Introduction
Apoptosis, also termed programmed cell death, is
involved in development, the elimination of damaged cells, and the
maintenance of cell homeostasis. Deregulation of apoptosis may
cause diseases, such as cancer, immune disorders and degenerative
diseases (1). Apoptosis is an
important cellular response to irreparable damage from exposure to
external stimuli (2). Apoptosis
can be induced by two different pathways; the extrinsic pathway,
which involves signaling from death receptors, such as tumor
necrosis factor receptor 1 (TNFR1), and the intrinsic pathway,
which centers around the mitochondria (3). In recent years, it has been
increasingly recognized that the mitochondria is pivotal in the
early events of apoptosis. A key process in the mitochondrial
apoptotic pathway is the release of a group of proteins, such as
cytochrome c, second mitochondria-derived activator of
caspases/direct IAP binding protein with low pI (Smac/DIABLO),
apoptosis-inducing factor (AIF) and endonuclease G from the
mitochondrial intermembrane space, which subsequently triggers a
cascade of cytoplasmic changes (4).
Protein function associated with apoptosis can be
modulated by post-translational modification, such as acetylation,
phosphorylation and nitrosylation. Currently, numerous studies
suggest that the function of various mitochondrial factors is
regulated by the interplay between kinases and phos-phatases, and
thereby promote or inhibit apoptosis (5–7). As
a pro-apoptotic kinase, c-Jun NH2-terminal kinase is associated
with Smac/DIABLO release from the mitochondria into the cytoplasm
during apoptosis in multiple myeloma cells (8). A complex of c-Abl and protein kinase
C δ migrates into the mitochondria in response to various oxidative
stresses, and induces cytochrome c release from the
mitochondria (9–11). Lck activity is required for
mitochondrial-dependent apoptosis following irradiation and
ceramide exposure (12,13).
By contrast, cumulative studies have suggested that
Akt inhibits apoptosis by inactivating Bad, Caspase-9 and Forkhead
box (FOX) proteins by direct phosphorylation (14–16).
Akt mediates cell proliferation, survival, migration, and glucose
homeostasis via the canonical phosphoinositide 3-kinase (PI3K)-Akt
signaling pathway (17).
Downstream targets of Akt for survival signaling include
transcription factor cAMP-response element binding protein or IκB
kinases that regulate the activity of nuclear factor-κB (18). Furthermore, Akt activation is
sufficient to inhibit the release of cytochrome c from the
mitochondria and the change in inner mitochondrial membrane
potential (19). Recent data,
however, indicated that Akt can act as a pro-apoptotic regulator
under specific conditions (20).
Numerous studies have demonstrated that Akt sensitized cells to
oxidative stress-mediated apoptosis (21,22)
while pharmacological inhibition of Akt reduced apoptosis mediated
by various cytotoxic or apoptotic intracellular molecules (23–26).
The pro-apoptotic effect of Akt was also demonstrated and it was
shown that Akt phosphorylates Smac/DIABLO and enhances its
interaction with XIAP in etoposide-induced apoptosis (27). Although increasing evidence
indicates that Akt has dual roles in the determination of cell fate
in a context-dependent way, the molecular mechanisms are not yet
well established.
In the present study, whether Akt is activated and
translo-cated into the mitochondria during etoposide induced
apoptosis was investigated. This appears to be a key step in the
progression of apoptotic, and regulation of this step may provide
effective as a novel strategy in the treatment of cancer in
humans.
Materials and methods
Materials
Etoposide, KCl, MgCl2, EDTA, EGTA, DTT
and PMSF was purchased from Sigma-Aldrich (St. Louis, MO, USA).
LY294002 was obtained from Calbiochem (San Diego, CA, USA).
Dulbecco's modified Eagle's medium (DMEM),
antibiotics-antimycotics, HEPES and fetal bovine serum (FBS) were
purchased from Invitrogen Life Technologies (Grand Island, NY,
USA). Protease inhibitor cocktail was purchased from Roche (Nutley,
NJ, USA). Antibodies against Akt (rabbit monoclonal IgG; cat. no.
4691; Cell Signaling Technology, Inc., Danvers, MA, USA),
phospho-Akt (Ser473; rabbit ant-human monoclonal IgG; cat. no.
4060; Cell Signaling Technology, Inc), α-tubulin (rabbit polyclonal
ant-human IgG; cat. no. 2144; Cell Signaling Technology, Inc.) and
cytochrome c oxidase subunit IV (COX IV; rabbit monoclonal
IgG; cat. no. 4850; Cell Signaling Technology, Inc.) were purchased
from Cell Signaling Technology Inc. (Beverly, MA, USA). Anti-poly
ADP-ribose polymerase (PARP) antibody (rabbit polyclonal anti-human
IgG; cat. no. sc-7150; Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA) was obtained from Santa Cruz Biotechnology Inc. (Santa
Cruz, CA, USA). All primary antibodies were diluted into 1:1,000
for western blotting and 1:100 for confocal imaging. Secondary
antibodies were diluted 1:5,000 for western blotting and 1:400 for
confocal imaging.
Cell culture and induction of
apoptosis
Human HeLa cervical carcinoma cells (Seoul National
University Cancer Center, Seoul, Republic of Korea) were cultured
in DMEM supplemented with 10% heat-inactivated FBS and
antibiotics-antimycotics in a humidified incubator with 5%
CO2 at 37°C. Apoptosis was induced by incubation with
10% FBS-supplemented DMEM containing 85 µl etoposide. To
inhibit the PI3K-Akt signaling pathway, the cells were pre-treated
with LY294002, a PI3K inhibitor, for 1 h prior to incubation with
the etoposide.
Subcellular fractionation
The etoposide-treated HeLa cells (1×105)
were collected and resuspended in buffer, containing 20 mM HEPES
(pH 7.4), 10 mM KCl, 1.5 mM MgCl2 1 mM EDTA, 1 mM EGTA,
1 mM DTT, 1 mM PMS and protease inhibitor cocktail. Following
incubation on ice for 15 min, the cells were homogenized and
centrifuged at 1,000 × g for 10 min at 4°C. The supernatant was
then centrifuged at 100,000 × g for 1 h at 4°C. The supernatant
(cytosol fraction) was then collected and the lysates
(mitochondrial fraction) were lysed using lysis buffer.
Immunoblot analysis
Cells were lysed in lysis buffer containing 0.5%
Triton X-100, 20 mM Tris (pH 7.5); 2 mM MgCl2, 1 mM DTT,
1 mM EGTA, 50 mM β-glycerophosphate, 25 mM NaF and 1 mM
Na3VO4, containing 1 mM PMSF and protease
inhibitor cocktail. The protein concentrations were determined
using the MicroBCA™ Protein Assay reagents (Pierce Biotechnology,
Inc., Rockford, IL, USA) according to the manufacturer's
instructions. Cell lysates were resolved by SDS-PAGE (12%) and
transferred to polyvinylidene difluoride membranes (EMD Millipore,
Billerica, MA, USA). Membranes were blocked with 5% non-fat dry
milk in TBST (TBS with 0.1% Tween-20; Sigma-Aldrich) buffer for 30
min at room temperature. The membranes were incubated with the
specific primary antibodies in 5% non-fat dry milk in TBST buffer
(1:1,000). The bands were visualized with enhanced
chemiluminescence (West-Zol plus®; Intron Biotechnology,
Sungnam, Republic of Korea).
Confocal microscopy
Cells on coverslips were washed twice with
phosphate-buffered saline (PBS) and fixed on ice with 3%
paraformaldehyde/PBS for 10 min, and then washed again with PBS.
Residual paraformaldehyde was quenched by incubation with 0.1 M
glycine for 10 min. After washing with PBS, cells were
permeabilized with 0.1% Triton X-100 in PBS for 3 min, washed with
PBS, and incubated in blocking solution (5% milk) for 10 min. Cells
were incubated with anti-Akt and COX IV antibodies (1:100) in
blocking buffer overnight at 4°C. Nuclei were counterstained with
4′,6-diamidino-2-phenylindole (DAPI). Subcellular localization of
the proteins was visualized using a Nikon Eclipse Ti inverted
microscope (Nikon, Tokyo, Japan) and analyzed using NIS-Elements
software (ver. 4.0; Nikon Corporation, Tokyo, Japan). Pearson's
correlation test was used to analyze the correlation between the
expression levels of Akt and COX IV in immunocytochemistry using
the NIS-Elements software.
Results
Akt translocates into the mitochondria
during etoposide-induced apoptosis
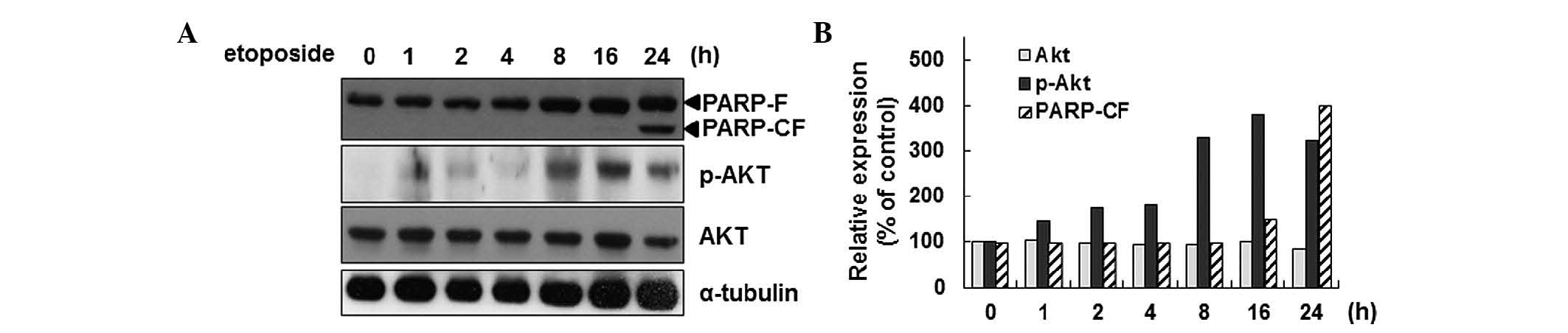
Previously, the activation of Akt during
etoposide-induced apoptosis in HeLa cells was reported (27). In the present study,
etoposide-mediated activation of Akt by phosphorylation of the
Ser473 residue in a time-dependent manner was confirmed (Fig. 1A and B). As an indicator of the
induction of apoptosis, PARP cleavage was detected. As Akt
activation is associated with Smac phosphorylation during apoptosis
in HeLa cells, it was surmised that Akt activation is associated
with the mitochondrial function during apoptosis. To test this
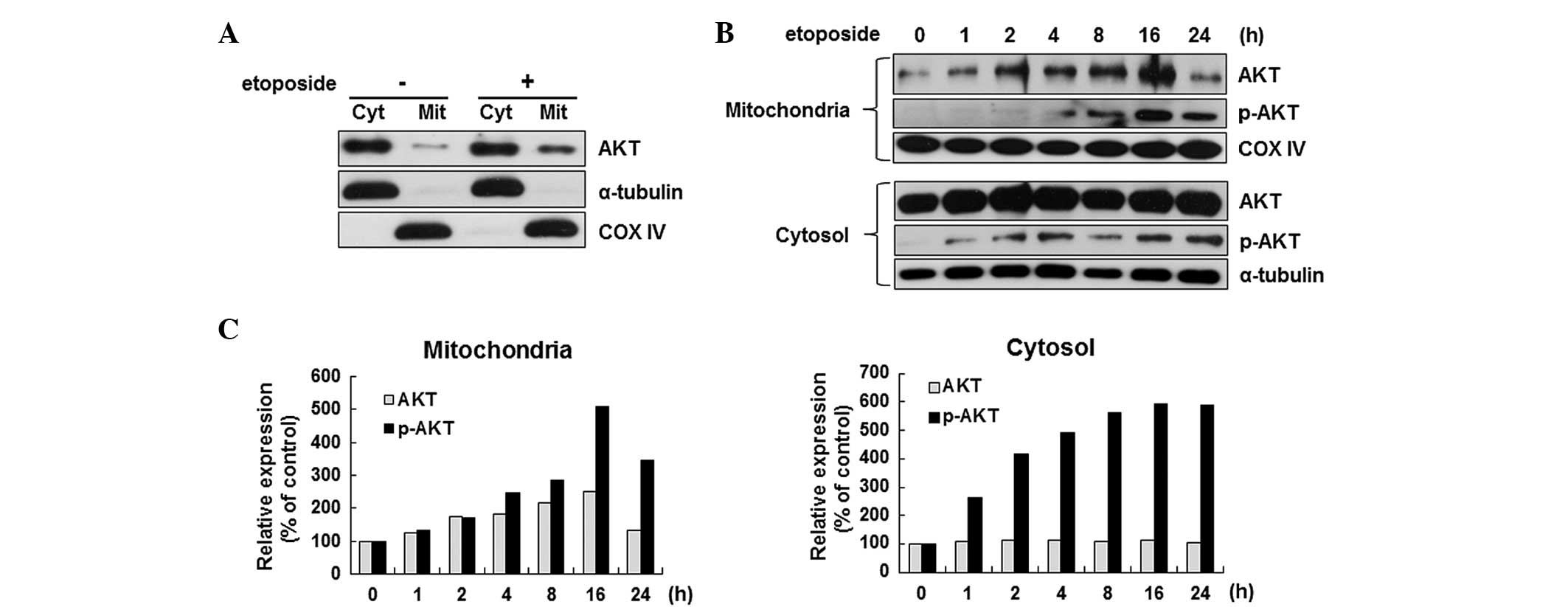
hypothesis, the localization of Akt in etoposide-treated HeLa cells
was examined by separating the mitochondrial fraction from the
cytoplasmic fraction. As shown in Fig.
2A, the Akt level in the mitochondria increased significantly
after the induction of apoptosis. The induction of Akt
translocation at various time points during apoptosis was further
confirmed. The Akt level in the mitochondria was increased
significantly as early as 2 h after etoposide stimulation, which
was further elevated with prolonged incubation by 2.5-fold at 16 h
(Fig. 2B upper panel). Similarly,
the level of phosphorylated Akt, the active form, was also
increased by etoposide in the mitochondria (Fig. 2B upper panel). However, the change
in the expression level of Akt of the cytosol was not significant
while clear induction of the p-Akt level was detected by etoposide
(Fig. 2B lower panel).
Nuclear Akt levels are reduced during
etoposide-induced apoptosis
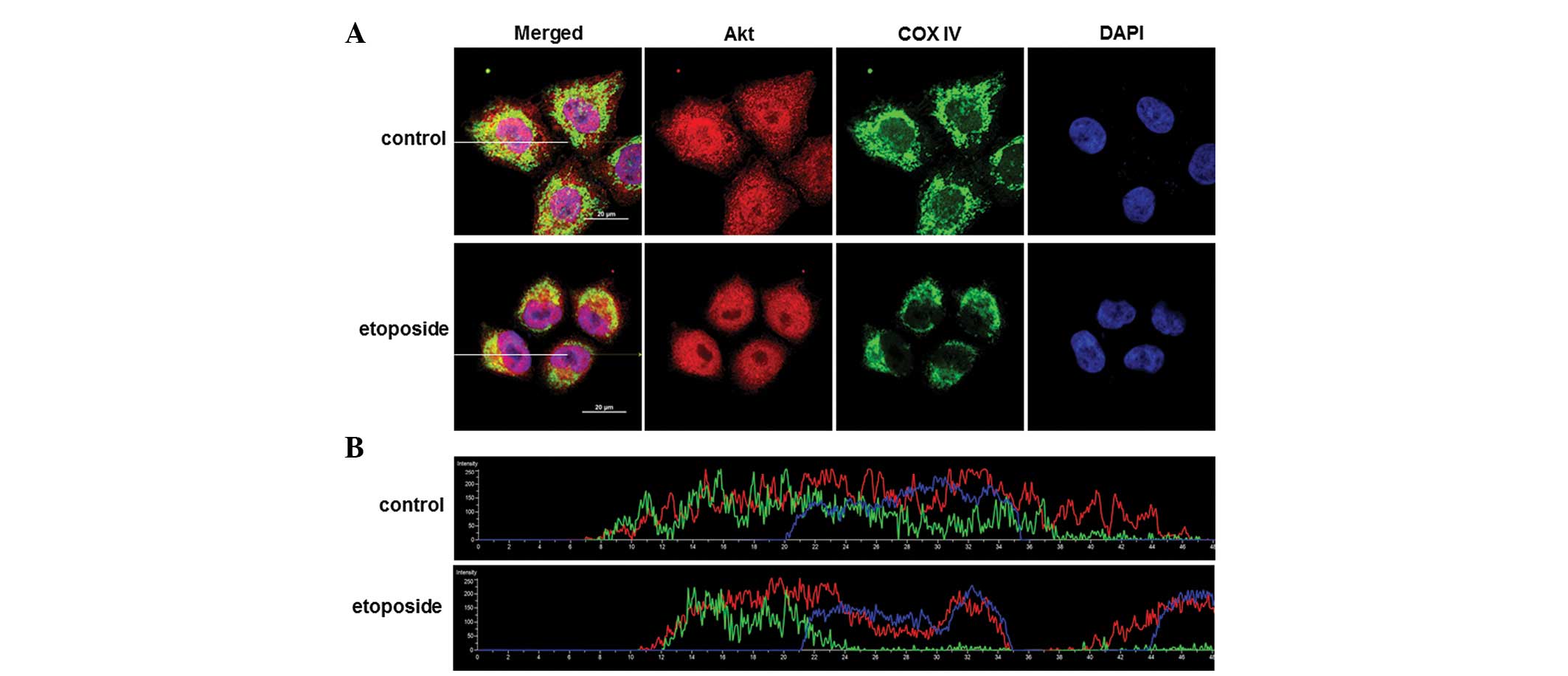
To confirm the mitochondrial migration of Akt during
etoposide-induced apoptosis, the subcellular localization of Akt
was examined using confocal microscopy. Akt was distributed in the
nucleus and the cytosol, and the partial co-localization of Akt and
COX IV, a mitochondrial marker, was identified in control cells
(Fig. 3A, upper). When cells are
treated with etoposide, cell size was reduced by ~50% (data not
shown) by the shrinkage of the cytoplasm. The co-localization of
Akt and COX IV was also detected in apoptosis-induced conditions
(Fig. 3A, lower). However, a
significant change in the parameter showing the levels of
co-localization between etoposide-treated cells and control was not
identified. The Pearson's correlation coefficient (PCC) was almost
the same (PCCControl=0.37; n=7 cells; SD=0.082 and
PCCEtoposide=0.37 ; n=12 cells; SD=0.092). This lack of
change in the PCC value suggests the possibility that Akt migration
into the mitochondria would be a local event during apoptosis, as
the majority were located in other areas, including the cytosol. To
further characterize the change in Akt distribution during
apoptosis, levels of Akt, COX IV and DAPI were measured across a
sectional plane (Fig. 3A, white
bar) in etoposide-treated cells. When cells were sectioned along
the cells (white bar), Akt (red) was located abundantly in the
cytosol of cells undergoing apoptosis, while depletion of Akt was
observed in the nucleolus suggesting that nuclear Akt was also
affected by apoptotic signals induced by etoposide (Fig. 3B). The change in the subcellular
distribution of Akt following etoposide treatment suggests that Akt
may exhibit compartment-specific roles during apoptosis.
Activation of Akt is required for
mitochondrial migration during apoptosis
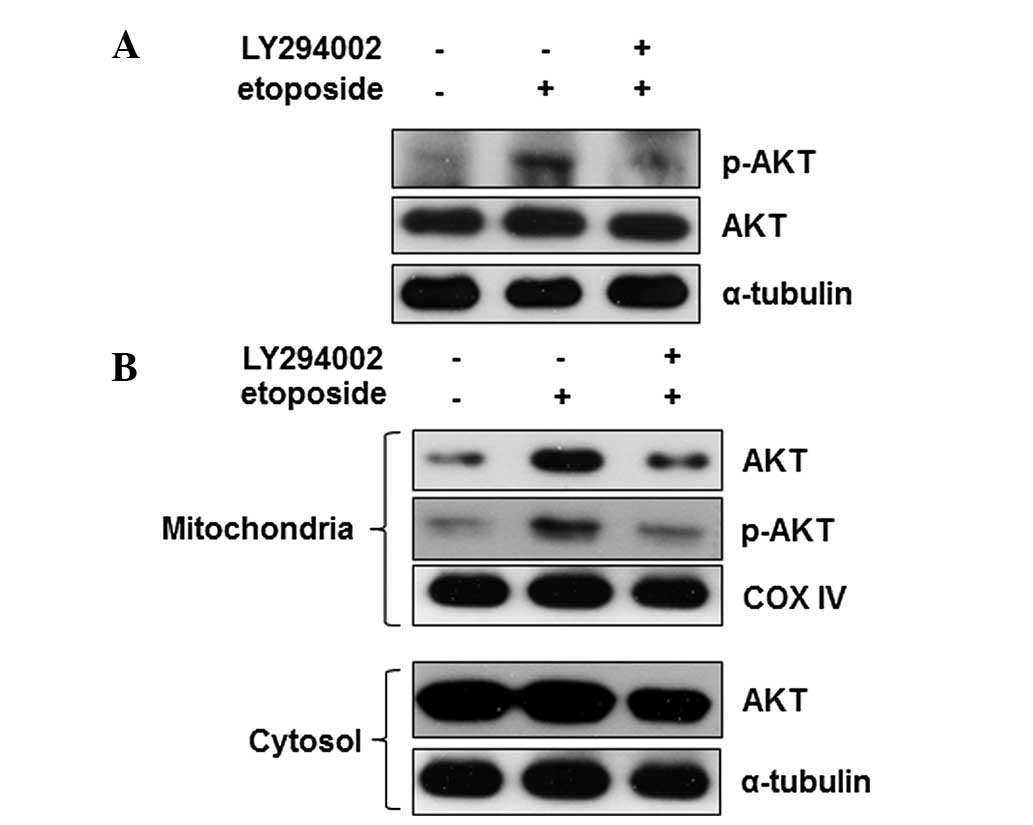
Numerous stimuli activate Akt via the PI3K-Akt
signaling pathway. To identify whether the etoposide-mediated
regulation of Akt is dependent on the canonical PI3K-Akt signaling
pathway, cells were pre-treated with LY294002, an inhibitor of
PI3K, and the effects on the activity of Akt were measured. When
cells were pretreated with LY294002, the etoposide-mediated Akt
phosphorylation was reduced to the basal level (Fig. 4A) indicating that PI3K is a key
regulator of Akt activation during etoposide-induced apoptosis.
Furthermore, the effect of inhibition of PI3K on Akt mitochondrial
translocation during apoptosis was identified. The Akt level in the
mitochondria was also reduced to the basal level by LY294002
(Fig. 4B). These data show that
PI3K may be an upstream regulator of the etoposide-mediated Akt
activation and mitochondrial translocation. These findings
demonstrate that Akt translocates into the mitochondria during
etoposide-induced apoptosis and PI3K activation is required for the
activity and localization of Akt in HeLa cells. This suggest that
Akt may act by regulating the mitochondrial factors associated with
the progression of apoptosis.
Discussion
Previously, it was demonstrated that Akt is
activated during etoposide-induced apoptosis in HeLa cells in which
Akt phosphorylates Smac and activates caspase-3 to promote
apoptosis. As Smac is a mitochondrial protein that promotes
cytochrome c release during apoptosis, it was suggested that
Akt exhibits a role in the mitochondria in the process of
apoptosis. In this study, it was found that the Akt level in the
mitochondria is low in a healthy normal state, but was increased
significantly by apoptotic stimuli (Fig. 2). This translocation was
accompanied by the elevation of Akt activity as shown by increased
phosphorylation. Furthermore, it was demonstrated that PI3K
activity is involved in these events during apoptosis (Fig. 4). To the best of our knowledge,
this is the first study to demonstrate the translocation of Akt
into the mitochondria during apoptosis.
Additionally, confocal microscopy further
demonstrated that Akt resides in the nucleus of HeLa cells under
normal conditions; however, Akt is depleted by etoposide treatment
(Fig. 3). In particular, the level
of Akt within the nucleolus was significantly diminished by
etoposide indicating that this migration is involved in the
pro-apoptotic function of Akt. This decrease of Akt from the
nucleus may be not a general event during apoptosis, considering
previous evidence that Akt translocation into the nucleus has a
pro-apoptotic effect in methotrexate and docetaxel-treated MCF7
breast cancer cells (23). Thus,
it appears that etoposide regulates distinct molecular targets from
methotrexate or docetaxel, although they require Akt activation to
induce apoptosis.
Akt has been known to be a survival factor and also
a tumor-promoting agent. However, recent studies have shown that
Akt is able to exhibit a pro-apoptotic effect under diverse
conditions, such as oxidative stress (hydrogen peroxide, arsenite)
(20,24), stimulation by cytokines [Fas ligand
(Fas-L), tumor necrosis factor α (TNFα)] (28,29),
and cytotoxic chemicals (staurosporine, methotrexate, docetaxel,
etoposide) (25,28).
Although the mechanisms underlying Akt activation
and its ability to elicit a pro-apoptotic effect has not been
elucidated clearly, several studies have suggested plausible
explanations. Nogueira et al (21) showed that Akt sensitizes cells to
oxidative stress-induced apoptosis by increasing reactive oxygen
species (ROS) production as well as by lowering the expression of
the oxygen scavenging enzymes, such as manganese superoxide
dismutase, catalase and sestrin 3. In C141 epidermal cells, Fas
ligand (Fas-L) caused Akt activation through the generation of the
hydroxyl radical (30). In
arsenite-, hydrogen peroxide-, TNFα- and staurosporine-induced
apoptosis, Akt phosphory-lates and inhibits glycogen synthase
kinase 3β [one of the first identified substrates of Akt (31)] and FoxO3a. Following inhibition of
FoxO3a, expression of oxygen scavenging enzymes is suppressed. In
addition, Akt can induce the expression of Fas receptor mRNA, thus
sensitizing cells to Fas-L. Furthermore, nuclear Akt was suggested
to phosphorylate cyclin-dependent kinase 2 (Cdk2) in methotrexate
and docetaxel-induced apop-tosis, thereby sustaining the
cytoplasmic location of Cdk2, which may be associated with G2/M
cell cycle arrest prior to apoptosis (25). Although a number of studies have
shown that Akt exerts pro-apoptotic effects through the
phosphorylation of target molecules, the function of Akt in
mitochondrial events have not been addressed. The present study
demonstrated the re-location of Akt among subcellular organelles,
which suggests that Akt may be important in mitochondria and may be
a promising target for cancer treatment.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea,
funded by the Ministry of Education, Science and Technology (grant
no. 2010–0025409).
References
|
1
|
Jiang X and Wang X: Cytochrome C-mediated
apoptosis. Annu Rev Biochem. 73:87–106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Green DR: Apoptotic pathways: Paper wraps
stone blunts scissors. Cell. 102:1–4. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khosravi-Far R and Esposti MD: Death
receptor signals to mitochondria. Cancer Biol Ther. 3:1051–1057.
2004. View Article : Google Scholar
|
|
4
|
van Gurp M, Festjens N, van Loo G, Saelens
X and Vandenabeele P: Mitochondrial intermembrane proteins in cell
death. Biochem Biophys Res Commun. 304:487–497. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thomson M: Evidence of undiscovered cell
regulatory mechanisms: Phosphoproteins and protein kinases in
mitochondria. Cell Mol Life Sci. 59:213–219. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boland ML, Chourasia AH and Macleod KF:
Mitochondrial dysfunction in cancer. Front Oncol. 3:2922003.
|
|
7
|
Yoo SH, Kim HY, Rho JH, Jeong SY, Yun J,
Yun I, Park HT and Yoo YH: Targeted inhibition of mitochondrial
Hsp90 induces mitochondrial elongation in Hep3B hepatocellular
carcinoma cells undergoing apoptosis by increasing the ROS level.
Int J Oncol. Sep;2015.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chauhan D, Li G, Hideshima T, Podar K,
Mitsiades C, Mitsiades N, Munshi N, Kharbanda S and Anderson KC:
JNK-dependent release of mitochondrial protein, Smac, during
apoptosis in multiple myeloma (MM) cells. J Biol Chem.
278:17593–17596. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ito Y, Pandey P, Mishra N, Kumar S, Narula
N, Kharbanda S, Saxena S and Kufe D: Targeting of the c-Abl
tyrosine kinase to mitochondria in endoplasmic reticulum
stress-induced apoptosis. Mol Cell Biol. 21:6233–6242. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar S, Bharti A, Mishra NC, Raina D,
Kharbanda S, Saxena S and Kufe D: Targeting of the c-Abl tyrosine
kinase to mitochondria in the necrotic cell death response to
oxidative stress. J Biol Chem. 276:17281–17285. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qi X and Mochly-Rosen D: The PKCdelta-Abl
complex communicates ER stress to the mitochondria-an essential
step in subsequent apoptosis. J Cell Sci. 121:804–813. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Belka C, Marini P, Lepple-Wienhues A,
Budach W, Jekle A, Los M, Lang F, Schulze-Osthoff K, Gulbins E and
Bamberg M: The tyrosine kinase lck is required for CD95-independent
caspase-8 activation and apoptosis in response to ionizing
radiation. Oncogene. 18:4983–4992. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hur YG, Yun Y and Won J: Rosmarinic acid
induces p56lck-dependent apoptosis in Jurkat and peripheral T cells
via mitochondrial pathway independent from Fas/Fas ligand
interaction. J Immunol. 172:79–87. 2004. View Article : Google Scholar
|
|
14
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nicholson KM and Anderson NG: The protein
kinase B/Akt signalling pathway in human malignancy. Cell Signal.
14:381–395. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kennedy SG, Kandel ES, Cross TK and Hay N:
Akt/Protein kinase B inhibits cell death by preventing the release
of cytochrome c from mitochondria. Mol Cell Biol. 19:5800–5810.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Benbrook DM and Masamha CP: The
pro-survival function of Akt kinase can be overridden or altered to
contribute to induction of apoptosis. Curr Cancer Drug Targets.
11:586–599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nogueira V, Park Y, Chen CC, Xu PZ, Chen
ML, Tonic I, Unterman T and Hay N: Akt determines replicative
senescence and oxidative or oncogenic premature senescence and
sensitizes cells to oxidative apoptosis. Cancer Cell. 14:458–470.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Gorp AG, Pomeranz KM, Birkenkamp KU,
Hui RC, Lam EW and Coffer PJ: Chronic protein kinase B (PKB/c-akt)
activation leads to apoptosis induced by oxidative stress-mediated
Foxo3a transcriptional up-regulation. Cancer Res. 66:10760–10769.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Galvez-Peralta M, Flatten KS, Loegering
DA, Peterson KL, Schneider PA, Erlichman C and Kaufmann SH:
Context-dependent antagonism between Akt inhibitors and
topoisomerase poisons. Mol Pharmacol. 85:723–734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shack S, Wang XT, Kokkonen GC, Gorospe M,
Longo DL and Holbrook NJ: Caveolin-induced activation of the
phosphati-dylinositol 3-kinase/Akt pathway increases arsenite
cytotoxicity. Mol Cell Biol. 23:2407–2414. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maddika S, Ande SR, Wiechec E, Hansen LL,
Wesselborg S and Los M: Akt-mediated phosphorylation of CDK2
regulates its dual role in cell cycle progression and apoptosis. J
Cell Sci. 121:979–988. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Andrabi S, Gjoerup OV, Kean JA, Roberts TM
and Schaffhausen B: Protein phosphatase 2A regulates life and death
decisions via Akt in a context-dependent manner. Proc Natl Acad Sci
USA. 104:19011–19016. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jeong CH, Chun KS, Kundu J and Park B:
Phosphorylation of Smac by Akt promotes the caspase-3 activation
during etoposide-induced apoptosis in HeLa cells. Mol Carcinog.
54:83–92. 2015. View
Article : Google Scholar
|
|
28
|
Ono K, Iwanaga Y, Hirayama M, Kawamura T,
Sowa N and Hasegawa K: Contribution of caveolin-1 alpha and Akt to
TNF-alpha-induced cell death. Am J Physiol Lung Cell Mol Physiol.
287:L201–L209. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suhara T, Mano T, Oliveira BE and Walsh K:
Phosphatidylinositol 3-kinase/Akt signaling controls endothelial
cell sensitivity to Fas-mediated apoptosis via regulation of
FLICE-inhibitory protein (FLIP). Circ Res. 89:13–19. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu B, Wang L, Stehlik C, Medan D, Huang C,
Hu S, Chen F, Shi X and Rojanasakul Y: Phosphatidylinositol
3-kinase/Akt positively regulates Fas (CD95)-mediated apoptosis in
epidermal Cl41 cells. J Immunol. 176:6785–6793. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|