulations of CaV1.2
Ca2+ channels. Biochem Biophys Res Commun. 391:1170–1176. 2010.
View Article : Google Scholar
|
|
16
|
Han DY, Minobe E, Wang WY, Guo F, Xu JJ,
Hao LY and Kameyama M: Calmodulin- and Ca2+-dependent facilitation
and inactivation of the Cav1.2 Ca2+ channels in guinea-pig
ventricular myocytes. J Pharmacol Sci. 112:310–319. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peterson BZ, DeMaria CD, Adelman JP and
Yue DT: Calmodulin is the Ca2+ sensor for Ca2+-dependent
inactivation of L-type calcium channels. Neuron. 22:549–558. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang W, Halling DB, Black DJ, Pate P,
Zhang JZ, Pedersen S, Altschuld RA and Hamilton SL: Apocalmodulin
and Ca2+ calmodulin-binding sites on the CaV1.2 channel. Biophys J.
85:1538–1547. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Erondu NE and Kennedy MB: Regional
distribution of type II Ca2+/calmodulin-dependent protein kinase in
rat brain. J Neurosci. 5:3270–3277. 1985.PubMed/NCBI
|
|
20
|
Zhang L, Kirschstein T, Sommersberg B,
Merkens M, Manahan-Vaughan D, Elgersma Y and Beck H: Hippocampal
synaptic metaplasticity requires inhibitory autophosphorylation of
Ca2+/calmodulin-dependent kinase II. J Neurosci. 25:7697–7707.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rich RC and Schulman H: Substrate-directed
function of calmodulin in autophosphorylation of
Ca2+/calmodulin-dependent protein kinase II. J Biol Chem.
273:28424–28429. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Welsby PJ, Wang H, Wolfe JT, Colbran RJ,
Johnson ML and Barrett PQ: A mechanism for the direct regulation of
T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J
Neurosci. 23:10116–10121. 2003.PubMed/NCBI
|
|
23
|
Dzhura I, Wu Y, Colbran RJ, Balser JR and
Anderson ME: Calmodulin kinase determines calcium-dependent
facilitation of L-type calcium channels. Nat Cell Biol. 2:173–177.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hudmon A, Schulman H, Kim J, Maltez JM,
Tsien RW and Pitt GS: CaMKII tethers to L-type Ca2+ channels,
establishing a local and dedicated integrator of Ca2+ signals for
facilitation. J Cell Biol. 171:537–547. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singleton MW, Holbert WH II, Lee AT,
Bracey JM and Churn SB: Modulation of CaM kinase II activity is
coincident with induction of status epilepticus in the rat
pilocarpine model. Epilepsia. 46:1389–1400. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Seki T, Matsubayashi H, Amano T, Kitada K,
Serikawa T, Sasa M and Sakai N: Adenoviral gene transfer of
aspartoacylase ameliorates tonic convulsions of spontaneously
epileptic rats. Neurochem Int. 45:171–178. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanaya R, Sasa M, Ujihara H, Fujita Y,
Amano T, Matsubayashi H, Serikawa T and Uozumi T: Effect of
antiepileptic drugs on absence-like seizures in the tremor rat.
Epilepsia. 36:938–942. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blair RE, Sombati S, Churn SB and
Delorenzo RJ: Epileptogenesis causes an N-methyl-d-aspartate
receptor/Ca2+-dependent decrease in Ca2+/calmodulin-dependent
protein kinase II activity in a hippocampal neuronal culture model
of spontaneous recurrent epileptiform discharges. Eur J Pharmacol.
588:64–71. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Serikawa T, Ohno Y, Sasa M, Yamada J and
Takaori S: A new model of petit mal epilepsy: Spontaneous spike and
wave discharges in tremor rats. Lab Anim. 21:68–71. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seki T, Matsubayashi H, Amano T, Kitada K,
Serikawa T, Sakai N and Sasa M: Adenoviral gene transfer of
aspartoacylase into the tremor rat, a genetic model of epilepsy, as
a trial of gene therapy for inherited epileptic disorder. Neurosci
Lett. 328:249–252. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sombati S and Delorenzo RJ: Recurrent
spontaneous seizure activity in hippocampal neuronal networks in
culture. J Neurophysiol. 73:1706–1711. 1995.PubMed/NCBI
|
|
32
|
Guo F, Xu X, Cai J, Hu H, Sun W, He G,
Shao D, Wang L, Chen T, Shaw C, et al: The up-regulation of
voltage-gated sodium channels subtypes coincides with an increased
sodium current in hippocampal neuronal culture model. Neurochem
Int. 62:287–295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li XM, Li JG, Yang JM, Hu P, Li XW, Wang
Y, Qin LN and Gao TM: An improved method for acute isolation of
neurons from the hippocampus of adult rats suitable for
patch-clamping study. Sheng Li Xue Bao. 56:112–117. 2004.PubMed/NCBI
|
|
34
|
Min D, Guo F, Zhu S, Xu X, Mao X, Cao Y,
Lv X, Gao Q, Wang L, Chen T, et al: The alterations of
Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of
Alzheimer's disease and vascular dementia. Neurosci Lett.
538:60–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuramoto T, Mori M, Yamada J and Serikawa
T: Tremor and zitter, causative mutant genes for epilepsy with
spongiform encephalopathy in spontaneously epileptic rat (SER), are
tightly linked to synaptobrevin-2 and prion protein genes,
respectively. Biochem Biophys Res Commun. 200:1161–1168. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cao HY, Jiang YW, Liu ZW and Wu XR: Effect
of recurrent epileptiform discharges induced by magnesium-free
treatment on developing cortical neurons in vitro. Brain Res Dev
Brain Res. 142:1–6. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Badea T, Goldberg J, Mao B and Yuste R:
Calcium imaging of epileptiform events with single-cell resolution.
J Neurobiol. 48:215–227. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Srinivas KV and Sikdar SK: Epileptiform
'activity induces distance-dependent alterations of the Ca2+
extrusion mechanism in the apical dendrites of subicular pyramidal
neurons. Eur J Neurosci. 28:2195–2212. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pal S, Sombati S, Limbrick DD Jr and
DeLorenzo RJ: In vitro status epilepticus causes sustained
elevation of intracellular calcium levels in hippocampal neurons.
Brain Res. 851:20–31. 1999. View Article : Google Scholar
|
|
40
|
Zhang J, Ding MP, Liu Z, Xiao B, Li GL and
Zhou FY: Dynamics of calcium in the hippocampal neuronal culture
model of epilepsy. Zhongguo Ying Yong Sheng Li Xue Za Zhi.
23:200–203. 2007.In Chinese. PubMed/NCBI
|
|
41
|
Amano T, Amano H, Matsubayashi H, Ishihara
K, Serikawa T and Sasa M: Enhanced Ca(2+) influx with mossy fiber
stimulation in hippocampal CA3 neurons of spontaneously epileptic
rats. Brain Res. 910:199–203. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Naderi N, Ahmad-Molaei L, Mazar-Atabaki A,
Ronaghi A, Shirazi-zand Z, Motiei-Langroudi SM and Eslahkar S:
L-type calcium channel mediates anticonvulsant effect of
cannabinoids in acute and chronic murine models of seizure.
Neurochem Res. 37:279–287. 2012. View Article : Google Scholar
|
|
43
|
Becker AJ, Pitsch J, Sochivko D, Opitz T,
Staniek M, Chen CC, Campbell KP, Schoch S, Yaari Y and Beck H:
Transcriptional upregulation of Cav3.2 mediates epileptogenesis in
the pilocarpine model of epilepsy. J Neurosci. 28:13341–13353.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yan HD, Ishihara K, Hanaya R, Kurisu K,
Serikawa T and Sasa M: Voltage-dependent calcium channel
abnormalities in hippocampal CA3 neurons of spontaneously epileptic
rats. Epilepsia. 48:758–764. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lie AA, Blümcke I, Volsen SG, Wiestler OD,
Elger CE and Beck H: Distribution of voltage-dependent calcium
channel beta subunits in the hippocampus of patients with temporal
lobe epilepsy. Neuroscience. 93:449–456. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Carter DS, Haider SN, Blair RE, Deshpande
LS, Sombati S and DeLorenzo RJ: Altered calcium/calmodulin kinase
II activity changes calcium homeostasis that underlies epileptiform
activity in hippocampal neurons in culture. J Pharmacol Exp Ther.
319:1021–1031. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang P, Wang WP, Sun-Zhang, Wang HX, Yan
Lou and Fan YH: Impaired spatial learning related with decreased
expression of calcium/calmodulin-dependent protein kinase IIalpha
and cAMP-response element binding protein in the
pentylenetetrazol-kindled rats. Brain Res. 1238:108–117. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zha XM, Dailey ME and Green SH: Role of
Ca2+/calmodulin-dependent protein kinase II in dendritic spine
remodeling during epileptiform activity in vitro. J Neurosci Res.
87:1969–1979. 2009. View Article : Google Scholar : PubMed/NCBI
|
















