Introduction
Gastric cancer (GC) is one of the leading causes of
cancer-associated mortality worldwide, and was the third leading
cause of cancer-associated mortality in China in 2012 (1,2).
However, the etiology of GC is complicated and remains to be fully
elucidated. Numerous receptors and downstream pathways are known to
be aberrantly activated in GC. Furthermore, several molecular
alterations involving various pathways have been linked to the
development and late-stage progression/metastasis of GC, and these
may present novel targets for therapeutic strategies. The most
important challenge for researchers in this field is to identify
how to exploit the knowledge embedded in these pathways concerning
the interactions among the various genes (3). Exploring pathway networks provides a
suitable means to investigate how the genes interact with and
regulate each other; however, the existing analytical approaches
consider only the sets of genes associated with these pathways and
do not take into consideration their topology, and consequently,
their positions in those pathways.
At present, systems biology offers an effective
approach to identify molecular mechanisms and connections between
genes, and their pathways of dynamic networks (4). Systems biology approaches, which
dissect the molecular mechanisms and pathways that regulate the
progression of GC, are still in their infancy. In the present
study, microarray data, pathway enrichment and network topological
analyses were used to identify major pathways associated with the
development of GC, and to construct pathway networks and functional
modules, which are based on the key pathways. The characterization
of genes and pathways associated with GC may prove to be useful for
identifying potential targets for the development of novel
strategies for the treatment of gastric carcinoma.
Materials and methods
Microarray data
The gene expression data set, GSE29272, was
downloaded from Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/). For GSE29272
analysis, two groups of gene expression profiles, gastric cardia
adenocarcinomas (GCAs; n=62) and matched normal tissues from
patients, and gastric non-cardia adenocarcinomas (GNCAs; n=72) and
matched normal tissues from patients, were included, based on the
GPL96 Affymetrix human genome U133A array (HG-U133A; Affymetrix,
Santa Clara CA, USA). The original CEL files and the platform probe
annotation information file were used for the bioinformatics
analysis.
Identification of differentially
expressed genes (DEGs)
The raw data were converted into the identifiable
format using the package affy of R 2.8.2, and missing values were
subsequently inserted (5). The
robust MultiArray average method (6) was applied to perform background
correction and data normalization, using defaulted parameters in
the affy package (7).
Subsequently, a differential analysis between GCA (or GNCA) and the
control was performed using the limma package (8), a modified version of the standard
t-test incorporating the Benjamini-Hochberg (BH) multiple
hypotheses correction technique (9). DEGs were defined as the false
discovery rate (FDR) q value <0.05, and adjusted P<0.01 and
fold change ≥2 were set as the cut-off parameters to screen out any
significant increases or decreases in gene expression levels.
Biological function annotation and
enrichment analysis
In order to identify biological functions, which
were disrupted in GC, a gene ontology (GO) functional enrichment
analysis was performed for the DEGs using BiNGO (version 3.0.2)
(10) in the open source
bioinformatics software platform, Cytoscape 2.8.0 (http://www.cytoscape.org/), with a threshold of
P<0.001. BiNGO is a tool to determine which GO categories are
statistically over-represented in a set of genes or a subgraph of a
biological network.
Pathway analysis
JEPETTO plug-in (version 1.3.1) in Cytoscape
(11) was used to analyze a set of
DEGs associated with GC. JEPETTO offers two types of analysis: The
enrichment analysis identifies pathways closely associated with a
query gene set in the context of an interaction network, whereas
topology analysis identifies pathways which share a similar set of
topological features. EnrichNet (12) and PathExpand (13) web servers were used for the
enrichment analysis. The relevant pathways from Kyoto Encyclopedia
of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) were identified using the
XD-score method, which uses a random walk to score how close a
pathway and the gene set are in the molecular interaction
network.
The enrichment analysis was complemented with a
network topology analysis, which was performed with TopoGSA
(http://www.topogsa.net/) (14). A network analysis of biological
systems revealed the connectivity and interactions of proteins
associated with different cellular processes. The topology analysis
identified pathways sharing a similar set of topological features.
The visualization of the reconstructed pathway network was
performed using the Cytoscape software.
Pathway-network construction and network
modularization
A pathway clustering analysis was performed on the
DEGs to construct a pathway network, using the (ClueGO + CluePedia)
plug-in (version 1.4) in Cytoscape (15,16),
according to KEGG (accessed on 14/3/2014). Using the plug-in,
MCODE, (version 1.4.0; http://apps.cytoscape.org/apps/MCODE) in Cytoscape
(17), the modules were identified
from the network. Each module was scored using Cytoscape, according
to density and size; a higher score represented a tighter module.
The threshold score was set at 3.0. Within the modules, path-ways
were analyzed using the ClueGO plug-in (version 1.4) in Cytoscape,
according to KEGG (accessed on 14/3/2014). Only pathways with the
overview 'true' were considered to be positive.
Results
Identification of DEGs
The original microarray data may be systematically
biased for various reasons, including the efficiency of the RNA
extraction process, reverse transcription, label incorporation,
exposure and spot detection (18).
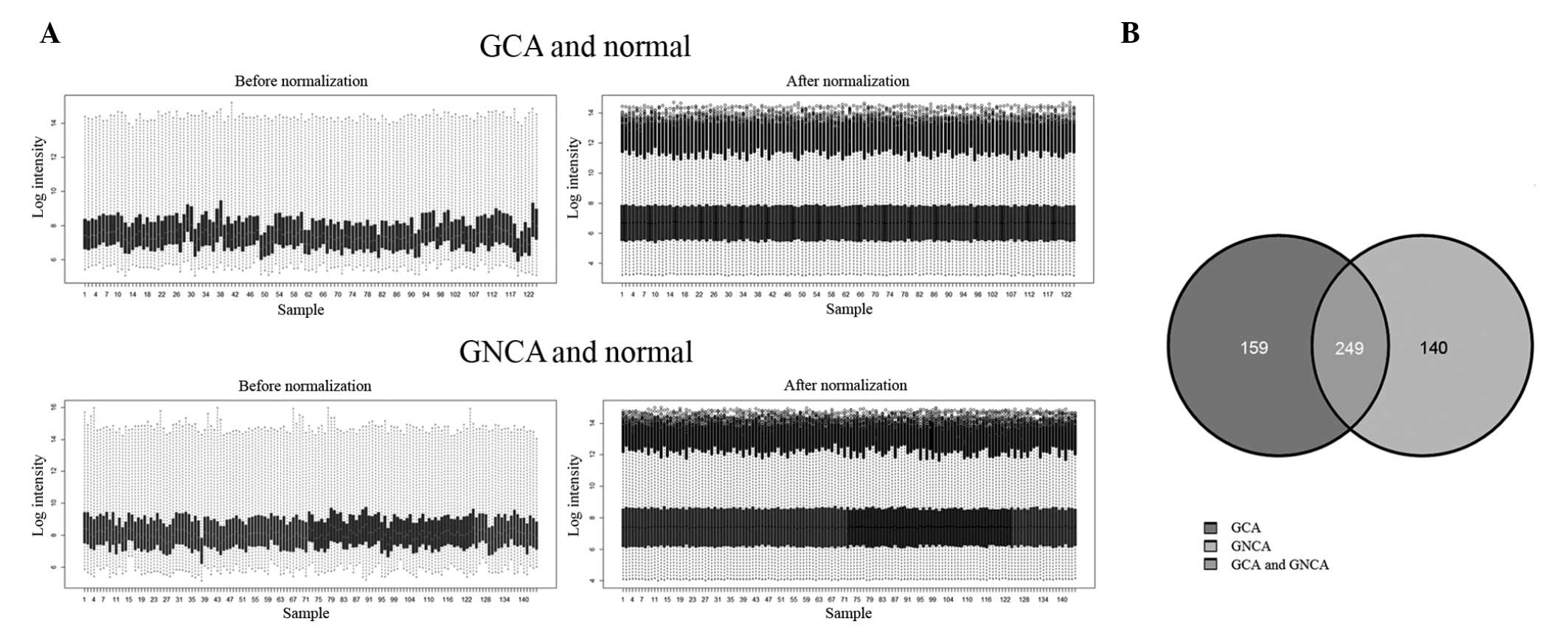
Therefore, data pre-processing was performed and this revealed that
the fluctuations following normalization were less marked compared
with those occurring prior to normalization (Fig. 1).
To obtain results with a high level of confidence,
the gene expression data were analyzed by a modified standard
t-test, based on the BH algorithm FDR method. The overlapping genes
from this set were subsequently selected for further analysis. At
an adjusted P=0.05, 249 genes revealed significantly different
levels of expression. Specifically, 150 genes were revealed to be
upregulated, whereas 99 genes were downregulated.
Pathway analysis
Pathways with the overview 'true' were considered to
be positive. Ultimately, the eight most significant pathways from
the groups with P<0.01 were identified using pathway clustering
analysis of the KEGG pathway datasets (data not shown). In order to
further screen the most relevant pathways, an enrichment analysis
was used to identify the key pathways. Three pathways or processes,
extracellular matrix (ECM)-receptor interaction, nitrogen
metabolism and linoleic acid metabolism, were selected, according
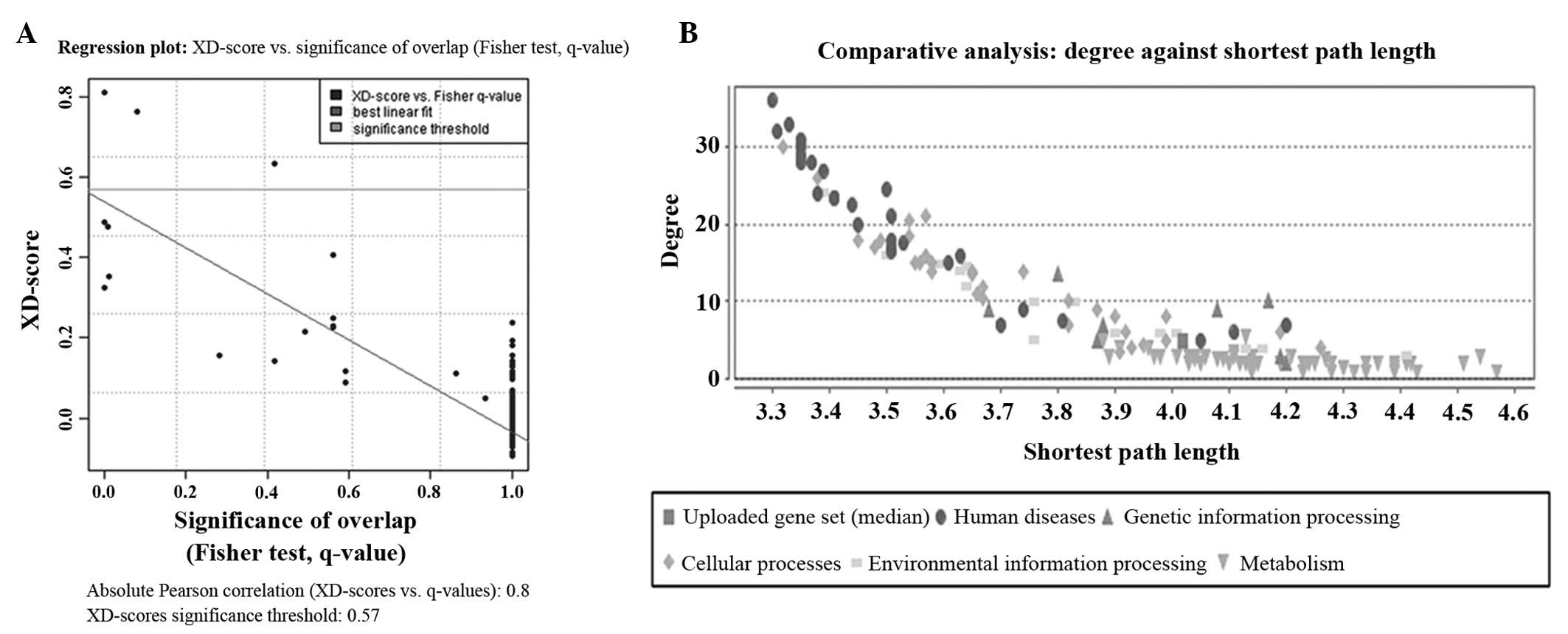
to the XD-score significance threshold (Table I). A Fisher test q-value (P-value
adjusted by false discovery rate) of 0.05 was considered as a
restriction to compensate for uncertainty in the model parameters
(Fig. 2A). The ECM-receptor
interaction pathway appeared to be a highly enriched pathway, based
on the results obtained (XD-score = 0.810; q-value = 0.000).
 | Table IMarkedly associated pathways
identified in KEGG. |
Table I
Markedly associated pathways
identified in KEGG.
| Pathway/process | XD-score | q-value | Overlap/Size |
|---|
| ECM-receptor
interaction | 0.810 | 0.000 | 16/77 |
| Nitrogen
metabolism | 0.763 | 0.081 | 3/14 |
| Linoleic acid
metabolism | 0.633 | 0.416 | 2/11 |
In the initial step of the enrichment analysis with
JEPETTO, the target gene set was mapped onto the string interaction
network. Of the 249 genes identified, 164 were successfully mapped,
and these were used in the further analyses.
As the input, the largest connected component (1,583
nodes) of the enriched network was analyzed. The topological
properties of the network were compared with the properties of
random interaction networks of an identical size. The results
revealed marked differences in the topological signatures (data not
shown).
The topological signature of the interactions in the
enriched network was subsequently compared with those of known
pathways and biological processes. The KEGG database was searched
for the closest topological matches (Fig. 2B). The most similar biological
mechanisms identified are listed in Table II. The closest topological matches
were identified for taste transduction and the regulation of
autophagy (score = 0.07).
| Table IITop 10 closest topological matches
found in KEGG. |
Table II
Top 10 closest topological matches
found in KEGG.
| Pathway/process | Class | Score |
|---|
| Regulation of
autophagy | Cellular process | 0.07 |
| Taste
transduction | Cellular process | 0.07 |
| Pyrimidine
metabolism | Metabolism | 0.13 |
| Calcium signaling
pathway | Environmental
information processing | 0.15 |
| PPAR signaling
pathway | Cellular process | 0.17 |
| Hedgehog signaling
pathway | Environmental
information processing | 0.17 |
| Cholera
infection | Human disease | 0.18 |
| Starch and sucrose
metabolism | Metabolism | 0.18 |
| Purine
metabolism | Metabolism | 0.19 |
| Neuroactive ligand
receptor interaction | Environmental
information processing | 0.22 |
Construction of the interaction pathway
network and module analysis
In view of the enrichment analysis, all pathways
associated with GC were used to build the network with DEGs. If a
gene was identified in ≥2 terms, it was assigned ≥2 colors. From
the network mapping, it was observed that the majority of the genes
were associated with different pathways. Genes, including those
encoding thrombospondin 1 (THBS1), fibronectin 1 (FN1), Myb
proto-oncogene protein, and collagen type 4 variants α (COL14A)1/2,
were involved in various processes. Furthermore, those encoding
interleukin 8, vascular cellular adhesion molecule 1, aldehyde
dehydrogenase 3 family member A1, aldehyde dehydrogenase 1C and
carbonic anhydrase 2, were involved in disparate pathways.
Using the MCODE plug-in in Cytoscape, the global
network was partitioned into seven modules, which were regarded as
the network core in terms of functionality (Table III). To determine the biological
function of each module, pathway enrichment from the seven modules
was processed using the ClueGO plug-in and the KEGG database.
Although numerous pathways in the KEGG database were detected, only
those with the overview 'true' (positive) were selected. A total of
22 pathways distributed in seven modules were classified as being
enriched. Since the pathway within a module represents only a part
of the signal cascade network, the module including the
ECM-receptor interaction pathway achieved the highest score
(8.222). The scores represent the module density, and the denser
modules are ranked more highly.
| Table IIINetwork modules and enriched signaling
pathways. |
Table III
Network modules and enriched signaling
pathways.
| Cluster | Scorea | Nodes | Edges | Node ID |
|---|
| 1 | 8.222 | 10 | 39 | FN1, COL4A2, THBS1,
SPP1, COL4A1 ECM-receptor interaction, focal adhesion, PI3K-Akt
signaling pathway, amoebiasis, protein digestion and
absorption |
| 2 | 5.250 | 9 | 21 | C1R, C1S, C2, C4A,
C4B Pertussis, prion diseases, complement and coagulation cascades,
Staphylococcus aureus infection |
| 3 | 4.501 | 5 | 9 | AKR1B10, AKR1B1
Fructose and mannose metabolism, glycerolipid metabolism, pentose
and glucuronate interconversions |
| 4 | 4.001 | 4 | 6 | Metabolism of
xenobiotics by cytochrome P450, retinol metabolism, fatty acid
degradation, chemical carcinogenesis |
| 5 | 3.600 | 6 | 9 | ALDH3A1, ALDH3B1,
ALDH1A3, ALDH3B2 drug metabolism, glycolysis/gluconeogenesis |
| 6 | 3.500 | 5 | 7 | MAOB, MAOA
Histidine metabolism, β-alanine metabolism, phenylalanine
metabolism |
| 7 | 3.000 | 3 | 3 | HBB, African
trypanosomiasis, malaria |
Cluster 1 from seven modules was demonstrated to be
highly enriched. The ECM-receptor interaction pathway had a
significant level of overlap with other pathways, focal adhesion,
phosphoinositide 3-kinase (PI3K)-Akt signaling pathway, amoebiasis,
protein digestion and absorption. Considerable crosstalk existed
among them and certain DEGs, FN1, secreted phosphoprotein 1,
COL4A1, COL4A2 and THBS1 (Fig.
3B). Numerous, close interactions were identified between
pathways and relevant genes in this module.
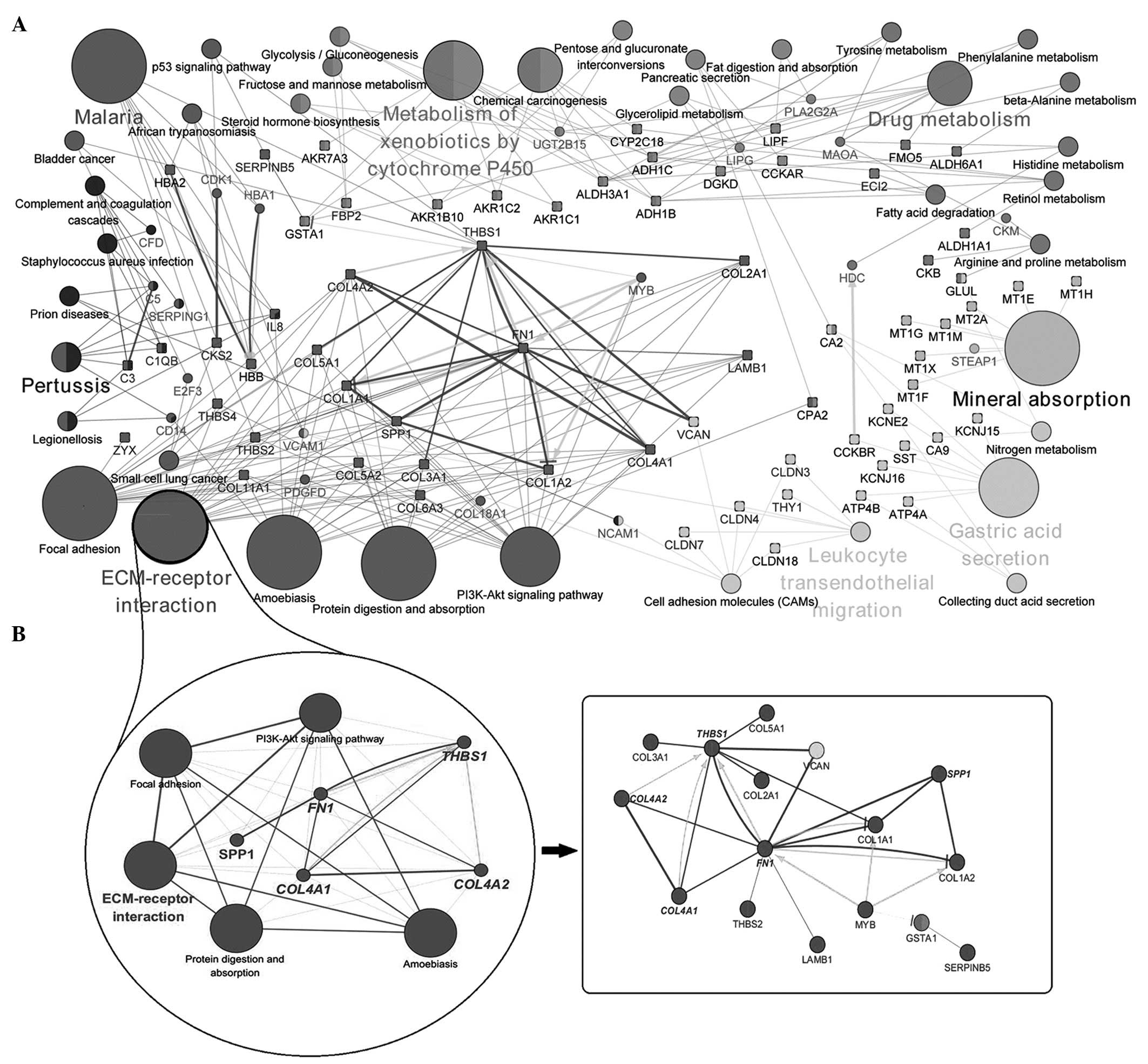 | Figure 3(A) A functionally grouped network
with pathways and differentially expressed genes is schematically
shown (genes are represented by their approved HUGO Gene
Nomenclature Committee symbols). (B) A network centred on the
ECM-receptor interaction pathway (cluster 1) is illustrated,
together with the gene inter-associations. The terms are linked
based on the κ score (≥0.3). The edge thickness is scaled between
the minimum and maximum scores demonstrated. The genes, which are
upregulated or downregulated are identified by squares. Arrows
represent the activation and the inhibition symbol represents
inhibition. Emboldened lines indicate a binding interaction.
COL1/2/3/4/5Ax, collagen type 1 (or type 2, 3, 4 or 5) family A
member x; PI3K, phosphoinositide 3-kinase; THBS, thrombospondin;
ECM, extracellular matrix; FN, fibronectin; SPP, secreted
phosphoprotein; MYB, Myb proto-oncogene protein; LAMB, laminin β;
GSTA, glutathione S-transferase α; VCAN, versican. |
Discussion
Systems biology, which is defined as the systematic
study of complex regulation and interaction in biological systems,
assesses cellular processes from a systematic, rather than a
reductionistic perspective. (19).
In the present study, a systems biology approach was performed to
identify any DEGs associated with GC, and to predict the underlying
molecular mechanisms.
In the initial stage of the enrichment analysis with
JEPETTO, ECM-receptor interaction pathways appeared at the top of
the pathways ranking. The high XD-score of 0.810 was >1.5 times
higher compared with the significance threshold of 0.57 identified
by the regression fit. Among the other top-ranked pathways and
processes were nitrogen metabolism and linoleic acid metabolism.
These two pathways had a Fisher test q-value >0.05 and a small
overlap size. The average shortest path length was identical with
the size in random networks, and the node degree was slightly
higher compared with the average node degree, which further
revealed certain dense interactions between the DEGs. However, the
results may also support the hypothesis that the target network is
specific, and that interactions between DEGs are unlike those
typically observed in the background or random network as a whole.
The closest topological match was identified for regulation of
autophagy. Autophagy is dysregulated in a wide spectrum of human
cancer types (20). There is
mounting evidence to suggest that alterations in
autophagy-associated genes are associated with the pathogenesis of
gastrointestinal cancer. Frameshift mutations in the
autophagy-related protein (ATG)2B, ATG5, ATG9B and ATG12 genes with
mono-nucleotide repeats are common, indicating that the genetic
disruption of autophagy may contribute to gastrointestinal
tumorigenesis (21). The decrease
in autophagic capacity may be associated with tumorigenesis and the
development of GC.
In the constructed pathway map, several genes were
involved in tumorigenesis. To assess the decomposition in the
clusters of a network, modularization methods were used to further
dissemble the networks. From cluster 1, certain path-ways
associated with GC, including the PI3K-Akt signaling pathway, were
also enriched. The ECM-receptor interaction pathway, in association
with the PI3K-Akt signaling, focal adhesion, amoebiasis, and
protein digestion and absorption pathways (Fig. 3), were considered as the key
pathways mediating the actions of GC. In addition, associated DEGs
were derived from an identical module. The genetic interactions
between the DEGs and these pathways were also displayed to confirm
possible connections.
ECM is a macromolecular network, comprising
collagens, non-collagenous glycoprotein, glycosaminoglycan,
proteoglycans, elastin and other components. The ECM was revealed
to influence cell survival, death, proliferation and
differentiation, as well as cancer metastasis (22). Collagen is the major constituent of
the tumor ECM, and several types of collagens have been implicated
in the focal adhesion and ECM-receptor interaction pathways in
gastric carcinoma (23).
Focal adhesions not only provide structural links
between the ECM and the actin cytoskeleton, they also comprise
important sites of signal transduction pathways leading to various
physiological and pathological processes, including cancer
(24). At the molecular level,
focal adhesions are predominantly mediated by integrins, which have
been identified as exerting a critical role in the invasion and
metastasis of cancer (25).
Kindlin-2 is a member of the focal adhesion protein family
recruited to integrin-containing adhesion sites. Kindlin-2 occupied
an important role in the progression of GC and was an independent
risk factor of progression-free survival (26). Few reports have focused on the
connections between the protein digestion and absorption pathway,
and GC. Although acidic and non-acidic gastric exocrine secretion
are required for micronutrient absorption and protein digestion,
ingested proteins undergo a complex series of degradative processes
following the action of gastric acid (27). Disorders of the protein digestion
and absorption pathway lead to a decrease in gastric exocrine
secretion with a possible progression to mucosal atrophy, which may
ultimately lead to cancer.
The majority of the unregulated genes in GC,
including COL1A1, COL2A1, COL4A1 and COL4A2, are associated with
cell adhesion or migration and the ECM-receptor interaction
pathways (28). Previous studies
have demonstrated that the overexpression of COL1A1 and COL1A2 is
associated with tumor invasion, metastasis and stage grouping,
indicating that these genes may be novel genetic markers for
high-grade malignancy (29,30).
In addition, THBS1 is a multifunctional protein implicated in
cancer cell adhesion, migration, invasion, inhibition of
angiogenesis and activation of latent trans-forming growth factor
β. Furthermore, THBS1 is an inhibitor of angiogenesis with tumor
suppressor properties (31,32).
A polymorphism in the 5′-untranslated region of THBS1 (rs1478604
A>G) is reported to be associated with lymph node metastasis of
GC (33). FN1 is an ECM protein,
which mediates the activation of focal adhesion kinase, which
promotes cell motility through the extracellular signal-regulated
kinase or PI3K/Akt signaling pathways to upregulate matrix
metalloproteinase 9 (MMP 9)/calpain-2 or MMP 9/RhoA activity
(34). Notably, several
downregulated genes are associated with GC, including COL3A1 and
COL5A1. The incidence of GC is likely to be associated with acute
changes in the expression of these genes.
In conclusion, the present study demonstrated that
changes in the primary pathways (i.e. the focal adhesion, PI3K-Akt
signaling, amoebiasis, protein digestion and absorption, and
ECM-receptor interaction pathways) may be associated with GC.
Extensive connections between these pathways and their candidate
genes, which may be associated with GC, were also identified by
modularity analysis. These findings shed new light on the biology
of GC, and provided novel insights into an improved understanding
of the molecular basis for identifying the associations between
candidate genes and key pathways.
Acknowledgments
This study was supported by the General Program of
the National Natural Science Foundation of China (no. 81373097).
The authors would like to thank the focus group participants and
volunteers from community organization partners for their time and
efforts.
References
|
1
|
Catalano V, Labianca R, Beretta GD, Gatta
G, de Braud F and Van Cutsem E: GC. Crit Rev Oncol Hematol.
71:127–164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.PubMed/NCBI
|
|
3
|
Draghici S, Khatri P, Tarca AL, Amin K,
Done A, Voichita C, Georgescu C and Romero R: A systems biology
approach for pathway level analysis. Genome Res. 17:1537–1545.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loscalzo J and Barabasi AL: Systems
biology and the future of medicine. Wiley Interdiscip Rev Syst Biol
Med. 3:619–627. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Troyanskaya O, Cantor M, Sherlock G, Brown
P, Hastie T, Tibshirani R, Botstein D and Altman RB: Missing value
estimation methods for DNA microarrays. Bioinformatics. 17:520–525.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
D'Souza M, Zhu X and Frisina RD: Novel
approach to select genes from RMA normalized microarray data using
functional hearing tests in aging mice. J Neurosci Methods.
171:279–287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davis JW: Bioinformatics and computational
biology solutions using R and bioconductor. J Am Statistical
Association. 102:388–389. 2007. View Article : Google Scholar
|
|
8
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benjamini Y, Drai D, Elmer G, Kafkafi N
and Golani I: Controlling the false discovery rate in behavior
genetics research. Behav Brain Res. 125:279–284. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maere S, Heymans K and Kuiper M: BiNGO: A
cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Winterhalter C, Widera P and Krasnogor N:
JEPETTO: A Cytoscape plugin for gene set enrichment and topological
analysis based on interaction networks. Bioinformatics.
30:1029–1030. 2014. View Article : Google Scholar :
|
|
12
|
Glaab E, Baudot A, Krasnogor N, Schneider
R and Valencia A: EnrichNet: Network-based gene set enrichment
analysis. Bioinformatics. 28:i451–i457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Glaab E, Baudot A, Krasnogor N and
Valencia A: Extending pathways and processes using molecular
interaction networks to analyse cancer genome data. BMC
Bioinformatics. 11:5972010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Glaab E, Baudot A, Krasnogor N and
Valencia A: TopoGSA: Network topological gene set analysis.
Bioinformatics. 26:1271–1272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bindea G, Galon J and Mlecnik B: CluePedia
cytoscape plugin: Pathway insights using integrated experimental
and in silico data. Bioinformatics. 29:661–663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bindea G, Mlecnik B, Hackl H, Charoentong
P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z and
Galon J: ClueGO: A cytoscape plug-in to decipher functionally
grouped gene ontology and pathway annotation networks.
Bioinformatics. 25:1091–1093. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu X and Sun S: Comparing a few SNP
calling algorithms using low-coverage sequencing data. BMC
Bioinformatics. 14:2742013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alakwaa FM, Solouma NH and Kadah YM:
Construction of gene regulatory networks using biclustering and
Bayesian networks. Theor Biol Med Model. 8:392011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mitra S, Das S and Chakrabarti J: Systems
biology of cancer biomarker detection. Cancer Biomark. 13:201–213.
2013.PubMed/NCBI
|
|
20
|
Zhang L, Sung JJ, Yu J, Ng SC, Wong SH,
Cho CH, Ng SS, Chan FK and Wu WK: Xenophagy in Helicobacter pylori-
and Epstein-Barr virus-induced gastric cancer. J Pathol.
233:103–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kang MR, Kim MS, Oh JE, Kim YR, Song SY,
Kim SS, Ahn CH, Yoo NJ and Lee SH: Frameshift mutations of
autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and
colorectal cancers with microsatellite instability. J Pathol.
217:702–706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bijian K, Takano T, Papillon J, Khadir A
and Cybulsky AV: Extracellular matrix regulates glomerular
epithelial cell survival and proliferation. Am J Physiol Renal
Physiol. 286:F255–F266. 2004. View Article : Google Scholar
|
|
23
|
Yin Y, Zhao Y, Li AQ and Si LM: Collagen:
A possible prediction mark for gastric cancer. Medical Hypotheses.
72:163–165. 2009. View Article : Google Scholar
|
|
24
|
Yam JW, Tse EY and Ng IO: Role and
significance of focal adhesion proteins in hepatocellular
carcinoma. J Gastroenterol Hepatol. 24:520–530. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ramos DM, But M, Regezi J, Schmidt BL,
Atakilit A, Dang D, Ellis D, Jordan R and Li X: Expression of
integrin beta 6 enhances invasive behavior in oral squamous cell
carcinoma. Matrix Biol. 21:297–307. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen Z, Ye Y, Dong L, Vainionpää S,
Mustonen H, Puolakkainen P and Wang S: Kindlin-2: A novel adhesion
protein related to tumor invasion, lymph node metastasis and
patient outcome in gastric cancer. Am J Surg. 203:222–229. 2012.
View Article : Google Scholar
|
|
27
|
Colacci E, Pasquali A and Severi C:
Exocrine gastric secretion and gastritis: Pathophysiological and
clinical relationships. Clin Ter. 162:e19–25. 2011.PubMed/NCBI
|
|
28
|
Jinawath N, Furukawa Y, Hasegawa S, Li M,
Tsunoda T, Satoh S, Yamaguchi T, Imamura H, Inoue M, Shiozaki H and
Nakamura Y: Comparison of gene-expression profiles between diffuse-
and intestinal-type gastric cancers using a genome-wide cDNA
microarray. Oncogene. 23:6830–6844. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ossandon FJ, Villarroel C, Aguayo F,
Santibanez E, Oue N, Yasui W and Corvalan AH: In silico analysis of
gastric carcinoma serial analysis of gene expression libraries
reveals different profiles associated with ethnicity. Mol Cancer.
7:222008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ramaswamy S, Ross KN, Lander ES and Golub
TR: A molecular signature of metastasis in primary solid tumors.
Nat Genet. 33:49–54. 2003. View
Article : Google Scholar
|
|
31
|
Wiedemann S, Wessela T, Schwarz K, Joachim
D, Jercke M, Strasser RH, Ebner B and Simonis G: Inhibition of
anti-apoptotic signals by Wortmannin induces apoptosis in the
remote myocardium after LAD ligation: Evidence for a protein kinase
C-delta-dependent pathway. Mol Cell Biochem. 372:275–283. 2013.
View Article : Google Scholar
|
|
32
|
Adams JC: Functions of the conserved
thrombospondin carboxy-terminal cassette in cell-extracellular
matrix inter-actions and signaling. Int J Biochem Cell Biol.
36:1102–1114. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin XD, Chen SQ, Qi YL, Zhu JW, Tang Y and
Lin JY: Polymorphism of THBS1 rs1478604 A>G in 5-untranslated
region is associated with lymph node metastasis of gastric cancer
in a Southeast Chinese population. DNA Cell Biol. 31:511–519. 2012.
View Article : Google Scholar
|
|
34
|
Rosman DS, Phukan S, Huang CC and Pasche
B: TGFBR1* 6A enhances the migration and invasion of
MCF-7 breast cancer cells through RhoA activation. Cancer Res.
68:1319–1328. 2008. View Article : Google Scholar : PubMed/NCBI
|