Introduction
Fibroblast growth factor 3 (FGF3), belonging to the
family of fibroblast growth factors (FGFs), regulates several
developmental processes, including brain patterning, branching
morphogenesis and limb development, by binding, dimerizing and
activating cell surface FGF receptors (FGFRs) (1). The extra-cellular ligand-binding
portion of FGFRs is composed of three immunoglobulin-like (Ig-like)
domains (D1–D3). The crystal structure of the ectodomain of the
FGFR complex with FGF demonstrated that the ligand-binding domain
of FGFR involves Ig-like domains II and III (D2 and D3,
respectively), as well as the linker between D2 and D3 (2). It has been reported that FGF3/FGFRs
are associated with multiple biological activities, including
cellular proliferation, differentiation, invasiveness and motility,
thus demonstrating the potential to initiate and promote
tumorigenesis (3).
Breast cancer is one of the most common types of
malignancy in females and the second most common cause of
cancer-associated mortality in females worldwide (4,5). The
literature devoted to prognostic factors for breast cancer is
extensive (6–8). Histology, tumor stage and lymph-node
status are now supplemented with measurements of ploidy, steroid
hormone receptors, S-phase fractions, growth factors, oncogenes and
oncogene products (9). Even though
significant progress has been achieved in developing early
diagnosis and treatment, acquired resistance to current
chemotherapies and failure of endocrine-targeted therapy in certain
patients have resulted in a great clinical requirement for new
therapeutic agents for breast cancer. The expression and associated
amplification of FGF3 and FGFRs have been identified in breast
cancer (10), and they correlate
with advanced-stage and high-grade tumors, as well as decreased
patient survival time (11).
Therefore, it is likely that the development of antagonists
targeting FGF3 and its receptors is a strategy that may assist in
overcoming resistance to current chemotherapy and
endocrine-targeted therapy. In the present study, a high affinity
FGF3-binding peptide was isolated from a phage display library and
the functions of the isolated peptide were further investigated to
evaluate its possible therapeutic potential in breast cancer.
Materials and methods
Cell lines and reagents
The cell lines MDA-MB-231, T47D and Cos-7 were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). They were all maintained at 37°C in
a humidified atmosphere of 5% CO2 and cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
(v/v) fetal bovine serum (FBS; Guangzhou Ruite Bio-tec Co., Ltd.,
Guangzhou, China). The Ph.D.-7™ Phage Display Peptide Library kit
and Escherichia coli ER2738 were purchased from New-England
Biolabs (Ipswich, MA, USA). Recombinant human FGF3 was obtained
from Uscn Life Science, Inc. (Export Processing Zone, Economic and
Technological Development Zone, Wuhan, China). The mouse
horseradish peroxidase (HRP) anti-M13 monoclonal antibody (cat. no.
27-9421-01; 1:5,000) was a product of GE Healthcare (Piscataway,
NJ, USA). Cell Signaling Technology Inc., (Danvers, MA, USA)
provided the monoclonal rabbit anti-phospho Erk1/2 (cat. no. 4370s;
1:2,000), monoclonal rabbit anti-Erk1/2 (cat. no. 9194s; 1:2,000),
monoclonal rabbit anti-phospho Akt (cat. no. 13038s; 1:1,000),
monoclonal rabbit anti-Akt (cat. no. 4685s; 1:1,000), monoclonal
rabbit anti-cyclin D1 (cat. no. 2978s; 1:1,000), monoclonal rabbit
anti-proliferating cell nuclear antigen (PCNA; cat. no. 13110s;
1:1,000) and the monoclonal rabbit anti-GAPDH antibodies (cat. no.
3907s; 1:1,000). The monoclonal rabbit anti-proliferation
associated protein 2G4 (PA2G4; cat. no. sc-292466; 1:1,000) was
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
goat-anti-rabbit secondary (cat. no. sc-2054; 1:1,000), and
goat-anti-mouse secondary (cat. no. sc-2005; 1:1,000) antibodies
were obtained from Santa Cruz Biotechnology, Inc.
In vitro phage-display biopanning
The 96-well microliter plates were coated with 10
µg/ml FGF3 in sodium carbonate buffer (0.1 M
NaHCO3, pH 8.6) at 4°C overnight, followed by blocking
with bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 2
h at 37°C and washing six times with 0.05% Tween-20 in TBS (0.05%
TBST). Subsequently, 10 µl of diluted original library
2×1011 plaque-forming units (pfus) with 100 µl
TBST was added to the coated well. Following incubation for 2 h at
room temperature, the plates were washed with 0.05% TBST. The bound
phages were eluted with 100 µl of 0.1 M glycine-HCl buffer
(pH 2.2) and were then neutralized by 15 µl of 1 M Tris-HCl
(pH 9.1). The eluted phages were amplified by propagation in E.
coli ER2738, purified and concentrated with polyethylene
glycol/NaCl, and titrated as described in the standard protocol
(NEB). Three additional rounds of selection were performed under
more stringent conditions. Briefly, the concentration and
incubation time of FGF3 was gradually reduced (5 µg for 1.5
h, 2.5 µg for 1 h, 1.25 µg for 0.5 h in the 2nd, 3rd
and 4th round, respectively), and the concentration of Tween-20 was
gradually increased (0.1, 0.3, 0.5% for the 2nd, 3rd and 4th round,
respectively).
Identification of positive phage clones
by enzyme-linked immunosorbent assay (ELISA)
The 96-well plates were coated at 4°C overnight with
FGF3 and blocked with 300 µl blocking buffer (5% BSA) for 2
h at 4°C. The wells coated without FGF3 with blocking buffer were
used as the negative control. Following washing with 0.05% TBST six
times, phage clones (1010 pfu/well) were added and
incubated for 1 h with gentle agitation at room temperature. The
plates were washed with TBST, then incubated with 200 µl of
HRP-anti-M13 (1:5,000) for 1 h and washed again. The plates were
developed with the substrate (50 µl/well of
3,3′,5,5′-tetramethylbenzidine), terminated by 50 µl/well of
2 M H2SO4 and the absorbance was measured at
450 nm using a microplate reader (3550; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
DNA sequencing and peptide synthesis
The DNA sequencing was performed by Invitrogen Life
Technologies (Shanghai, China) and sequences were analyzed using
DNAssist software (version 2.2; Softonic, Barcelona, Spain).
Peptide FP16 (VLWLKNR, translated from the selected F16 phage clone
DNA sequence) and an irrelevant control ZP8 peptide (RPNPTLS,
obtained from another screening strategy) were synthesized by
Beijing SBS Genetech Co., Ltd. (Beijing, China).
Cell proliferation assessment
Effects of FP16 on cancer cell viability and
proliferation were assessed by a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
American Type Culture Collection, Rockville, MD, USA) assay. Cells
were seeded in 96-well plates (5×103 cells/well) in DMEM
with 10% FBS at 37°C for 24 h. Subsequently, the cells were starved
for 24 h and treated with 50 ng/ml FGF3 alone or FGF3 plus serially
diluted peptides for 48 h. Cells treated with DMEM with 0.4% FBS
alone were used as controls. A total of 20 µl of MTT was
added to each well and incubated for 4 h. The crystals were
dissolved in 150 µl dimethyl sulfoxide (DMSO) and the
absorbance was measured at 490 nm with the aforementioned
microplate reader.
Flow cytometric analysis of the cell
cycle
Cells were seeded in 6-well plates (3×105
cells/well) for 24 h, starved for another 24 h and treated with 50
ng/ml FGF3 alone or FGF3 plus serially diluted peptides for 48 h.
Cells were collected and fixed in 70% ice-cold ethanol for 30 min
at 4°C. Following washing with phosphate-buffered saline (PBS), the
cells were stained with propidium iodide (PI) in the dark at room
temperature for 30 min. The percentage of cells at various phases
of the cell cycle was analyzed using the FlowJo analysis program
(FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA).
Western blot analysis of
mitogen-activated protein kinase (MAPK), Akt activation and
expression of PA2G4, PCNA and cyclin D1
Cells were seeded in 6-well plates (4×106
cells/well) for 24 h, starved for another 24 h and treated with
serially diluted FP16 for 30 min prior to stimulation with FGF3 for
3 h. Cells were lysed in SDS-PAGE loading buffer following being
washed with cold PBS. The samples were separated by 10% SDS-PAGE
and transferred onto a polyvinylidene fluoride membrane (350 mA, 70
min; Sigma-Aldrich, Shanghai, China). The membrane was blocked with
5% non-fat dry milk in TBS buffer for 1 h and incubated with the
primary antibody (an anti-phospho-Erk1/2 rabbit mAb or an
anti-phospho-Akt rabbit mAb) overnight at 4°C, followed by a goat
anti-rabbit IgG, HRP-linked antibody (1:1,000 dilution) at room
temperature for 1 h. Western blot analysis of PA2G4, cyclin D1 and
PCNA expression was performed using the same method, with the
following exceptions: To detect PA2G4 and PCNA expression, the
starved cells were treated with 50 ng/ml FGF3 alone or 50 ng/ml
FGF3 plus 4 µM FP16 for 48 h, and to detect cyclin D1
expression, the starved cells were treated with serially diluted
FP16 for 30 min prior to stimulation with FGF3 for 6 h.
Results
In vitro screening
The Ph.D.-7™ Phage Display Peptide Library kit was
used to isolate high-affinity phages that could specifically bind
to FGF3. As shown in Table I, with
gradually increased stringency of selection, the recovery rate was
124-fold higher (between 4.67×10−2 and
3.75×10−4) following four rounds of screening compared
with after the first round, suggesting that the phages specifically
bound to FGF3 were successfully enriched.
 | Table IEnrichment of phages for each round
of selection from the phage display library. |
Table I
Enrichment of phages for each round
of selection from the phage display library.
| Round | FGF3
(µg) | Concentration of
Tween 20 (v/v) | Input phage
(pfu) | Output phage
(pfu) | Recovery
(output/input) |
|---|
| 1 | 10 | 0.05 |
2.00×1011 |
7.50×107 |
3.75×10−4 |
| 2 | 5 | 0.10 |
3.50×1011 |
2.80×108 |
8.00×10−4 |
| 3 | 2.5 | 0.30 |
1.60×1011 |
8.20×108 |
5.13×10−3 |
| 4 | 1.25 | 0.50 |
1.20×1011 |
5.60×109 |
4.67×10−2 |
Identification of the affinity of
selected phage clones by ELISA
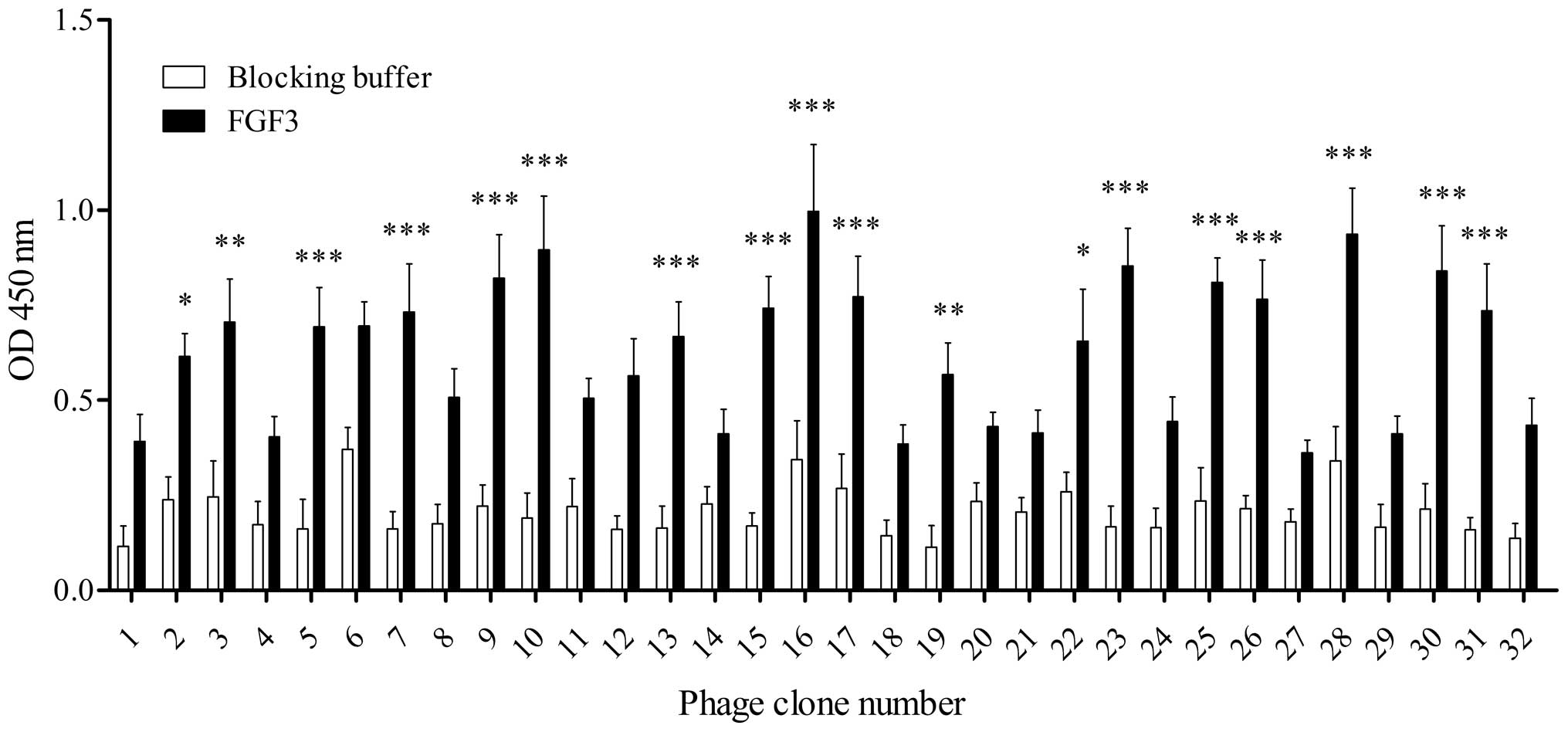
High-affinity FGF3-binding clones were further
identified from the recovered phage clones by ELISA. As shown in
Fig. 1, following four rounds of
selection, 32 individual phage clones were randomly selected and
individually amplified. Of the 32 phage clones, 14 clones exhibited
relatively high binding capabilities to FGF3 (clones 5, 7, 9, 10,
13, 15, 16, 17, 23, 25, 26, 28, 30 and 31) compared with the
control blocking buffer, and the 16th clone demonstrated the
highest, suggesting that it may have a greater affinity for FGF3
than the other clones. Subsequently, these 14 positive phage clones
were selected for sequencing.
Sequence analysis and property prediction
of positive phages
FGF3 binds to and executes its pleiotropic
biological actions in cells expressing FGFR1, FGFR2, FGFR3 or
FGFR4. The affinity of FGF3 for binding to FGFR2 is significantly
higher compared with binding to the other three isoforms (2). The crystal structure of the FGF3
complex with FGFR demonstrated that the ligand-binding domains of
FGFR involve the highly conserved Ig-like D2 and D3, and the linker
region between D2 and D3 (12).
Therefore, the amino acid sequences of the selected peptides were
compared with the motif (151–355 aa) located at D2–D3 of FGFR2. As
shown in Table II, phage clones
16 (FP16) and 7 (FP7) demonstrated the highest sequence similarity
to D2–D3 of FGFR2 (0.0147059, PAM250 Matrix). The FP16 peptide
(VLWLKNR) contains four (WLKN) amino acids that are identical to
the peptides of the 188–194 (TMRWLKN) of FGFR2 (GenBank ID
CAA96492.1). In the physiological state, FP16,
FGFR2188–194 and FGFR2151–355 all carry
positive charges. In addition, their grand averages of
hydropathicity (GRAVY) are all negative. Taken together, these data
suggest that the FP16 peptide, sharing four amino acids (WLKN)
identical to the ligand-binding motif in D2 of FGFR2, may bind FGF3
via electrostatic interactions and therefore may have the potential
to interrupt FGF3 binding to its receptor. Thus, peptide FP16 was
selected for further investigation.
| Table IIAmino acid sequences of specific
FGF3-binding peptides compared with FGFR2. |
Table II
Amino acid sequences of specific
FGF3-binding peptides compared with FGFR2.
| Clone | Peptide | Sequence
(N-C)a | Similarities to
FGFR2151–355 | Theoretical PI | GRAVY |
|---|
| F5 | FP5 | NITPWDT | 0.0049020 | 3.80 | −0.914 |
| F7/13 | FP7 | QPMLKIS | 0.0147059 | 8.75 | 0.057 |
|
F9/10/16/23/28/30 | FP16 | VLWLKNR | 0.0147059 | 11.00 | −0.143 |
| F15 | FP15 | ESKVGAP | 0.0098039 | 6.10 | −0.600 |
| F17/26 | FP17 | WLGHRVP | 0.0098039 | 9.76 | −0.371 |
| F25 | FP25 | KEHDPSR | 0.0098039 | 6.75 | −3.010 |
| F31 | FP31 | SQPAWLP | 0.0098039 | 5.24 | −0.400 |
|
FGFR2188–194 | TMRWLKN | | 11.00 | −1.114 |
|
FGFR2151–355 | | | 8.60 | −0.490 |
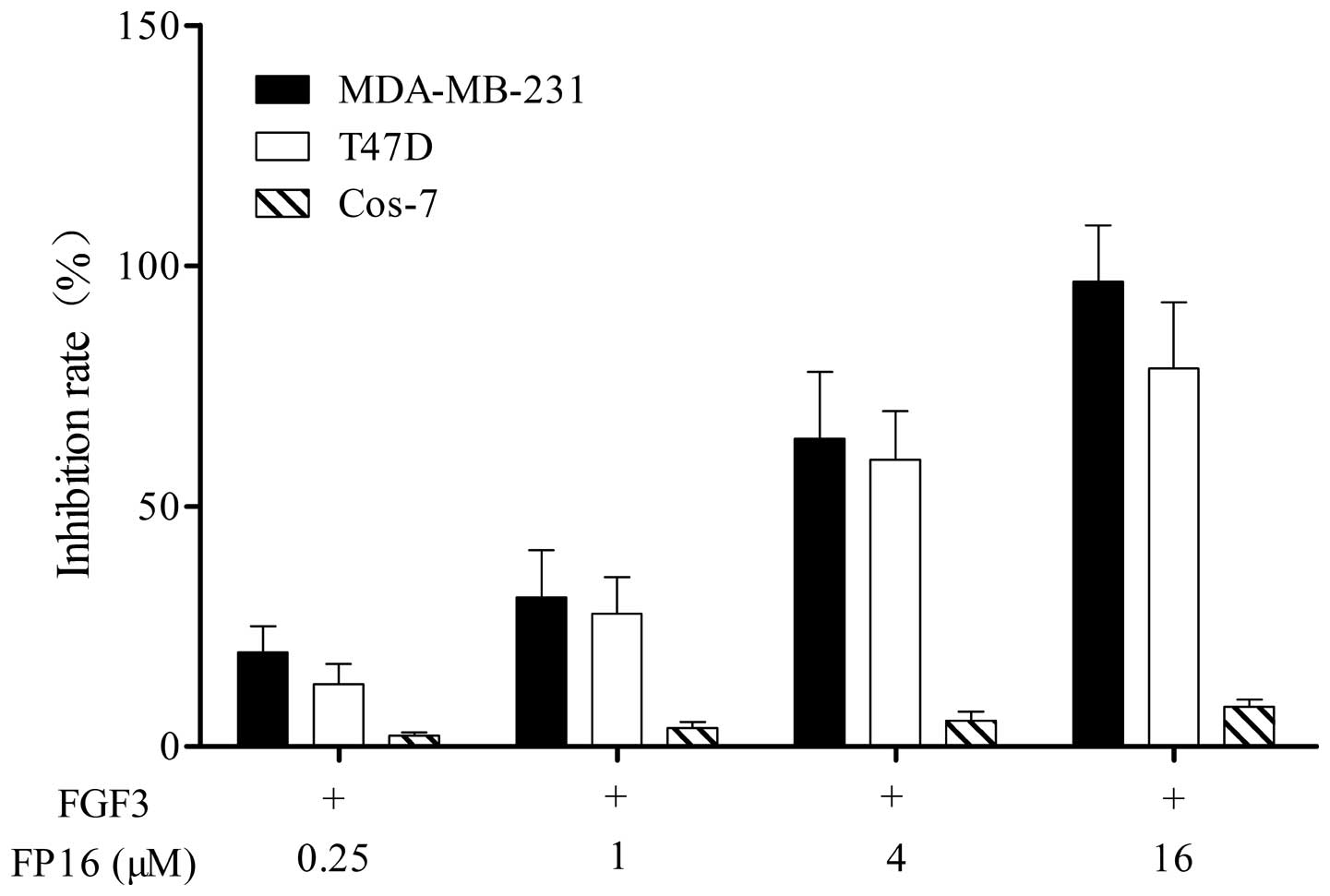
FP16 inhibits FGF3-stimulated cell
proliferation
Cell lines expressing a high level of FGFRs,
including MDA-MB-231 and T47D breast cancer cells, and Cos-7 cells,
which do not express FGFRs were used in the study (13,14).
The efficacy of the synthetic FP16 peptides in inhibiting
FGF3-stimulated tumor cell proliferation was determined by an MTT
assay. As shown in Fig. 2, the
FP16 peptides inhibited MDA-MB-231 and T47D cell proliferation in a
dose-dependent manner, while FP16 had little inhibitory effect on
Cos-7 cells that do not express FGF3 receptors.
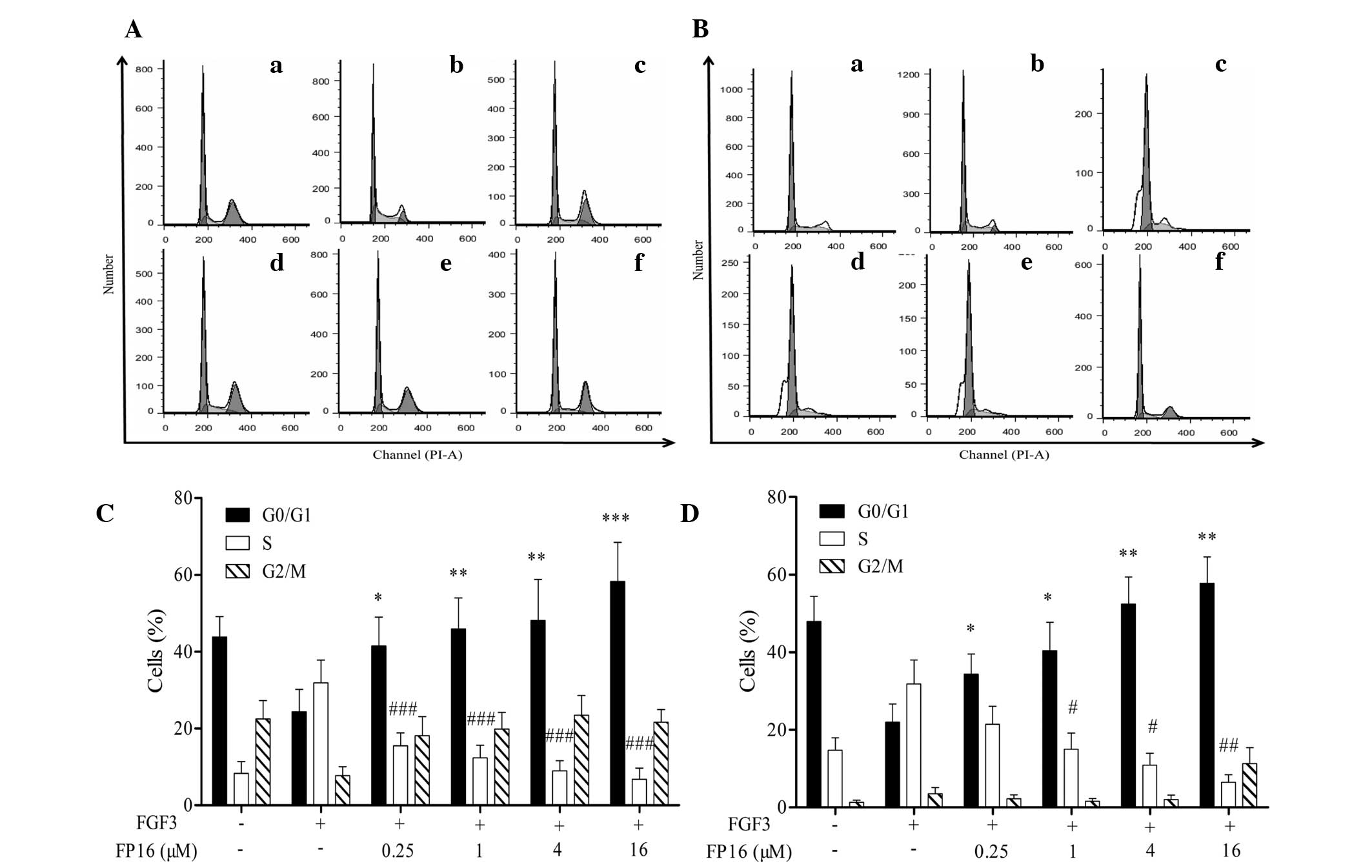
FP16 arrests FGF3-induced cells at the
G0/G1 phase via cyclin D1
PI staining combined with flow cytometric analysis
was performed to investigate the effect of FP16 on the cell-cycle
progression of MDA-MB-231 and T47D cells induced by FGF3. As shown
in Fig. 3, FGF3 increased the
percentage of S-phase cells and decreased the ratio of G0/G1 phase
cells compared with the control. By contrast, cells treated with
FGF3 plus FP16 presented a higher G0/G1-phase population and a
decreased S-phase population compared with those treated with FGF3
alone. These results suggest that FP16 specifically inhibits
FGF3-stimulated cell proliferation by arresting the cells at the
G0/G1 phase.
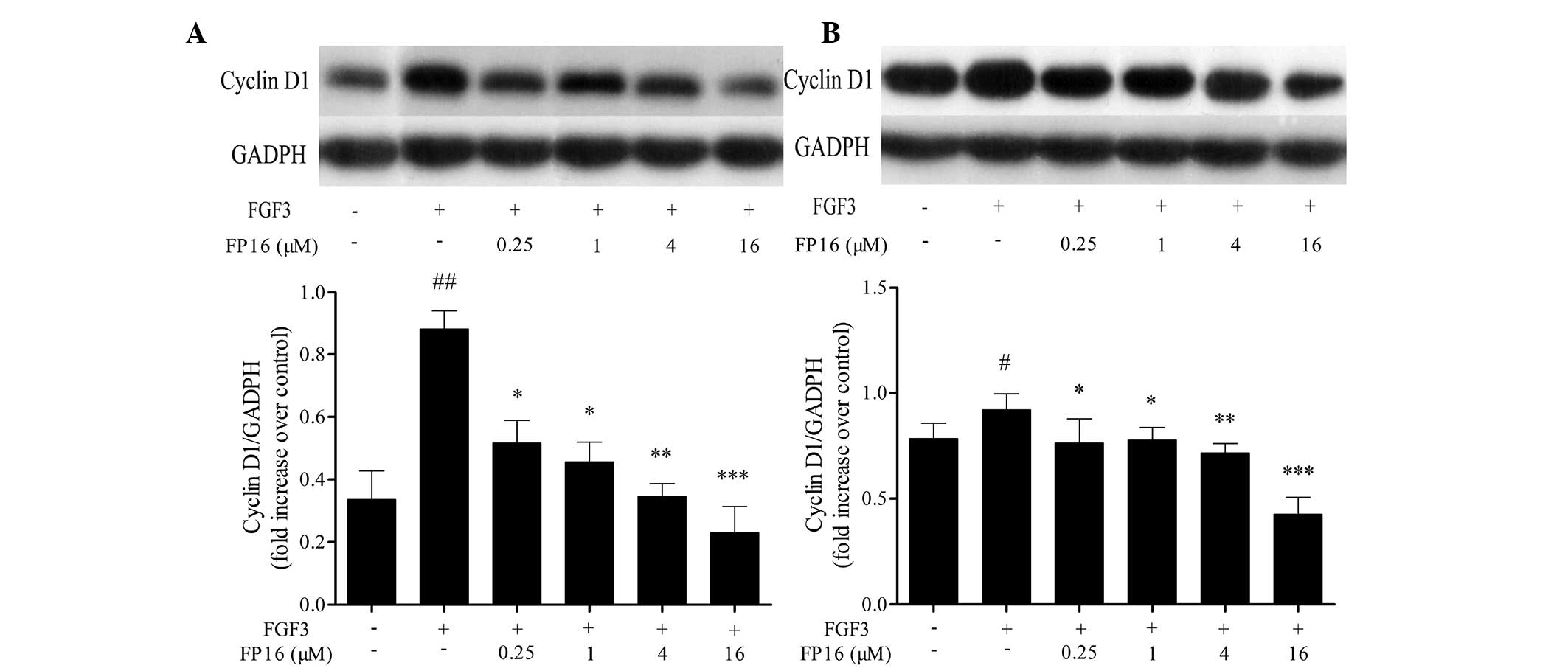
It has been reported that cyclin D1 is a
G1/S-specific regulating protein that controls cell cycle
progression. The active cyclin D1-CD4/6 complexes release E2F
transcription factors and induce specific gene expression required
for G1- to S-phase progression (15). The effect of FP16 on the expression
of cyclin D1 was determined by western blot analysis. The results
shown in Fig. 4 indicate that FGF3
significantly increased the expression of cyclin D1 in MDA-MB-231
and T47D cells, whereas pretreatment with P12 peptides prior to
stimulation with FGF3 led to a significant decrease in the
expression of cyclin D1, suggesting that the mechanisms by which
FP16 peptides arrest cells at the G0/G1 phase may partially be
through downregulation of the expression of the G1/S-specific
protein cyclin D1.
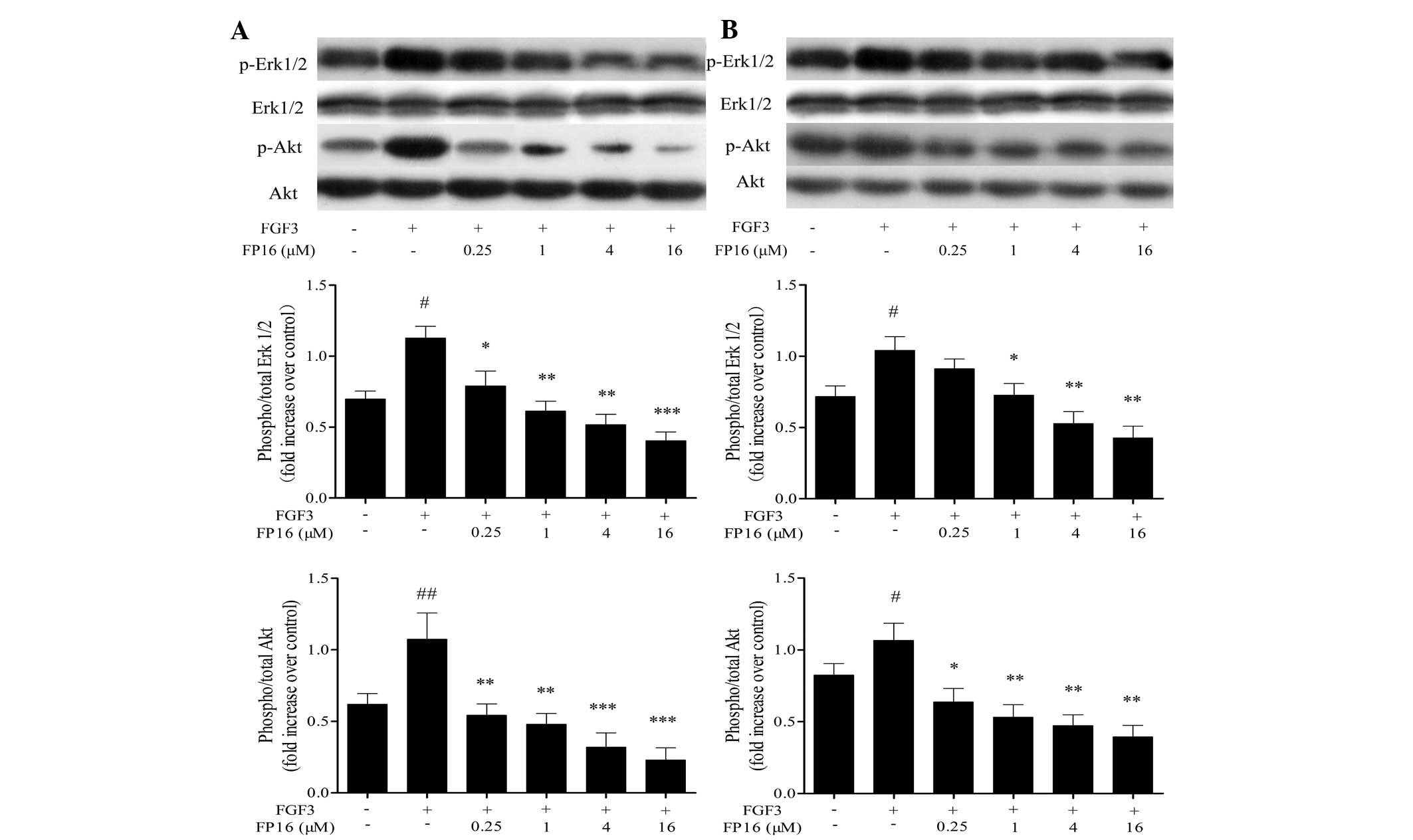
Synthetic FP16 peptides inhibit
FGF3-induced phosphorylation of Akt and MAP kinases
The tyrosine kinase-activated Ras/MEK/Erk pathway
and the Ras/PI3K pathway have a pivotal role in cell proliferation
by regulating cyclin D1 expression in the middle of the G1 phase,
and by driving cells past the G1-restriction point (16). In order to examine the potential of
FP16 in cancer treatment, the present study further investigated
the effects of FP16 on MAPK and Akt signal transduction in
MDA-MB-231 and T47D cells. As shown in Fig. 5, exogenous FGF3 significantly
stimulated the phosphorylation of Erk1/2 and Akt, while
pretreatment of the cells with various concentrations of FP16
(0.25–16 µM) for 30 min prior to stimulation with FGF3
resulted in significant inhibition of the activation of these
signaling molecules in a dose-dependent manner. Taken together,
these observations suggest that FP16 could irreversibly suppress
FGF3-induced activation of MAPK and Akt.
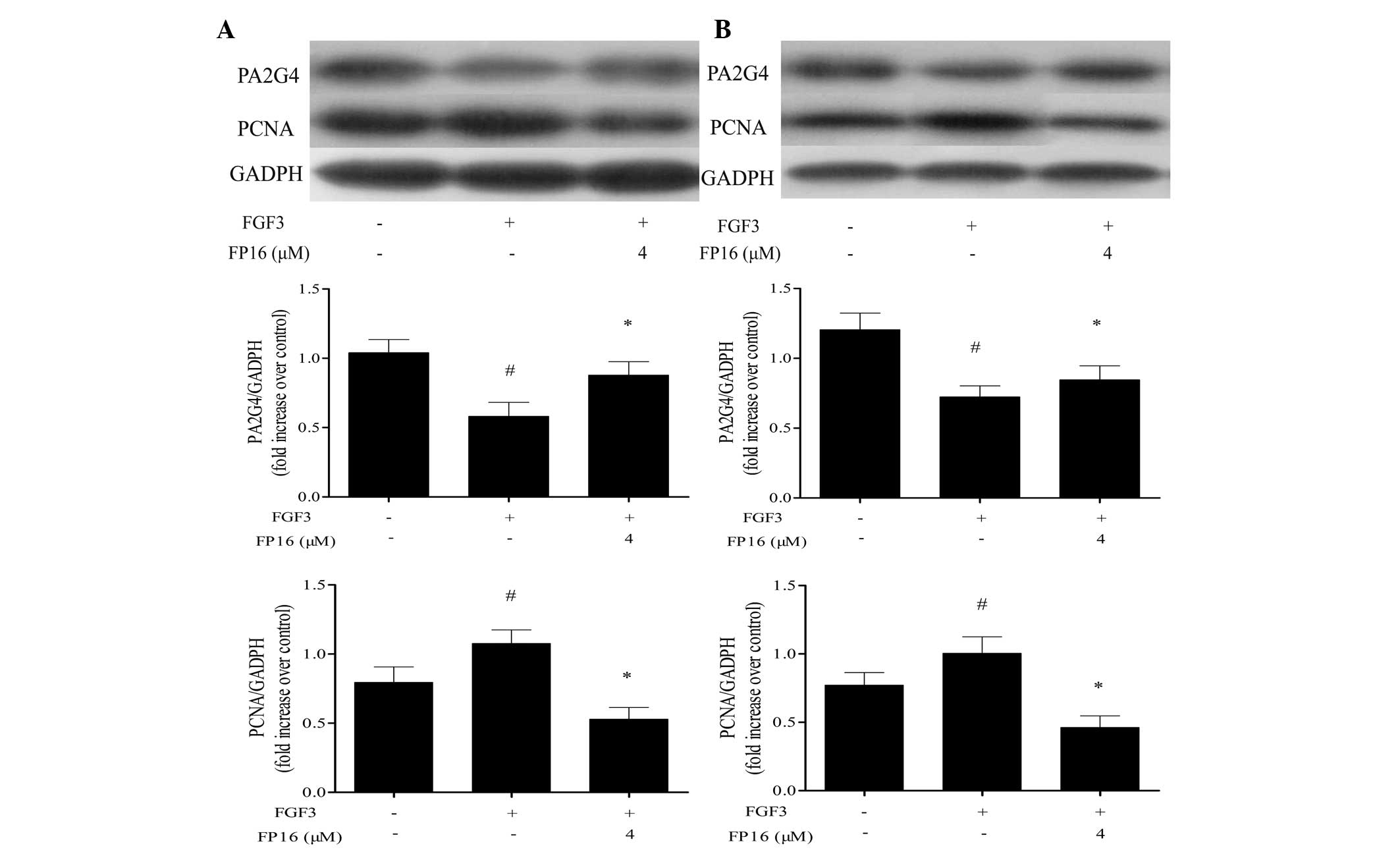
FP16 counteracts the regulatory effect of
FGF3 on PA2G4 and PCNA expression
It has been reported that PA2G4 is a cellular
proliferation-inhibited DNA-binding protein, which can inhibit the
proliferation and induce the differentiation of human breast cancer
cells (17). PCNA is known as a
DNA polymerase accessory protein involved in DNA replication, DNA
repair and cell cycle control, and is considered to be a marker of
cell proliferation in various types of cancer (18). In order to determine whether FP16
inhibited FGF3-stimulated cell proliferation by affecting PA2G4 and
PCNA signal transduction, western blot analysis was performed. As
shown in Fig. 6, PA2G4 expression
was downregulated by FGF3 stimulation and enhanced by FP16
treatment, whereas FP16 treatment downregulated the expression of
PCNA induced by FGF3, suggesting that PA2G4 and PCNA have an
important role in how P12 peptides counteract FGF3-stimulated
proliferation in MDA-MB-231 and T47D cells.
Discussion
Drug-resistant metastatic tumors and side effects
are major limitations of conventional cancer therapy, including
irradiation and chemotherapy (19), necessitating the search for novel
tumor-targeting agents. Phage display technology provides an
efficient tool to identify the desirable sequences binding
specifically to targets, and has been widely used in diagnostic and
therapeutic protein developments in previous years (20,21).
These small molecules, which are characterized from phage display
libraries, exhibit great efficiency in penetrating into the
targeted sites with low immunogenicity (22) and high concentration and validity
(23). FGF/FGFR complexes have
been demonstrated to act as driving oncogenes in certain types of
cancer to maintain the malignant properties of tumor cells in a
cell autonomous manner (24).
Accumulating studies have demonstrated that FGF3 is upregulated in
breast cancer and accelerates tumor growth and angiogenesis
(10,25–27).
Therefore, FGF3 has been considered as a potential target for
breast cancer therapy.
In the present study, a phage-displayed heptapeptide
library was used for identifying FGF3-binding peptides antagonists
and the conditions of the selection were strictly limited to
enrichment specific FGF3-binding phages. Following four rounds of
panning, FP16 demonstrated a significantly positive signal and
exhibited the strongest binding according to ELISA. In addition,
FP16 peptide has four amino acids identical to the ligand-binding
motif in D2 of FGFR2. Thus, it is reasonable to hypothesize that
FP16 may have the capability to bind FGF3 and inhibit the
biological activity of FGF3 by interrupting its interactions with
FGFR2.
Cell proliferation is regulated during the G0/G1
phase in the cell cycle. Cyclin-dependent kinase (CDK)4 and CDK6
interacting with the cyclin D family of proteins drive G0/G1 phase
into the DNA synthesizing S-phase. The active cyclin D1-CD4/6
complexes release E2F transcription factors and motivate the
specific gene expression required for G1 to S phase progression
(28). The results of an MTT assay
demonstrated that FP16 was effective in inhibiting MDA-MB-231 and
T47D cell proliferation in a dose-dependent manner, while FP16
partially suppressed the growth of Cos-7 cells that do not express
FGFRs. Furthermore, the results revealed that FP16 arrested
FGF3-stimulated cells at the G0/G1 phase and downregulated the
expression of cyclin D1 and PCNA induced by FGF3. These data
indicate that FP16 may inhibit FGF3-stimulated cell proliferation
by restricting FGF3-induced G1 to S phase progression and
downregulating the expression of G1/S-specific proteins cyclin D1
and PCNA.
The tyrosine kinase-activated Ras/MEK/Erk pathway
and Ras/PI3K pathway are important in cell proliferation by
regulating cyclin D1 expression during mid-G1 and driving cells
past the G1-restriction point (29). It has been reported that PA2G4
inhibits the proliferation and induces the differentiation of human
breast cancer cells (17). Western
blot analysis demonstrated that AP8 suppressed FGF3-induced Erk1/2
and Akt phosphorylation in a dose-dependent manner, while
increasing PA2G4 expression in MDA-MB-231 and T47D cells. It is
reasonable to hypothesize that FP16 may have counteracted
FGF3-stimulated proliferation and the cell cycle by inhibiting
activation of MAPK and Akt, increasing PA2G4 expression and
suppressing the expression of cyclin D1 and PCNA.
In conclusion, an FGF3-binding peptide FP16 was
successfully screened from a phage display heptapeptide library. It
was found that FP16 can counteract FGF3-stimulated proliferation
and the cell cycle by inhibiting activation of MAPK and Akt,
increasing PA2G4 expression and suppressing the expression of
cyclin D1 and PCNA. Thus, FP16 may have potential application for
the treatment of various types of cancer, including breast cancer,
characterized by the upregu-lation of FGF3/FGFRs.
Acknowledgments
This study was supported by the City and academy
cooperation projects of Foshan (grant no. 2012HY100611); the major
National Science and Technology project: Major infectious diseases
such as AIDS and Viral Hepatitis Prevention and treatment in the
area of Baoan district; Shenzhen and Panyu district of Guangzhou
(project no. 2012ZX10004903); and the research on the
Nanotechnology-based Diagnostics of interleukin-22 (project no.
2014ZX10003002-003). This study was edited and modified by the
English Language Service.
References
|
1
|
Beenken A and Mohammadi M: The FGF family:
Biology, pathophysiology and therapy. Nat Rev Drug Discov.
8:235–253. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mohammadi M, Olsen SK and Ibrahimi OA:
Structural basis for fibroblast growth factor receptor activation.
Cytokine Growth Factor Rev. 16:107–137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rusnati M and Presta M: Fibroblast growth
factors/fibroblast growth factor receptors as targets for the
development of anti-angiogenesis strategies. Curr Pharm Des.
13:2025–2044. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saxena R and Dwivedi A: ErbB family
receptor inhibitors as therapeutic agents in breast cancer: Current
status and future clinical perspective. Med Res Rev. 32:166–215.
2012. View Article : Google Scholar
|
|
5
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamamoto M, Hosoda M, Nakano K, et al: p53
accumulation is a strong predictor of recurrence in estrogen
receptor-positive breast cancer patients treated with aromatase
inhibitors. Cancer Sci. 105:81–88. 2014. View Article : Google Scholar
|
|
7
|
Choi SY, Chang YW, Park HJ, Kim HJ, Hong
SS and Seo DY: Correlation of the apparent diffusion coefficiency
values on diffusion-weighted imaging with prognostic factors for
breast cancer. Brit J Radiol. 85:E474–E479. 2012. View Article : Google Scholar
|
|
8
|
van de Vijver MJ: Molecular tests as
prognostic factors in breast cancer. Virchows Arch. 464:283–291.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Donegan WL: Tumor-related prognostic
factors for breast cancer. CA Cancer J Clin. 47:28–51. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dickson C, Spencer-Dene B, Dillon C and
Fantl V: Tyrosine kinase signalling in breast cancer: Fibroblast
growth factors and their receptors. Breast Cancer Res. 2:191–196.
2000. View Article : Google Scholar
|
|
11
|
Grose R and Dickson C: Fibroblast growth
factor signaling in tumorigenesis. Cytokine Growth Factor Rev.
16:179–186. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ibrahimi OA, Zhang F, Eliseenkova AV, Itoh
N, Linhardt RJ and Mohammadi M: Biochemical analysis of pathogenic
ligand-dependent FGFR2 mutations suggests distinct
pathophysiological mechanisms for craniofacial and limb
abnormalities. Hum Mol Genet. 13:2313–2324. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Penault-Llorca F, Bertucci F, Adélaïde J,
Parc P, Coulier F, Jacquemier J, Birnbaum D and deLapeyrière O:
Expression of FGF and FGF receptor genes in human breast cancer.
Int J Cancer. 61:170–176. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu XP, Huang HX, Wang C, Lin S, Huang Y,
Wang Y, Liang G, Yan Q, Xiao J, Wu J, et al: Identification of a
novel peptide that blocks basic fibroblast growth factor-mediated
cell proliferation. Oncotarget. 4:1819–1828. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fu M, Wang C, Li Z, Sakamaki T and Pestell
RG: Minireview: Cyclin D1: Normal and abnormal functions.
Endocrinology. 145:5439–5447. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pagès G, Lenormand P, L'Allemain G,
Chambard JC, Meloche S and Pouysségur J: Mitogen-activated protein
kinases p42mapk and p44mapk are required for fibroblast
proliferation. Proc Natl Acad Sci USA. 90:8319–8323. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia X, Cheng A, Lessor T, Zhang Y and
Hamburger AW: Ebp1, an ErbB-3 binding protein, interacts with Rb
and affects Rb transcriptional regulation. J Cell Physiol.
187:209–217. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maga G and Hubscher U: Proliferating cell
nuclear antigen (PCNA): A dancer with many partners. J Cell Sci.
116:3051–3060. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang JT and Liu Y: Use of comparative
proteomics to identify potential resistance mechanisms in cancer
treatment. Cancer Treat Rev. 33:741–756. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kandel E: Tumor-associated oncogenes go on
(phage) display. Oncotarget. 1:84–85. 2010. View Article : Google Scholar
|
|
21
|
Ionov Y: A high throughput method for
identifying personalized tumor-associated antigens. Oncotarget.
1:148–155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bae DG, Kim TD, Li G, Yoon WH and Chae CB:
Anti-Flt1 peptide, a vascular endothelial growth factor receptor
1-specific hexapeptide, inhibits tumor growth and metastasis.
Clinical Cancer Res. 11:2651–2661. 2005. View Article : Google Scholar
|
|
23
|
Yardley DA, Hart L, Bosserman L, Salleh
MN, Waterhouse DM, Hagan MK, Richards P, DeSilvio ML, Mahoney JM
and Nagarwala Y: Phase II study evaluating lapatinib in combination
with nab-paclitaxel in HER2-overexpressing metastatic breast cancer
patients who have received no more than one prior chemotherapeutic
regimen. Breast Cancer Res Treat. 137:457–464. 2013. View Article : Google Scholar :
|
|
24
|
Knights V and Cook SJ: De-regulated FGF
receptors as therapeutic targets in cancer. Pharmacol Ther.
125:105–117. 2010. View Article : Google Scholar
|
|
25
|
Li Y, Hively WP and Varmus HE: Use of
MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast
cancer. Oncogene. 19:1002–1009. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Turnbull C, Ahmed S, Morrison J, et al:
Genome-wide association study identifies five new breast cancer
susceptibility loci. Nat Genet. 42:504–547. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roy D and Calaf GM: Allelic loss at
chromosome 11q13 alters FGF3 gene expression in a human breast
cancer progression model. Oncol Rep. 32:2445–2452. 2014.PubMed/NCBI
|
|
28
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Welsh CF, Roovers K, Villanueva J, Liu YQ,
Schwartz MA and Assoian RK: Timing of cyclin D1 expression within
G1 phase is controlled by Rho. Nat Cell Biol. 3:950–957. 2001.
View Article : Google Scholar : PubMed/NCBI
|