Introduction
Osteosarcoma (OS) is considered to be one of the
most frequent types of primary malignant bone tumor in children and
adolescents (1). The underlying
mechanisms of the formation and development of OS have been
investigated for a long time. OS is a high-grade neoplasm with
rapid growth and early metastasis (2), and lung metastasis is the predominant
cause of OS-associated mortality. At present, high-dose
chemotherapy and surgery are often adopted for the treatment of OS,
however, this often results in resistance to chemotherapy, side
effects and drug resistance, which influence therapeutic efficacy
(3). Therefore, the priority is to
identify more reliable markers for predicting treatment outcomes,
therapeutic targets and effective therapeutic agents for
suppressing OS metastasis.
As with other types of cancer, OS is often
considered to be associated with the dysregulation of tumor
suppressor genes and oncogenes, for example, p53 (4,5) and
retinoblastoma tumor suppressor (Rb) (6). Notably, p21 signaling is considered
to be a pathway that regulates the progress of OS (7–9). The
CDNK1A gene, which encodes p21(Cip1/Waf1), is a
cyclin-dependent kinase inhibitor and is considered to be a major
transcriptional target of the p53 protein (10). Various studies have demonstrated
that p21 has an important role in the induction of normal and
transformed cell differentiation (11–13).
In tumor initiation, p21 suppresses the growth of malignant cells
in vitro and in vivo (11–13)
by inducing G1- and G2/M-phase cell cycle
arrest (14–17). Furthermore, there is evidence that
mice lacking p21 are predisposed to developing a wide spectrum of
tumor types, which implies that p21 acts as a tumor suppressor
(18). In a previous study, p21
was shown to act as an oncogene, although this was dependent on the
type of culture (13). In
addition, it has been identified that p21 depletion promotes
sarcoma formation in mice (19)
and numerous different clinical studies demonstrated in various
types of cancer that the downregulation of p21 was associated with
a poor prognosis (20–22). This indicates that p21 may act as a
gene target, which regulates the response of tumor cells to
chemotherapy and radiotherapy (23).
The mammalian polo-like kinase (Plk) family consists
of five members: Plk1, Plk2, Plk3, Plk4 and Plk5 (24). It is a family of conserved protein
serine/threonine kinases, which exhibit a polo-box domain (PBD) at
the carboxyl-terminus (25,26)
(except for human Plk5, the kinase domain of which lacks kinase
activity) (24–26). The founding member of the Plk
family is Drosophila polo, which has regulatory involvement
in mitosis and meiosis (25,27).
In addition, the biological functions of mammalian Plks are more
diverse than those in lower eukaryotes (25). The focus of the present study is on
Plk3, as recently various studies have indicated that Plk3 is a
multifunctional protein that is involved in numerous different
biological events, such as DNA damage responses, cell cycle control
and apoptosis (28,29). Plk3 shares high homology with
Drosophila polo kinases, and possesses a kinase domain at
the N-terminus (the function of which is phosphorylation of
downstream serine/threonine proteins) and a Polo-box domain at the
C-terminus (to bind interactive proteins). During cell cycle
progression, marked changes occur regarding Plk3 abundance, kinase
activity and subcellular distribution. A previous study
demonstrated that Plk3 ablation in aged mice enhanced tumor
angiogenesis (30). However, the
function of Plk3 in OS remains poorly understood. The aim of the
present study was to identify the role of Plk3 in osteosarcoma, to
confirm the mechanism for Plk3 in facilitating tumorigenesis
potential of osteosarcoma cells.
Materials and methods
Cell culture
The Saos-2, U2OS and HEK-293T human OS cell lines
were obtained from the American Type Culture Collection (Manassas,
VA, USA). The cells were cultured in Dulbecco's modified Eagle's
medium (GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (GE Healthcare Life Sciences), 100
units of penicillin/streptomycin (Invitrogen Life Technologies,
Carlsbad, CA, USA) and maintained at 37°C in air with a 5%
CO2 humidified atmosphere.
Patients and specimens
The 30 OS samples and their adjacent healthy tissues
were obtained from the surgical specimens of patients treated in
Yuhuangding Hospital of Yantai (Yantai, China). Written informed
consent was obtained from the patient. The samples were selected
from patients with complete clinicopathologic information, who had
been diagnosed and treated between June 2009 and November 2013. Of
the 30 cases, there were 17 males and 13 females aged between 32
and 70 years (median, 55.3 years). The study was approved by the
ethics committee of Qingdao University Medical College (Yantai,
China). All patients were followed up for survival. The survival
time was followed up from the diagnosis date up until
cancer-associated death every 6 months. Only 2 patients were
excluded from the study due to loss of contact, therefore these two
cases were excluded from the statistical analysis (n=28).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total cellular RNA was isolated from the Saos-2
cells using TRIzol reagent and amplified using an RT-qPCR kit
(Invitrogen Life Technologies). The first strand of cDNA was
synthesized using 1 µg total RNA with M-MLV Reverse
Transcriptase and the following primers were used for the detection
of Plk3: Forward, 5-TCTGTTTGCCAAAGTTACCA-3 and reverse,
5-TTCTTCAAATCCATCTCCACTG-3. β-actin served as the internal control
and was amplified using the following primers: Forward,
5-GTGGACATCCGCAAAGAC-3 and reverse, 5-AAAGGGTGTAACGCAACTA-3. qPCR
was conducted using an ABI PRISM® 7500 sequence
detection system (Applied Biosystems Life Technologies, Foster
City, CA, USA) and the relative expression levels of Plk3 in
non-transduced and transduced Saos-2 cells were determined. The
cycling conditions were as follows: 93°C for 2 mins, (93°C for 1
min, 55°C for 1 min, 72°C for 1 min, 40 cycles) 72°C for 10 min,
then held at 4°C. A Plk3 plasmid was generated by inserting a full
length of Plk3 into a pCMV-Tag2B vector (Invitrogen Life
Technologies).
Western blot analysis
Non-transduced and transduced Saos-2 cells were
lysed in 50 ml lysis buffer (100 mM Tris-HCl (pH 7.4), 10 mM EDTA,
4% SDS and 10% glycine) on ice for 45 min. The supernatants were
collected by centrifugation at 12,000 × g for 30 min at 4°C, and
performance of a bicin-choninic acid method determined the protein
concentration (BCA kit; GE Healthcare Life Sciences). Equal
quantities (30 µg protein) of lysate were run on 10%
SDS-PAGE gels (Invitrogen Life Technologies). Following
electrophoresis, the protein blots were transferred onto
nitrocellulose membranes (GE Healthcare Life Sciences) using
electroblotting apparatus (Bio-Rad Mini-PROTEAN Tetra
Electrophoresis system, Bio-Rad Laboratories, Inc., Hercules, CA,
USA) and were then blocked with 5% non-fat milk in Tris-buffered
saline and Tween-20 (TBST) solution (Sigma-Aldrich, St. Louis, MO,
USA). The membrane was incubated overnight with rabbit anti-Plk3
antibody (cat. no. SAB1300123), which was obtained from
Sigma-Aldrich (dilution, 1:1,000) and rabbit anti-P21 antibody
(cat. no. sc-397) obtained from Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA; dilution, 1:500);. After three washes with TBST
solution, the membrane was incubated with a secondary antibody
(Santa Cruz Biotechnology, Inc.; dilution, 1:5,000) at room
temperature for 60 min. Immunoreactive bands were visualized using
Western Blotting Luminol Reagent (Santa Cruz Biotechnology, Inc.)
according to the manufacturer's instructions. The β-actin protein
level served as a control (cat. no. sc-8432; mouse IgG1; Santa Cruz
Biotechnology, Inc.).
The recombinant lentivirus and lentivirus vectors
containing Plk3 short hairpin (sh)RNA#1
(5-CCGGGCCCTTGCCTTTGTGGCCTTCCTCGAGGAAGGCCACAAAGGCAAGGGCTTTTTTG-3)
or Plk3 shRNA#2
(5-CCGGGATTTGAAGAAGGTCTGACTGCTCGAGCAGTCAGACCTTCTTCAAATCTTTTTTG-3)
were constructed and inserted into the PCMV-t green fluorescent
protein (GFP) plasmid (Sigma-Aldrich). Non-silencing shRNA
(5-CTAGCCCGGTTCTCCGAACGTGTCACGTATCTCGAGATACGTGACACGTTCGGAGAATTTTTTTAAT-3)
served as a control, as it does not target any genes in humans.
Digestion analysis using a restriction endonuclease (Beijing
TransGen Biotech Co., Ltd., Beijing China) confirmed the formation
of the recombinant vector and all of the inserted sequences were
verified by DNA sequencing. Lentiviruses were developed by triple
transfection of 80% confluent 293T cells with Plk3 shRNA-expressing
vector or the control shRNA, using Lipofectamine 2000 (Invitrogen
Life Technologies). The cells were harvested in serum-free medium
after 3 days and filtered through a 0.45-µm filter (Merck
Millipore, Bedford, MA, USA).
Transduction was conducted by seeding Saos-2 cells
(5×104 cells/well) in 6-well plates along with shRNA
encoded recombinant lentiviruses against Plk3 (Lv-shPlk3) at a
multiplicity of infection of 60 in serum-free growth medium. When
serum-containing growth medium was added to the cells after 4 h,
there had been complete replacement of the growth medium with
recombinant lentivirus. After 3 days of transfection, expression of
the reporter gene, GFP was examined using fluorescence microscopy
(Olympus BX53; Olympus Corporation).
Immunoprecipitation (IP)
For IP assays, cells were washed with cold
phosphate-buffered saline (PBS) and lysed with cold lysis buffer at
4°C for 45 min. Whole cell lysates were incubated with Plk3 or P21
antibodies or normal rabbit/mouse immunoglobulin G on a rotator
(Liuyi Instrument Factory, Beijing, China), overnight at 4°C,
followed by the addition of protein A/G Sepharose CL-4B beads (GE
Healthcare Life Sciences) for 2 h at 4°C. The beads were then
washed five times with lysis buffer [50 mM Tris-Cl (pH 7.4), 150 mM
NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate and protease
inhibitor mixture]. The immune complexes were subjected to
SDS-PAGE, followed by immunoblotting (IB) with secondary
antibodies.
Flow cytometry
Saos-2 cells stably transfected with shPlk3 or
control shRNA were synchronized by serum starvation for 24 h, cells
(density, >90%) were then trypsinized (Sigma-Aldrich),
collected, washed with PBS and fixed in 70% ethanol at 4°C
overnight. After washing with PBS, cells were incubated with RNase
A (Sigma-Aldrich) in PBS for 30 min at 37°C and stained with 50
mg/ml propidium iodide (Sigma-Aldrich). Cell cycle data were
collected using a FACSCalibur™ flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA) and analyzed with FlowJo 7.6.2 (FlowJo,
LLC, Ashland, OR, USA), which was repeated three times.
Colony formation assay
Following infection with the lentivirus for 3 days,
a total of 1,000 Saos-2 cells were seeded into 6-well plates, and
the medium was changed at 3-day intervals. G418 was used to select
the cells transfected with the recombinant lentivirus and
lentivirus vectors. After 10 days of culturing, the colonies formed
were washed with PBS and fixed in 4% paraformaldehyde at 37°C for
30 min, after which the colonies were stained with Coomassie
(Sigma-Aldrich) for 15 min, washed and air-dried. The colonies were
counted under a microscope (Olympus BX46; Olympus Corporation).
This experiment was performed in triplicate.
EdU assay
Saos-2 cells were seeded in 6-well plates for 24 h
and transfected with the indicated shRNAs using Lipofectamine 2000.
After 48 h of transfection, cells were incubated with EdU
(Guangzhou Ribobio, Co., Ltd., Guangzhou, China) for 4 h, fixed
with 4% formaldehyde for 30 min and incubated with 2 mg/ml glycine
for 5 min. The assay was performed according to the manufacturer's
instructions, using a fluorescence method. Furthermore, the
positive and negative controls for the EdU assay were applied
according to the manufacturer's instructions. Finally,
6-diamidino-2-phenylindole (Sigma-Aldrich) was used to stain the
nuclei. The Olympus BX53 was used to observe the fluorescence.
Statistical analysis
SPSS version 17.0 was used for statistical analysis
(SPSS, Inc., Chicago, IL, USA). Comparisons between cancer tissue
and adjacent normal tissue were performed using a paired samples
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Abnormal expression of Plk3 in the OS
cell lines and tissues
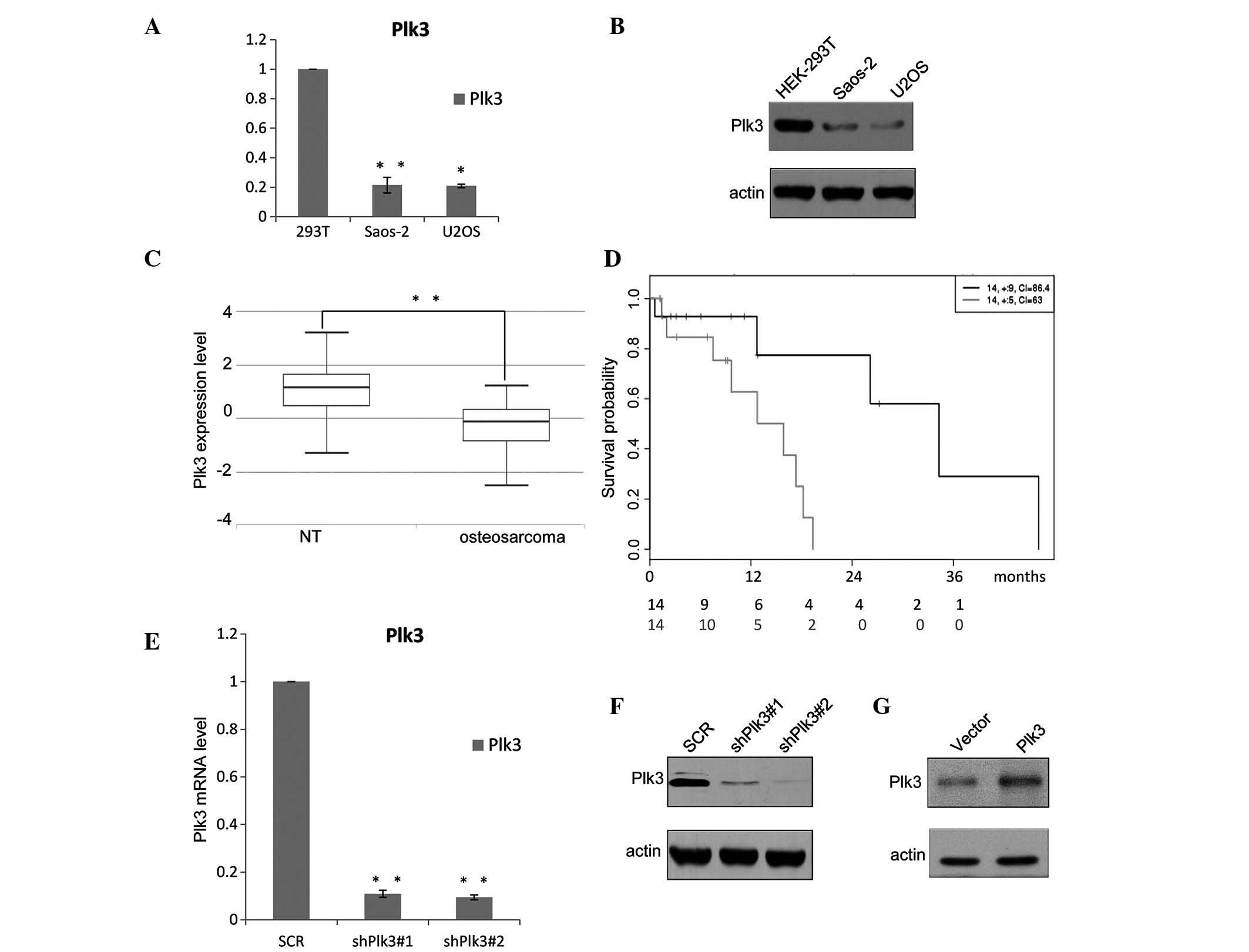
The expression level of Plk3 was analyzed in
different OS cell lines. As shown in Fig. 1A, downregulation of Plk3 was
observed in the two examined OS cell lines, Saos-2 and U2OS, when
compared with the HEK293 cells; the protein levels were also
detected to be deceased (Fig. 1B).
In addition, the expression levels of Plk3 were examined in 30 OS
samples collected at the Yuhuangding Hospital of Yantai (Yantai,
China) and the paired non-cancerous tissue (NT) samples were
analyzed using RT-qPCR. The results demonstrate that the mean
expression levels of Plk3 in the 30 OS samples (median, 1.4250;
max., 3.1859; min., −1.3429) are significantly lower than those in
the pairs of adjacent non-cancerous tissue (median, −0.1489; max.,
1.2074; min., −2.5398; P=0; Fig.
1C). The expression level of Plk3 decreased in the OS samples,
indicating that downregulation of Plk3 may be significant in OS.
The OS samples obtained from the patients were divided into two
groups, high and low, based on the median Plk3 expression level.
Kaplan-Meier survival analyses of the different groups (Fig. 1D) demonstrated that higher Plk3
expression was associated with a longer overall survival time for
patients with OS (hazard ratio=6.09; P=0.0239).
shRNA inhibits the expression of Plk3 in
OS cells
The efficiency of the recombinant lentivirus was
measured by RT-qPCR and western blotting. The mRNA expression level
of shRNA-Plk3#1 was ~10% that of the control group and shRNA-Plk3#2
was ~8% of the control (P<0.001; Fig. 1E). The transfection ratio was
observed to be ~100%. Western blot analysis demonstrated the
protein levels (Fig. 1F) and
indicated that the inhibitory ratios of shRNA-Plk3#1 and
shRNA-Plk3#2 were 12.5 and 8.5%, respectively. The efficiency of
the Plk3 overexpression was also detected by western blotting
(Fig. 1G).
Plk3 modulates the human OS cell
cycle
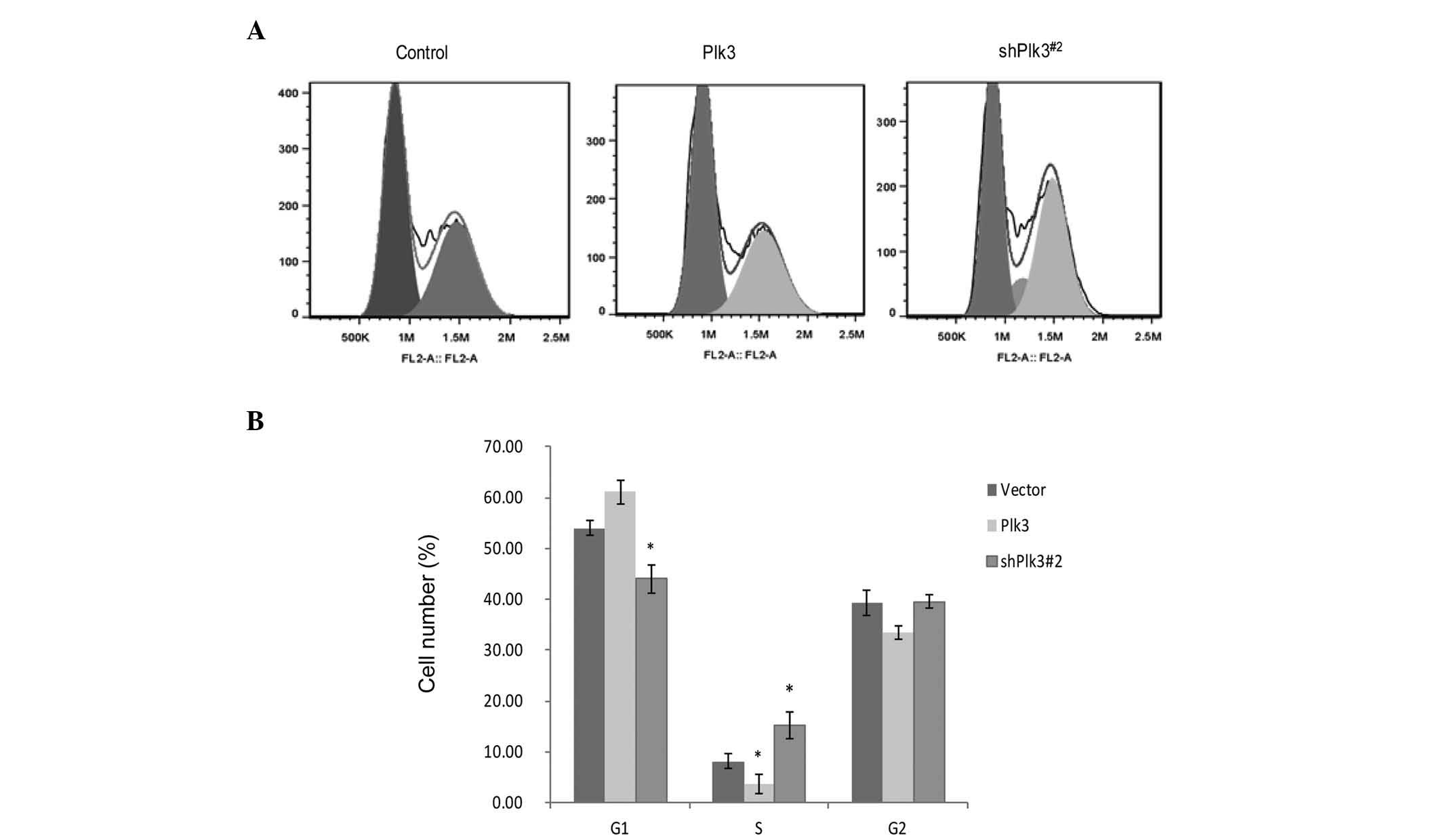
To investigate the function of Plk3 in cell
proliferation, the cell cycle distribution following transfection
of shRNA-Plk3#2, or the recombinant plasmid with Plk3
overexpression, was examined in Saos-2 cells by flow cytometry.
Compared with mock-transfected cells, shPlk3-transfected cells
revealed a substantial increase in S-phase populations and a
decrease in G1 phase populations, with Plk3
overexpression causing a block at the G1 to S phase
(Fig. 2A and B). These data
indicate that inhibition of OS cell growth by Plk3 may be mediated
via induction of G1 arrest.
Plk3 modulates the proliferation and
tumorigenicity of human OS cells in vitro
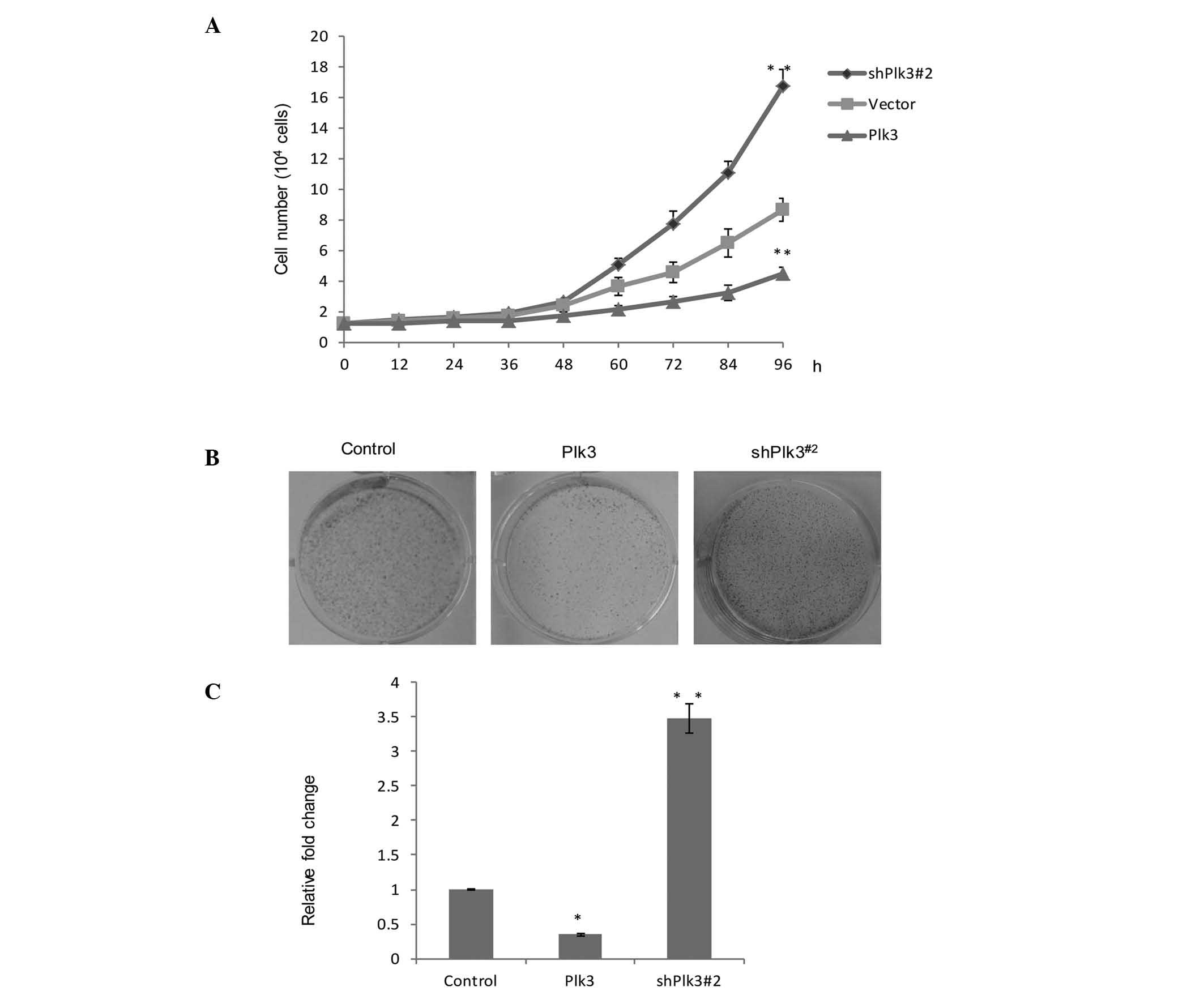
In order to further understand the role of Plk3 in
tumorigenicity, growth assays were performed in different cell
vectors, Plk3 overexpression and Plk3 knock down. Saos-2 cells
exhibiting Plk3 overexpression demonstrated marked growth
inhibition, while the Plk3 knockdown vector (shPlk3#2) revealed
evident growth promotion, when compared with the untreated group
(Fig. 3A). In addition, a colony
formation assay was used (Fig.
3B). The average number of colonies in the control group was
standardized to 1. The data are presented as the fold change over
vector for a better comparison. The number of colonies in the Plk3
overexpression group was found to be 35.3% compared with the
control group. The shPlk3#2 group was 3.48 fold greater than the
control group. (P<0.05). The data indicated there was a
significant increase in colony formation as a result of the low
expression of Plk3 (Fig. 3C). An
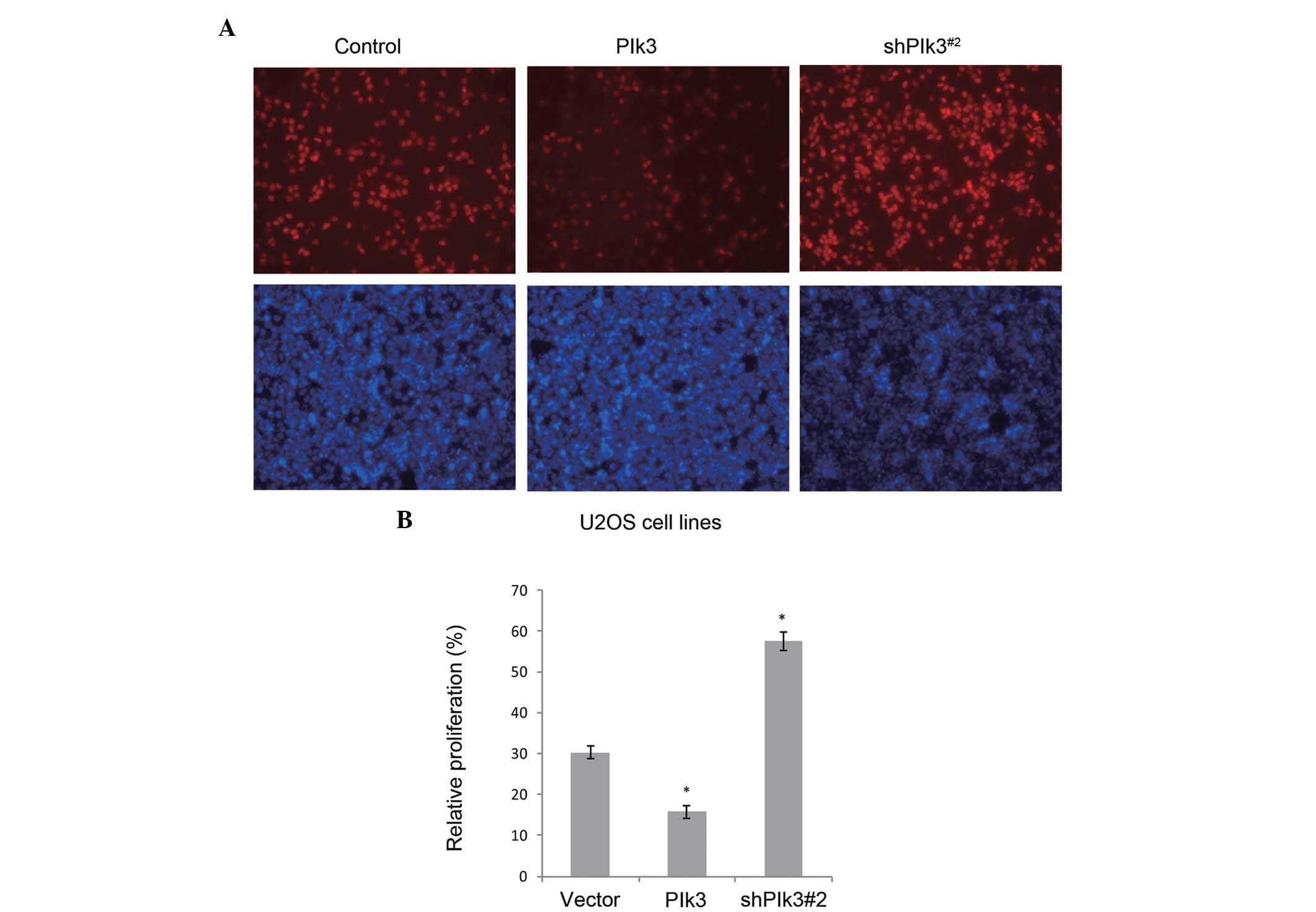
EdU assay using U2OS cell lines further demonstrated the function
of Plk3 in proliferation, in vitro. When Plk3 was over
expressed by the transfected recombinant plasmid, or knocked down
by the shRNA, a similar tendency was detected (Fig. 4).
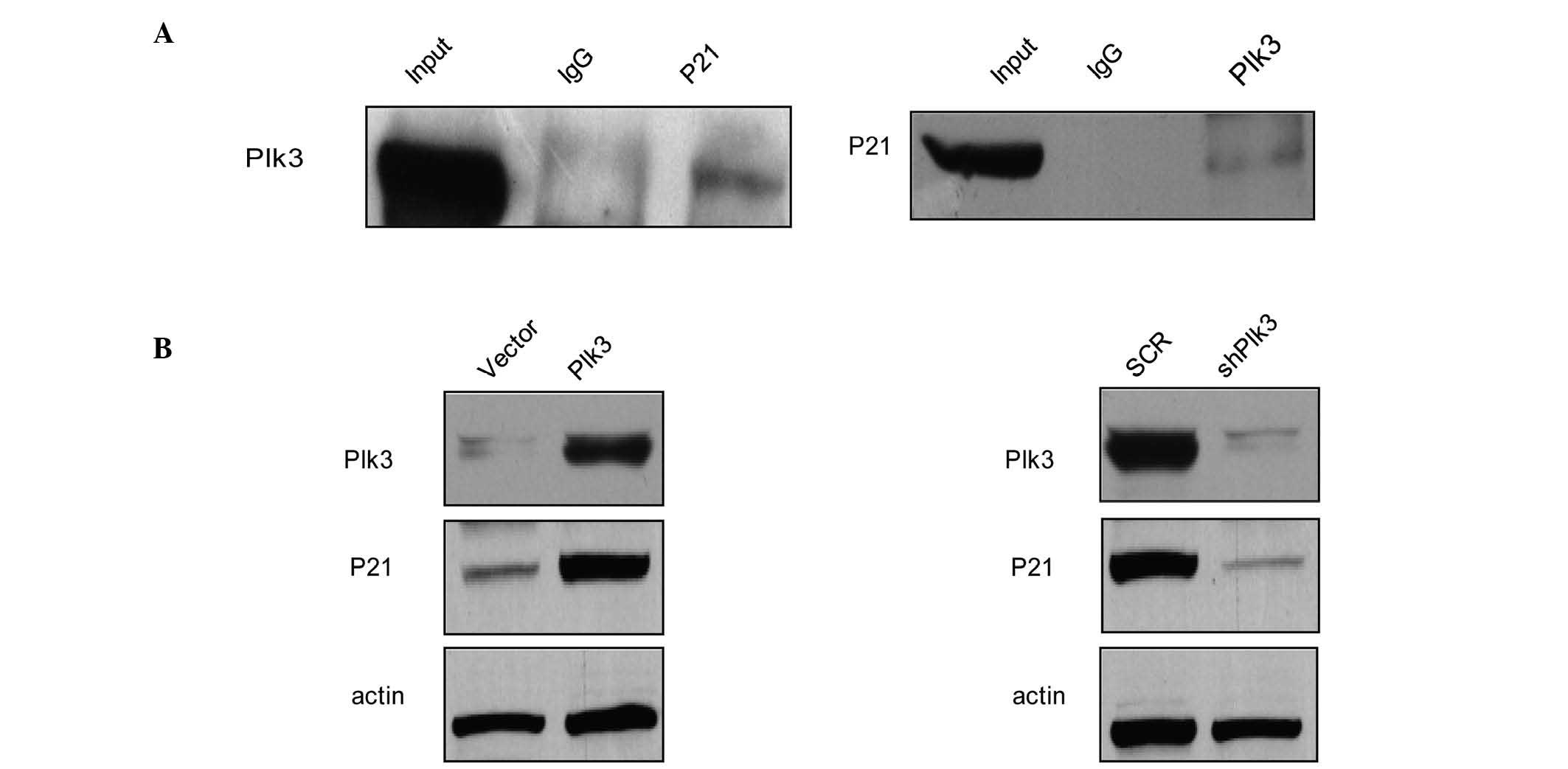
P21 is physically associated with Plk3
and the protein level varies with Plk3
A previous study demonstrated that Ect2 promotes
cell proliferation by regulating p21, a cell cycle-related gene,
which is required for Rb protein-mediated G1 arrest
(31). To further investigate the
in vivo interaction between Plk3 and p21, total proteins
from Saos-2 cells were extracted and co-IP experiments were
performed. IP was performed with antibodies against p21 followed by
IB with antibodies against Plk3 (Fig.
5A, left panel). Simultaneously, IP was performed with
antibodies against Plk3 followed by IB with antibodies against p21
(Fig. 5A, right panel). The co-IP
assay indicated that P21 was physically associated with Plk3 in
vivo. To consider the role of p21 in OS, the effects of Plk3 on
the expression levels of p21 were examined. The results revealed
that protein levels of p21 increased in Saos-2 cells that had been
transfected with Plk3 when compared with the control cells
(Fig. 5B, left panel). When Plk3
was knocked down, the protein levels of p21 decreased (Fig. 5B, right panel). These results
indicate that Plk3 interacts with p21 and the p21 protein level
changes along with the Plk3 level, indicating that Plk3 may cause
post-translational modification regulation of the P21 protein level
and p21 activity may lead to cell cycle arrest.
Discussion
OS often originates in the metaphysis of long bones
and has a high tendency for distant metastasis (32–34).
Patients with metastasizing OS are often associated with a
poor-prognosis signature, with <20% mean 5-year survival
(35,36). As a result, more detailed knowledge
on the key mechanisms of the early stages of OS, as well as more
reliable molecular markers for early diagnosis are urgently
required. It is important to develop a more effective treatment
strategy for OS patients with novel therapeutic agents that can
target the tumor cells at an early stage (37).
In a previous study, data demonstrated that
activated Plk3 leads to enhanced apoptosis in targeted cells; this
effect was observed in cells that overexpress Plk3 (25). Additional evidence exists
indicating that Plk3 ablation in mice does not noticeably perturb
normal animal development (30).
Therefore, targeting Plk3 may result in fewer side effects in
vivo.
In the present study, Plk3 was observed to be
downregulated in OS tissue and cell lines, with higher Plk3
expression indicating improved overall survival. During the
progress of OS, when Plk3 was knocked down with the appropriate
shRNA, OS cell proliferation was enhanced; a similar phenomenon was
also identified by observation of colony formation (using a soft
agar assay) and by performance of an EdU assay. By contrast,
overexpression of Plk3 reduced OS cell proliferation.
P21 is a cyclin-dependent kinase inhibitor, and a
major transcriptional target of the p53 protein (10). As a tumor suppressor, it was
proved, using mice models, that mice without the p21(Cip1/Waf1)
protein are more sensitive to tumorigenesis (18). The absence of p21 protein enables
the proliferation of cells with damaged DNA and promotes tumor
progression (13). Notably, p21 is
phosphorylated by various kinases, and its phosphorylation of
serine or threonine residues (including Thr57, Thr145, Ser146 and
Ser130) is considered to be particularly important. The exact roles
of particular phosphorylation sites continue to be investigated.
However, a previous study found that certain phosphorylation sites,
such as at Thr57 (by mammalian sterile 20-like 1), at Thr145 and
Ser146 (by AKT), or Ser130 (by p38 and c-Jun N-terminal kinase),
increase the stability of the p21 protein (38). Additional studies have demonstrated
that phosphorylation of p21 by AKT kinase leads to stabilization,
as well as to distribution of p21 into the cytoplasm (39,40).
In conclusion, the present results indicate that the
dysregulation of Plk3 in OS affects cell proliferation in
vitro. Furthermore, the function of Plk3 may depend on the
interaction with p21, as the protein level of p21 varies with Plk3
expression. This may be due to the fact that p21 has numerous
phosphorylation sites and Plk3 may phosphorylate the downstream
proteins at the serine/threonine residues. However, the underlying
mechanism requires further investigation.
References
|
1
|
Mirabello L, Troisi RJ and Savage SA:
Osteosarcoma incidence and survival rates from 1973 to 2004: Data
from the surveillance, epidemiology and end results program.
Cancer. 115:1531–1543. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marina N, Gebhardt M, Teot L and Gorlick
R: Biology and therapeutic advances for pediatric osteosarcoma.
Oncologist. 9:422–441. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takeshita H, Gebhardt MC, Springfield DS,
Kusuzaki K and Mankin HJ: Experimental models for the study of drug
resistance in osteosarcoma: P-glycoprotein-positive, murine
osteosarcoma cell lines. J Bone Joint Surg Am. 78:366–375.
1996.PubMed/NCBI
|
|
4
|
Lin PP, Pandey MK, Jin F, Raymond AK,
Akiyama H and Lozano G: Targeted mutation of p53 and Rb in
mesenchymal cells of the limb bud produces sarcomas in mice.
Carcinogenesis. 30:1789–1795. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rubio R, Gutierrez-Aranda I, Sáez-Castillo
AI, Labarga A, Rosu-Myles M, Gonzalez-Garcia S, Toribio Ml,
Menendez P and Rodriguez R: The differentiation stage of
p53-Rb-deficient bone marrow mesenchymal stem cells imposes the
phenotype of in vivo sarcoma development. Oncogene. 32:4970–4980.
2013. View Article : Google Scholar
|
|
6
|
Sosa-Garcia B, Gunduz V, Vázquez-Rivera V,
Cress WD, Wright G, Bian H, Hinds PW and Santiago-Cardona PG: A
role for the retinoblastoma protein as a regulator of mouse
osteoblast cell adhesion: Implications for osteogenesis and
osteosarcoma formation. PLoS One. 5:e139542010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shen H and Maki CG: p53 and p21(Waf1) are
recruited to distinct PMl-containing nuclear foci in irradiated and
Nutlin-3a-treated U2OS cells. J Cell Biochem. 111:1280–1290. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lossaint G, Besnard E, Fisher D, Piette J
and Dulic V: Chk1 is dispensable for G2 arrest in response to
sustained DNA damage when the ATM/p53/p21 pathway is functional.
Oncogene. 30:4261–4274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu J, Yao Q, Hou Y, Xu M, Liu S, Yang L,
Zhang L and Xu H: MiR-223/Ect2/p21 signaling regulates osteosarcoma
cell cycle progression and proliferation. Biomed Pharmacother.
67:381–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cmielová J and Rezáčová M: p21Cip1/Waf1
protein and its function based on a subcellular localization
[corrected]. J Cell Biochem. 112:3502–3506. 2011. View Article : Google Scholar
|
|
11
|
Matsumoto T, Sowa Y, Ohtani-Fujita N,
Tamaki T, Takenaka T, Kuribayashi K and Sakai T: p53-independent
induction of WAF1/Cip1 is correlated with osteoblastic
differentiation by vitamin D3. Cancer Lett. 129:61–68. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gartel AL, Serfas MS and Tyner AL:
p21-negative regulator of the cell cycle. Proc Soc Exp Biol Med.
213:138–149. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harper JW, Adami GR, Wei N, Keyomarsi K
and Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent
inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dulić V, Kaufmann WK, Wilson SJ, Tlsty TD,
Lees E, Harper JW, Elledge SJ and Reed SI: p53-dependent inhibition
of cyclin-dependent kinase activities in human fibroblasts during
radiation-induced G1 arrest. Cell. 76:1013–1023. 1994. View Article : Google Scholar
|
|
16
|
Ando T, Kawabe T, Ohara H, Ducommun B,
Itoh M and Okamoto T: Involvement of the interaction between p21
and proliferating cell nuclear antigen for the maintenance of G2/M
rrest after DNA damage. J Biol Chem. 276:42971–42977. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dash BC and El-Deiry WS: Phosphorylation
of p21 in G2/M promotes cyclin B-Cdc2 kinase activity. Mol Cell
Biol. 25:3364–3387. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martin-Caballero J, Flores JM,
Garcia-Palencia P and Serrano M: Tumor susceptibility of
p21(Waf1/Cip1)-deficient mice. Cancer Res. 61:6234–6238.
2001.PubMed/NCBI
|
|
19
|
Young NP, Crowley D and Jacks T:
Uncoupling cancer mutations reveals critical timing of p53 loss in
sarcomagenesis. Cancer Res. 71:4040–4047. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Yang WT and Zheng PS: Msi1 promotes
tumor growth and cell proliferation by targeting cell cycle
checkpoint proteins p21, p27 and p53 in cervical carcinomas.
Oncotarget. 5:10870–10885. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo DH, Zhou Q, Hu SK, et al: Differential
expression of Notch1 intracellular domain and p21 proteins, and
their clinical significance in gastric cancer. Oncol Lett.
7:471–478. 2014.PubMed/NCBI
|
|
22
|
Zhang X, Liu J, Zang D, et al:
Upregulation of miR-572 transcriptionally suppresses SOCS1 and p21
and contributes to human ovarian cancer progression. Oncotarget.
6:15180–15193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Sturgis EM, Zafereo ME, Wei Q and
Li G: p14ARF genetic polymorphisms and susceptibility to second
primary malignancy in patients with index squamous cell carcinoma
of the head and neck. Cancer. 117:1227–1235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Andrysik Z, Bernstein WZ, Deng L, Myer DL,
Li YQ, Tischfield JA, Stambrook PJ and Bahassi el M: The novel
mouse Polo-like kinase 5 responds to DNA damage and localizes in
the nucleolus. Nucleic Acids Res. 38:2931–2943. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dai W: Polo-like kinases, an introduction.
Oncogene. 24:214–216. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lowery DM, Lim D and Yaffe MB: Structure
and function of Polo-like kinases. Oncogene. 24:248–259. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fenton B and Glover DM: A conserved
mitotic kinase active at late anaphase-telophase in syncytial
Drosophila embryos. Nature. 363:637–640. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Conn CW, Hennigan RF, Dai W, Sanchez Y and
Stambrook PJ: Incomplete cytokinesis and induction of apoptosis by
overexpression of the mammalian polo-like kinase, Plk3. Cancer Res.
60:6826–6831. 2000.
|
|
29
|
Wang Q, Xie S, Chen J, Fukasawa K, Naik U,
Traganos F, Darzynkiewicz Z, Jhanwar-Uniyal M and Dai W: Cell cycle
arrest and apoptosis induced by human Polo-like kinase 3 is
mediated through perturbation of microtubule integrity. Mol Cell
Biol. 22:3450–3459. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang Y, Bai J, Shen R, Brown SA,
Komissarova E, Huang Y, Jiang N, Alberts GF, Costa M, Lu L, et al:
Polo-like kinase 3 functions as a tumor suppressor and is a
negative regulator of hypoxia-inducible factor-1 alpha under
hypoxic conditions. Cancer Res. 68:4077–4085. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Iyoda M, Kasamatsu A, Ishigami T,
Nakashima D, Endo-Sakamoto Y, Ogawara K, Shiiba M, Tanzawa H and
Uzawa K: Epithelial cell transforming sequence 2 in human oral
cancer. PLoS One. 5:e140822010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Poletajew S, Fus L and Wasiutyński A:
Current concepts on pathogenesis and biology of metastatic
osteosarcoma tumors. Ortop Traumatol Rehabil. 13:537–545. 2011.In
English and Polish. View Article : Google Scholar
|
|
33
|
Endo-Munoz L, Evdokiou A and Saunders NA:
The role of osteoclasts and tumour-associated macrophages in
osteosarcoma metastasis. Biochim Biophys Acta. 1826:434–442.
2012.PubMed/NCBI
|
|
34
|
Rainusso N, Wang LL and Yustein JT: The
adolescent and young adult with cancer: State of the art-bone
tumors. Curr Oncol Rep. 15:296–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gorlick R and Meyers PA: Osteosarcoma
necrosis following chemotherapy: Innate biology versus
treatment-specific. J Pediatr Hematol Oncol. 25:840–841. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hawkins DS and Arndt CA: Pattern of
disease recurrence and prognostic factors in patients with
osteosarcoma treated with contemporary chemotherapy. Cancer.
98:2447–2456. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klein MJ and Siegal GP: Osteosarcoma:
Anatomic and histologic variants. Am J Clin Pathol. 125:555–581.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hwang CY, Lee C and Kwon KS: Extracellular
signal-regulated kinase 2-dependent phosphorylation induces
cytoplasmic localization and degradation of p21Cip1. Mol Cell Biol.
29:3379–3389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Blagosklonny MV: Are p27 and p21
cytoplasmic oncoproteins? Cell Cycle. 1:391–393. 2002. View Article : Google Scholar
|
|
40
|
Sohn D, Essmann F, Schulze-Osthoff K and
Jänicke RU: p21 blocks irradiation-induced apoptosis downstream of
mitochondria by inhibition of cyclin-dependent kinase-mediated
caspase-9 activation. Cancer Res. 66:11254–11262. 2006. View Article : Google Scholar : PubMed/NCBI
|