Introduction
Heart failure (HF) is a clinical syndrome, which can
result from any heart diseases that lead to inability of the
ventricle to pump sufficient blood to meet the body's metabolic
demands. HF is associated with a high incidence of sudden death,
primarily from ventricular arrhythmia (1). Although the mechanisms underlying
ventricular arrhythmia in HF remain to be fully elucidated, it is
known that gap junctions, which are unique intercellular channels
that directly connect adjacent cardiac myocytes by providing
electrical and chemical communication, contribute to various
cardiac arrhythmia (2). Gap
junctions are composed of connexins, and the number, distribution,
and composition of the connexins are important in regulation of the
conductive properties of cardiac myocytes (3).
Connexin 43 (Cx43) is the major connexin protein in
the ventricle, which is predominantly located in the intercalated
disk region between cardiac myocytes in the adult heart. Several
lines of evidence have demonstrated that the expression and
localization of Cx43 are altered in the diseased myocardium
(2). Downregulation of Cx43 has
been observed in an animal model of HF and in the failing human
heart (4–6). In hypertrophic cardiomyopathy and
myocardial ischemia, the expression of Cx43 is downregulated
(4), and shifts between an
end-to-end location and a lateral location (7,8).
Furthermore, the heterogeneous loss of Cx43 in non-ischemic dilated
cardiomyopathy is associated with ventricular tachycardia (9).
Zonula occludens (ZO)-1 is a scaffolding protein,
which provides a structural basis for assembling protein complexes
and maintaining the polarity of epithelial cells (10). In cardiac myocytes, ZO-1 is located
in the intercalated disc and the lateral membrane (11–13).
Several studies have reported that ZO-1 is directly associated with
Cx43 (14), and regulates the
number, distribution and function of Cx43 (15–17).
Furthermore, it has been reported that the expression of ZO-1 is
reduced in the failing human heart, accompanied by a decrease in
the expression of Cx43 (18,19),
suggesting that ZO-1 is important in the downregulation of Cx43 in
HF.
It has been reported that the ubiquitin-proteasome
system is involved in the degradation of Cx43 (20). Several studies have revealed that
the internalization of Cx43 is dependent on the proteasomal
activities (20–22). Furthermore, ubiquitination of Cx43
at the cytoplasmic membrane promotes the internalization and
subsequent degradation of Cx43 in a proteasome-dependent manner
(21,23). In addition, proteasomes can
regulate the interaction between Cx43 and ZO-1, thus leading to an
increase in Cx43 degradation (24). The proteasome inhibitor, MG132, has
been observed to inhibit the internalization and degradation of
Cx43 (24,25). However, whether proteasome
inhibition can alter the expression of Cx43 in failing hearts
remains to be elucidated.
The present study aimed to investigate the effect of
the proteasome inhibitor MG132 on the expression levels of Cx43,
ZO-1, 20S proteasome and ubiquitin in a rat model of HF, induced by
adriamycin. The purpose of this investigation was to examine the
role of inhibition of the ubiquitin-proteasome system by MG132 in
the expression of Cx43 in the HF rat model. The results of this
investigation may determine whether inhibition of the
ubiquitin-proteasome system offers a potential therapeutic strategy
for improving the expression of Cx43 and preventing Cx43-mediated
ventricular arrhythmia in HF.
Materials and methods
Animals
All experimental procedures were approved by the
ethics committee of Harbin Medical University (Harbin, China).
Animals were obtained from the Animal Care Center of Harbin Medical
University. A total of 70 male Wistar rats (weighing 180–300 g)
were housed separately in an atmosphere containing 60% humidity,
with a 12 h light/dark cycle at 23±2°C. The rats were fed standard
chow and water ad libitum.
Adriamycin-induced heart failure
model
HF was induced via intraperitoneal injections of
adriamycin (2.5 mg/kg; Hisun Pharmacuetical Co. Ltd., Taizhou,
China) to a cumulative dose of 15 mg/kg in six injections. A total
of 70 rats received the first three injections of adriamycin every
3 days, with one injection per day followed by three injections
every week. Prior to each drug administration, the body weight of
the animal was measured for recalculating dosage. Following the
last adriamycin injection, 14 rats had died, with 56 rats remaining
alive. The 56 rats were randomly assigned into two groups: Heart
failure (HR) group (n=28) and MG132 group (n=28). The rats in the
MG132 group received an intraperitoneal injection of MG132 (0.1
mg/kg; Sigma-Aldrich, St. Louis, MO, USA) for 14 days. The rats in
the HR group were injected at the same time with the same volume of
saline. In addition, normal rats (n=28) without adriamycin
injections were used as controls.
Echocardiographic examination
Echocardiographic examination was performed two
weeks following the injections of MG132 or saline, using a VIVID 7
dimension system (General Electric, Milwaukee, WI, USA). The rats
(n=14 per group) were anesthetized by intraperitoneal injection of
10% chloral hydrate (300 mg/kg; Sigma-Aldrich, St. Louis, MO, USA)
and the animals were placed in a supine position. The 10 S
transducer (8 MHz) was placed directly on the shaved chest wall and
the image depth was adjusted 2–4 cm. The M-mode echocardiogram was
used to measure the left ventricle end-systolic diameter (LVESD)
and left ventricle end-diastolic diameter (LVEDD) of three
consecutive heart cycles, and the average was used for calculation
of the ejection fraction (EF) and fractional shortening (FS), as
previously reported (26).
Sample collection
Following the final injections of MG132 and saline,
the rats (n=14 per group) were sacrificed by intra-peritoneal
injection of 10% chloral hydrate (300 mg/kg), and the hearts were
immediately removed and rinsed in saline. Subsequently, two tissue
blocks were obtained from the free wall of the left ventricle,
along the long axis of the heart, one of which was fixed in 4%
paraformaldehyde (Sigma-Aldrich) and embedded in paraffin
(Sigma-Aldrich). Transverse sections (5 µm thick) were
obtained from paraffin-embedded tissue blocks for hematoxylin and
eosin (H&E) staining (Shanghai Ruiqi Bio-Technology Co., Ltd.,
Shanghai, China) and immunohistochemistry. The other tissue block
was fixed in 3% glutaraldehyde (Sigma-Aldrich) and used for
electron microscopy. The remaining left ventricle tissues were
stored at −80°C and used for western blot analysis.
H&E staining and immunohistochemical
analysis
The paraffin-embedded tissue sections were washed in
xylene (Sigma-Aldrich) to remove the paraffin, and rehydrated with
serial dilutions of alcohol, followed by a wash in
phosphate-buffered saline solution (GE Healthcare Life Sciences,
Shanghai, China). For the H&E staining, the sections were
stained with H&E (Beyotime Institute of Biotechnology, Haimen,
China). For immunohistochemistry, rthe sections were treated with
3% H2O2 (Sigma-Aldrich) at room temperature
for 10 min to block endogenous peroxidase activity. The sections
were then incubated in 5% normal goat serum to block nonspecific
protein binding sites. The sections were then incubated with
primary antibodies against Cx43 (polyclonal rabbit anti-rat Cx43;
1:50; cat. no. SC-9059; Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA), ZO-1 (polyclonal goat anti-rat ZO-1; 1:50; cat. no.
SC-8146; Santa Cruz Biotechnology, Inc.), 20S Proteasome α3
(polyclonal rabbit anti-rat 20S Proteasome α3; 1:5; cat. no.
SC-67340; Santa Cruz Biotechnology, Inc.) and ubiquitin (polyclonal
rabbit anti-rat ubiquitin; 1:50; cat. no. SC-9133; Santa Cruz
Biotechnology, Santa Cruz Biotechnology, Inc.) overnight at 4°C.
Subsequently, the primary antibody was washed off, and the sections
were incubated with goat anti-rabbit (HaiGene Biotechnology,
Harbin, China) or donkey anti-goat (Santa Cruz Biotechnology, Inc.)
biotin-conjugated secondary antibodies (1:1,000) for 20 min at
37°C. The sections were then incubated with streptavidin
horseradish peroxidase for 20 min at 37°C. The 3,
3-diamino-benzidine (Sigma-Aldrich) substrate was then applied to
the sections, which were then counterstained with hematoxylin.
Sections in which the primary antibodies were omitted were used as
negative controls. The immunostained sections were examined under a
light microscope (Olympus IX-51; Olympus Corporation, Tokyo,
Japan).
Electron microscopy
The tissue blocks of the left ventricle were fixed
with 3% glutaraldehyde. They were dehydrated in a graded series of
ethanol (50, 70, 90 and 100% for 10 min each), and then embedded in
a 1:1 mixture of Epon812 (Sigma-Aldrich) and acetone
(Sigma-Aldrich) for 1 h at room temperature, followed by embedment
in Epon812 for 2 h. Ultrathin sections (50–100 nm) were cut,
stained with lead citrate (Polysciences Inc., Warrington, PA, USA)
and uranyl acetate (Sigma-Aldrich). The sections were then viewed
and images were captured using an electron microscope (JEM-1200EX;
JEOL, Ltd., Tokyo, Japan).
Western blot analysis
The ventricles were homogenized on ice in lysis
buffer (Nanjing Sunshine Biotechology Co., Ltd., Nanjing, China).
The proteins were resolved by 15% SDS-PAGE, and transferred onto
polyvinylidene fluoride membranes (Sigma-Aldrich) by
electroblotting. The membranes were incubated with primary
antibodies against Cx43 (polyclonal rabbit anti-rat Cx43, 1:500;
Santa Cruz Biotechnology, Inc.), ZO-1 (polyclonal goat anti-rat
ZO-1; 1:400; Santa Cruz Biotechnology, Inc.), 20S Proteasome α3
(polyclonal rabbit anti-rat 20S Proteasome α3; 1:40; Santa Cruz
Biotechnology, Inc) and ubiquitin (polyclonal rabbit anti-rat
ubiquitin; 1:400; Santa Cruz Biotechnology, Inc.) at room
temperature for 30 min. β-actin was used as a loading control. The
membranes were then incubated with horseradish peroxidase-linked
goat anti-rabbit (haiGene Biotechnology) or donkey anti-goat (Santa
Cruz Biotechnology, Inc,.) secondary antibodies at room temperature
for 20–30 min. The bands were visualized using a chemiluminescence
detection system (Invitrogen Life Technologies, Carlsbad, CA, USA)
and analyzed using BandScan5.0 (Glyko, Inc., Novato, CA, USA). The
expression levels of Cx43, ZO-1, 20S Proteasome and ubiquitin were
normalized to that of β-actin.
Statistical analysis
Analyses were performed using SPSS 17.0 (SPSS Inc.,
Chicago, IL, USA). All values are presented as the mean ± standard
deviation. One-way analysis of variance was used to compare the
differences among groups. P<0.05 were considered to indicate a
statistically significant difference.
Results
MG132 treatment reduces
adriamycin-induced cardiotoxicity
The EF and FS were significantly lower in the HF and
MG132 groups, compared with the control group (P<0.05; Table I). Although the EF and FS were
marginally higher in the MG132 group, compared with the HF group,
no significant difference was observed between the two groups
(P>0.05; Table I). These data
indicated the presence of myocardial damage and suggested that the
cardiac contractile function was reduced in adriamycin-induced
HF.
 | Table IEF and FS in the control, HF and MG132
groups. |
Table I
EF and FS in the control, HF and MG132
groups.
| Factor | Control (%) | HF (%) | MG132 (%) |
|---|
| EF | 82.15±3.57 | 69.65±4.65a | 67.21±7.13a |
| FS | 45.25±3.12 | 33.78±3.35a | 31.85±4.15a |
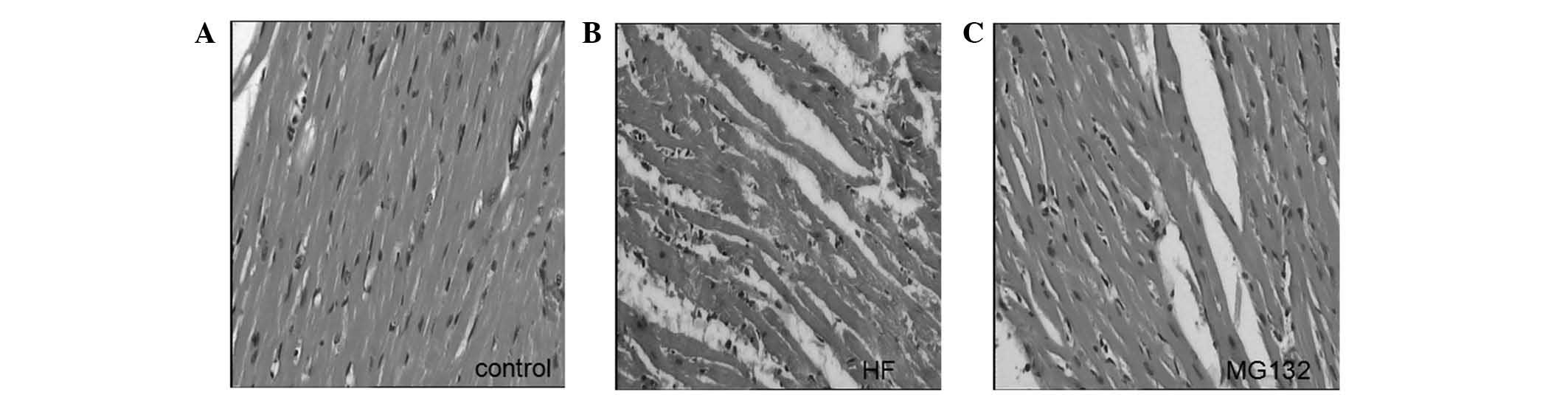
Histological examination of the left ventricle in
the control group demonstrated that the myocardial fibers were
orderly arranged and exhibited homogenously-stained cytoplasm. No
damaged fibers were observed, and no edema or exudation were
observed. By contrast, vacuolar degeneration and lysis of cardiac
myocytes, rupture of myocardial fibers, interstitial edema and
infiltration of inflammatory cells were observed in the HF group.
Adriamycin-induced damage was reduced in the MG132 group (Fig. 1).
The electron microscopic findings of cardiac
myocytes in the control, HF, and MG132 groups were compared
(Fig. 2). In the control group,
the fine band structure of the sarcomere was clearly identified.
The mitochondria were intact and exhibited clear and dense cristae.
Desmosomes, gap junctions and intermediate junctions were clearly
visible (Fig. 2A and B). By
contrast, in the HF group, lysis of the myocardial fibers and
disappearance of the band structure of the sarcomere were observed.
The remaining myofibrils were disorganized, and the lysed
myofibrils were filled with a numerous cell organelles. An
increased number of lysosomes were present between the
mitochondria, and swollen mitochondria with loss of cristae were
found. High electron-dense particles were observed in the nucleus,
with disappearance of organelles at the paranuclear region. The gap
junctions were disorganized and formed a tube-like structure. In
the MG132 group, the band structure of the sarcomere was clearly
visible. The sarcoplasmic reticulum was marginally enlarged. The
mitochondria were intact, however, the number of mitochondria
containing flocculent densities inside was decreased. Desmosomes,
gap junctions and intermediate junctions were intact.
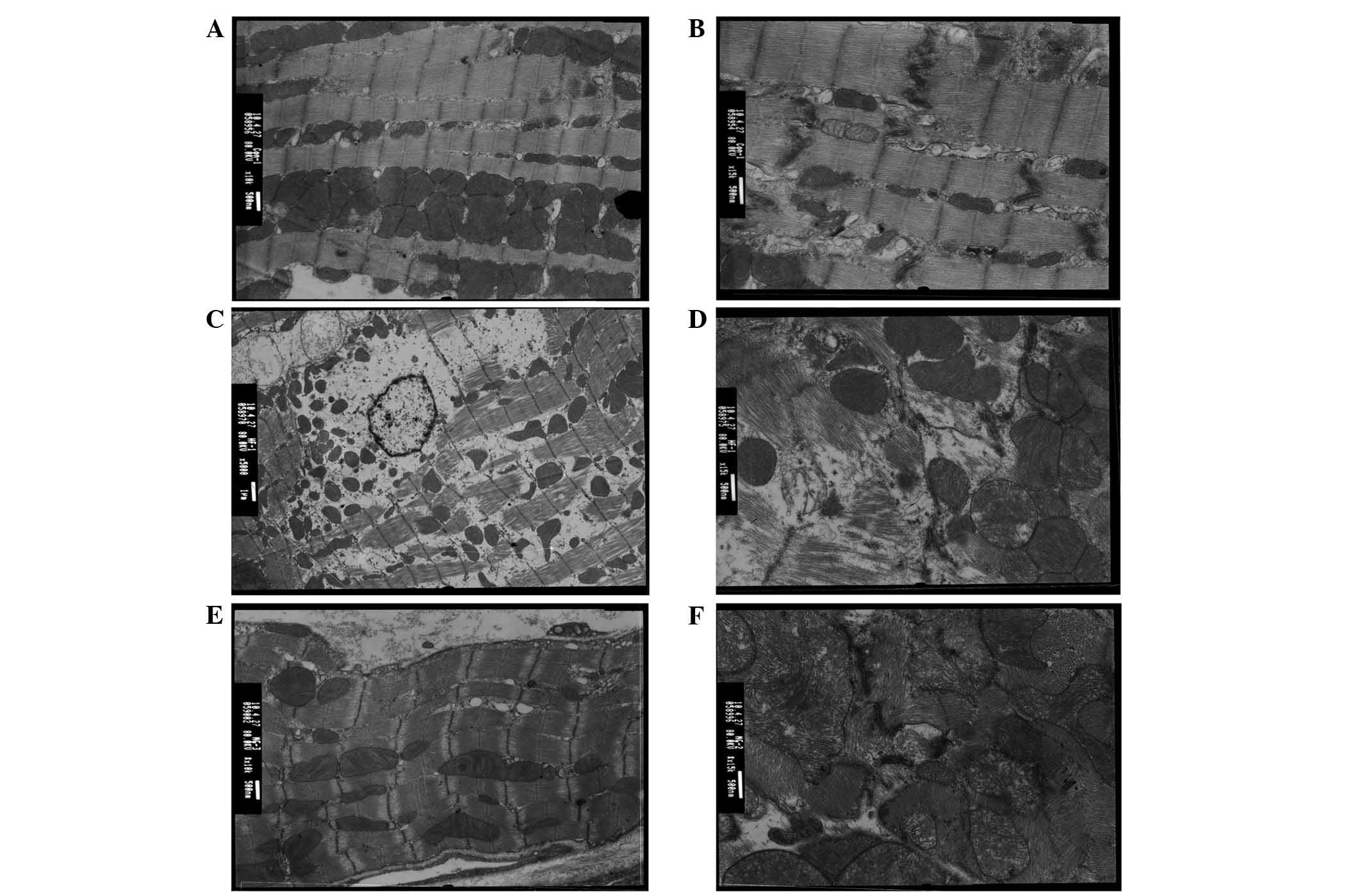 | Figure 2Electron microscopy images of cardiac
myocytes in the (A and B) control, (C and D) HF, and (E and F)
MG132 groups. (A an B) The fine band structure of the sarcomere is
clearly visible with intact mitochondria, desmosomes, gap
junctions, and intermediate junctions. (C and D) Lysis of
myocardial fibers, disappearance of sarcomere band structure, and
disorganized myofibrils are visible with a high number of
lysosomes, swollen mitochondria and high electron-dense particles.
In addition, the gap junctions are disorganized. (E and F) The band
structure of the sarcomere is clearly visible with partially
enlarged sarcoplasmic reticulum and intact mitochondria,
desmosomes, gap junctions, and intermediate junctions.
Magnification: A,C and E, x10,000; B, D, and F, x15,000. HF, heart
failure; Cx43, connexin 43. |
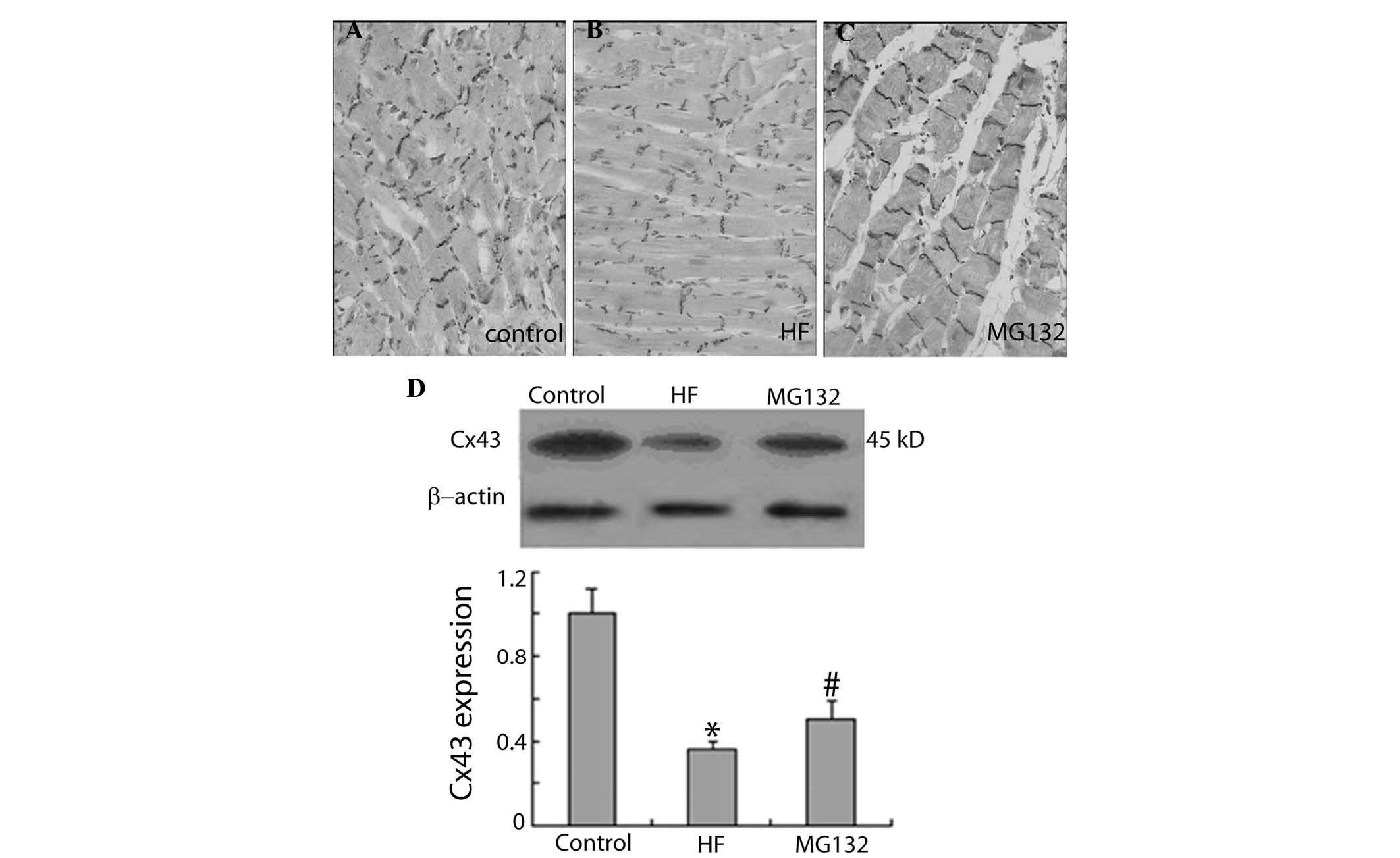
MG132 increases the expression of Cx43
and ZO-1 in HF rats
The present study further examined the effect of
MG132 on the expression levels of Cx43 and ZO-1 in HF rats, using
immunohistochemistry and western blot analysis. Immunohistochemical
investigations demonstrated that the expression of Cx43 was lower
in the HF group, compared with the control group. MG132 treatment
increased the expression of Cx43 in the HF rats (Fig. 3A–C). Consistent with the
immunohistochemical results, the results of the western blot
analysis demonstrated that the expression of Cx43 was significantly
decreased in the HF group, compared with the control group, and
MG132 treatment increased the expression of Cx43 in the HF rats
(Fig. 3D).
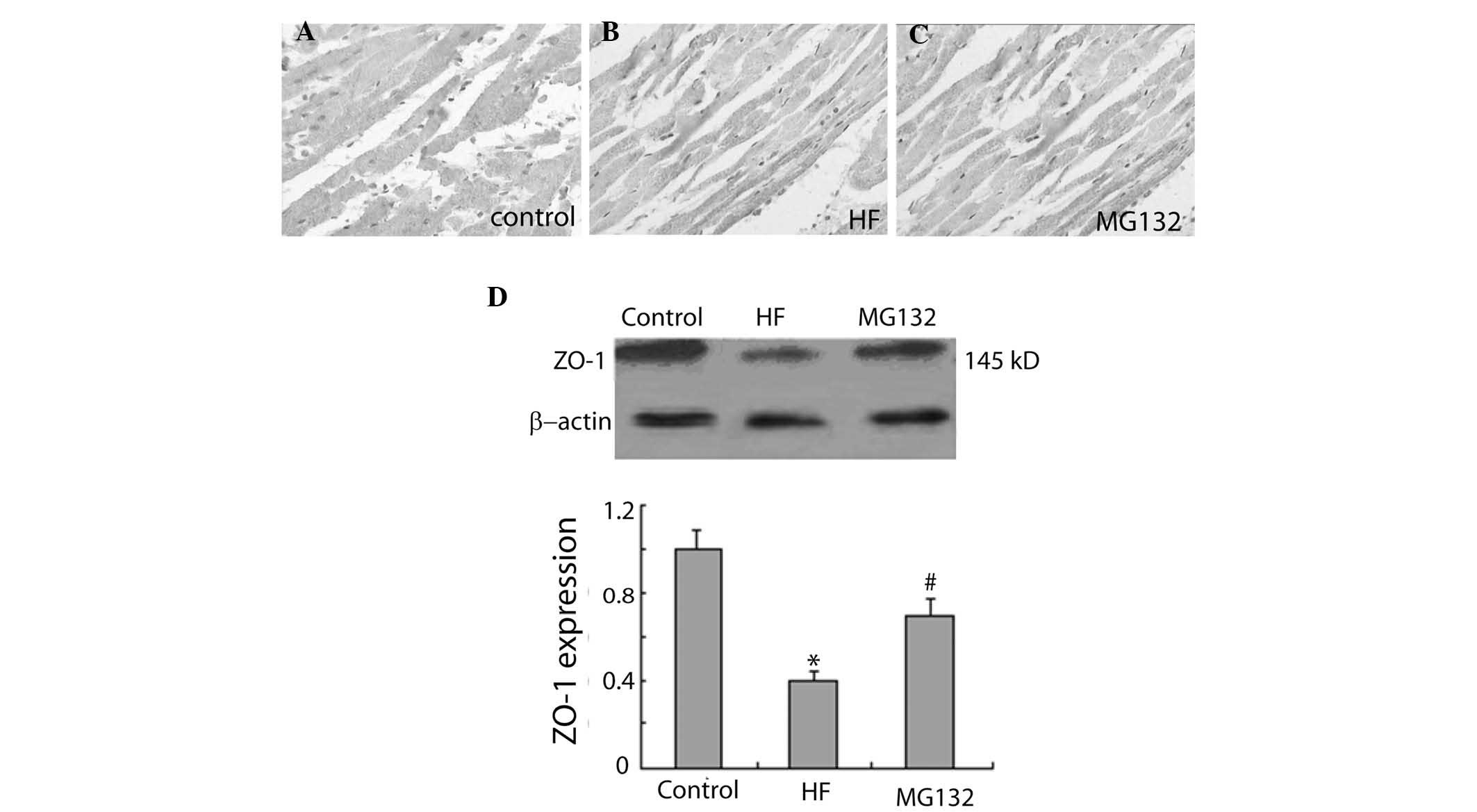
The immunohistochemical results revealed that ZO-1
was widely expressed in each group (Fig. 4). The intensity of ZO-1
immunostaining was weaker in the HF group, compared with the
control group, and was more marked in the MG132 group, compared
with the HF group (Fig. 4A–C).
Consistent with these findings, western blot analysis revealed that
the expression of ZO-1 was significantly decreased in the HF group,
compared with the control group, and significantly increased in the
MG132 group, compared with the HF group (Fig. 4D).
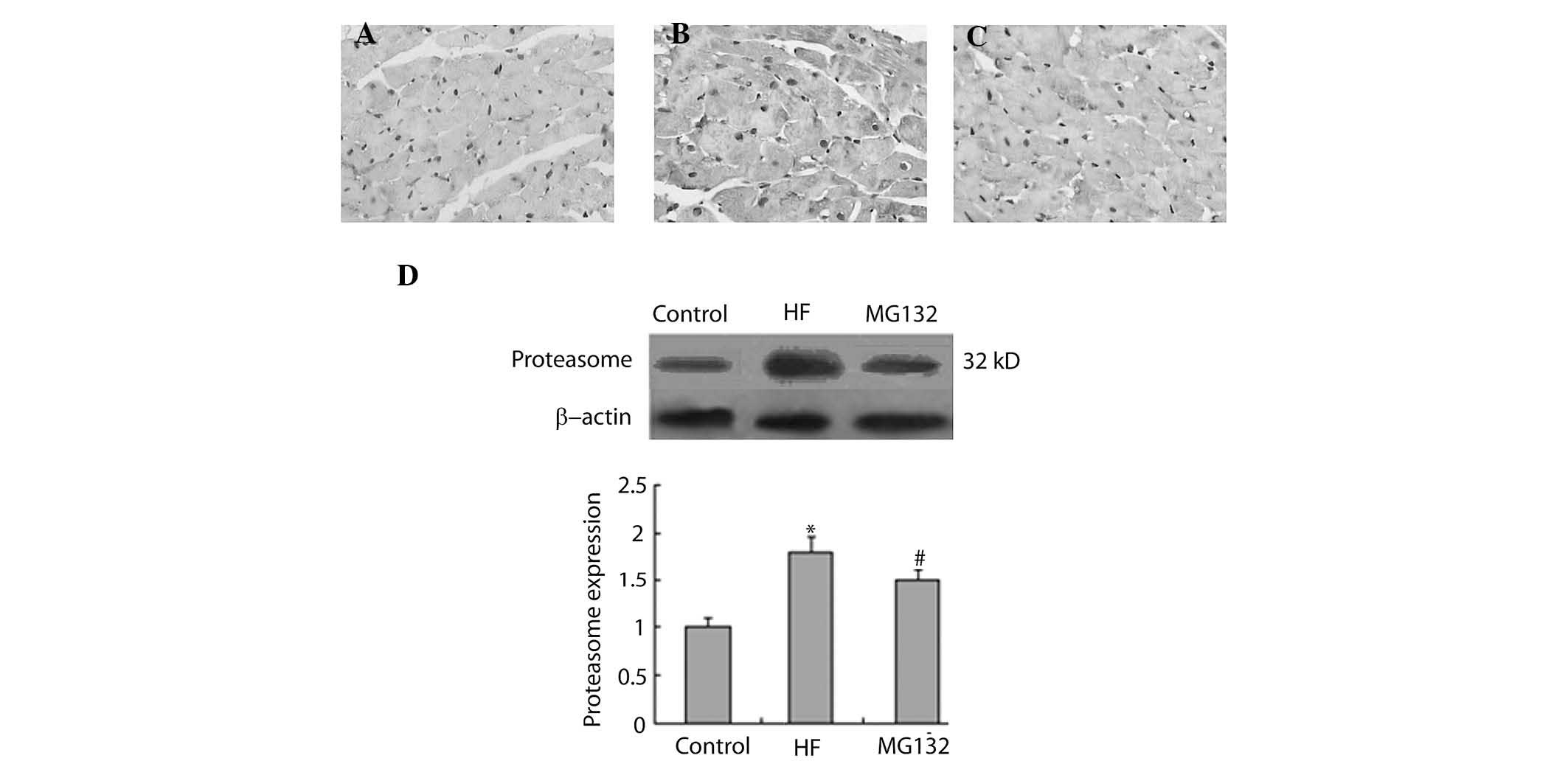
MG132 donwregulates the expression levels
of 20S proteasome and ubiquitin in HF rats
The present study then investigated the effect of
MG132 on the expression levels of 20S proteasome and ubiquitin in
the HF rats, using immunohistochemistry and western blot analysis.
The immunohistochemical analysis demonstrated that 20S proteasome
was widely expressed in each group (Fig. 5A–C). The expression of
proteasome-positive cells was higher in the HF group, compared with
the control group, and MG132 treatment downregulated the expression
of proteasome in the HF rats (Fig.
5A–C). Consistent with the immunohistochemistry, western blot
analysis revealed that the expression of proteasome was
significantly increased in the HF group, compared with the control
group, and that MG132 treatment decreased the expression of
proteasome in the HF rats (Fig.
5D).
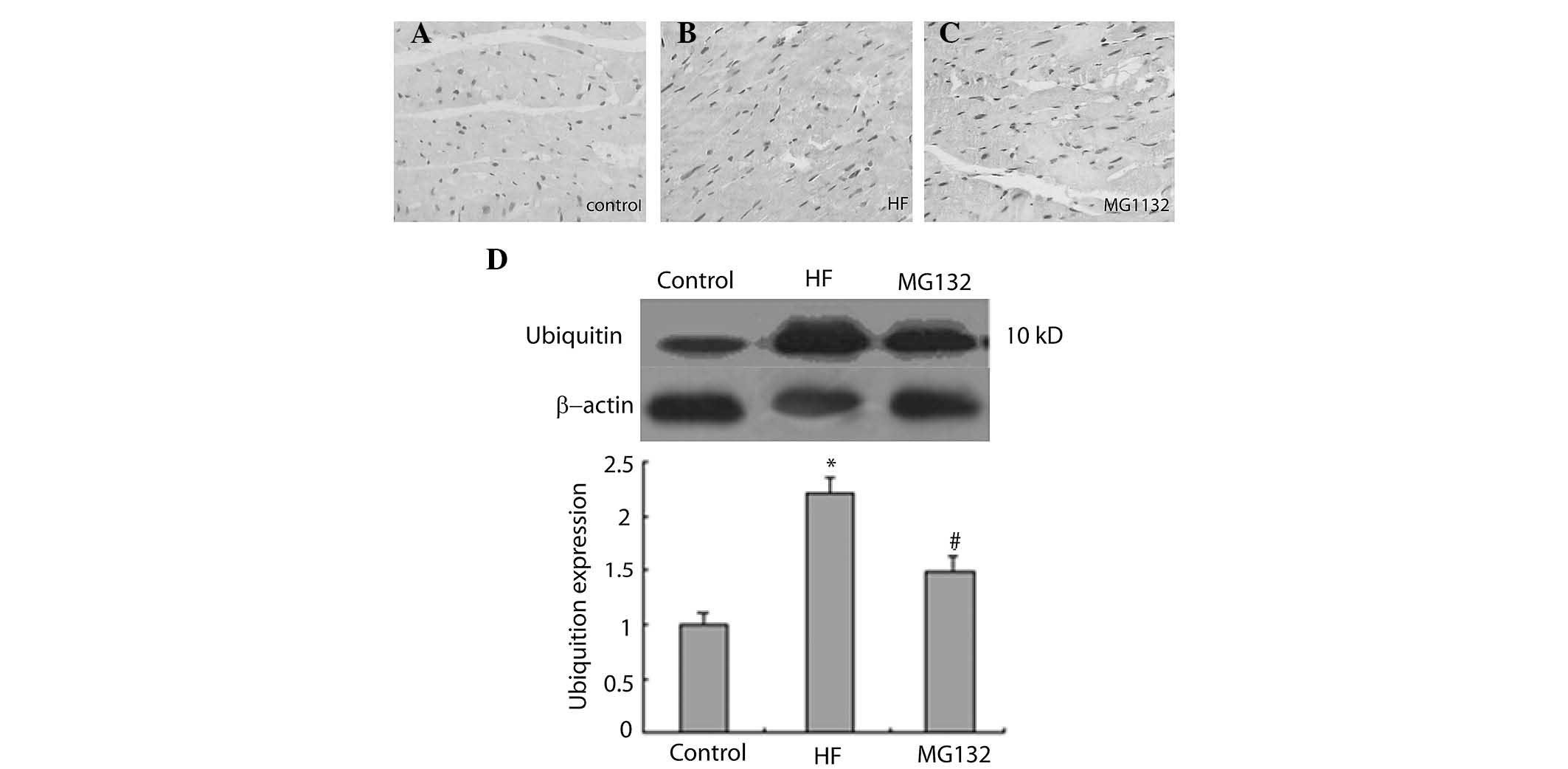
The immunohistochemical results revealed that
ubiquitin was predominantly expressed in the nuclei of the cells in
each group (Fig. 6A–C). The
intensity of ubiquitin immunostaining was more marked in the HF
groupn compared with the control group, and was weaker in the MG132
group, compared with the HF group (Fig. 6A–C). Consistent with this, western
blot analysis revealed that the expression of ubiquitin was
significantly increased in the HF group, compared with the control
group, and was decreased in the MG132 group, compared with the HF
group (Fig. 6D).
Discussion
Gap junctions have been well known to contribute to
cardiac arrhythmia (2), which is a
predominant cause of sudden death in HF. Previous animal and
clinical studies have found downregulation of Cx43 in failing
hearts (4–6). Although several lines of evidence
have demonstrated that proteasomal activities are required for the
degradation of Cx43, whether proteasome activities contribute to
the downregulation of Cx43 in HF remain to be elucidated. In the
present study, the role of proteasome in regulation of the
expression of Cx43 in rats with HF, induced by adriamycin, was
investigated using the MG132 proteasome inhibitor. The results
revealed that MG132 inhibited the expression of 20S proteasome and
ubiquitin, and reduced adriamycin-induced injury in the heart,
accompanied by upregulation in the expression of Cx43 and ZO-1.
These findings suggested that the inhibition of proteasome
upregulates the expression Cx43 in HF, and thus may prevent
Cx43-mediated arrhythmia.
In the present study, a rat model of heart failure
was produced by intraperitoneal injections of adriamycin at a
cumulative dose of 15 mg/kg. This treatment resulted in a decrease
in the cardiac contractile function, as indicated by a reduction in
EF and FS, and an increase in myocardial cell damage, as indicated
by H&E staining and electron microscopy, suggesting that HF was
successfully induced by adriamycin. This experimental model has
been used previously for investigating HF in rats, and the
myocardial lesion induced by adriamycin exhibits histological and
hemodynamic features similar to those reported in failing human
hearts (27–29). In the present study, MG132
treatment reduced myocardial damage in the HF rats, suggesting that
proteasome inhibition may be a therapeutic strategy for the
treatment of HF.
Several previous studies have demonstrated that the
expression of Cx43 shifts to the lateral membrane in hypertrophic
cardiomyopathy and myocardial ischemia (7,8).
Furthermore, it has been reported that Cx43 is downregulated in an
animal model of HF and in the failing human heart (4–6).
Consistent with these studies, the present study found that the
expression of Cx43 was significantly decreased in the HF rats. In
addition, it has been reported that the heterogeneous reduction of
Cx43 contributes to the development of malignant ventricular
tachycardia (9). In the present
study, MG132 increased the expression of Cx43 in HF rats, which was
demonstrated using immunohistochemistry and western blot analysis.
This observation that MG132 increased the expression of Cx43 in the
HF rats suggested that proteasome may mediate the heterogeneous
reduction of Cx43, and that inhibition of proteasome may prevent
ventricular tachycardia in HF.
The present study further investigated the
expression of ZO-1 in HF rats, and found that the expression of
ZO-1 was not restricted to the intercalated disc. Consistent with
this finding, several studies have reported that the expression of
ZO-1 is located in the intercalated disk and the lateral membrane
(11–13). Furthermore, it has been reported
that the expression of ZO-1 is reduced in the failing human heart,
accompanied by a decrease in the expression of Cx43 (18,19).
Consistent with these studies, the present study demonstrated that
ZO-1 was downregulated in the HF rats, accompanied by a
downregulation of Cx43, suggesting that ZO-1 may contribute to the
downregulation of Cx43 in HF. Several studies have reported that
ZO-1 can regulate the number, distribution and function of Cx43
(15–17). MG132 treatment significantly
increased the expression levels of ZO-1 and Cx43 in the HF rats,
suggesting that the proteasome-mediated regulation of the
interaction between Cx43 and ZO-1 may contribute to changes in the
expression of Cx43 in HF. In agreement with this hypothesis, it has
been reported that proteasome can regulate the expression of Cx43
via modulating the interaction of Cx43 with ZO-1 (24).
The ubiquitin-proteasome system is involved in the
internalization and degradation of Cx43 (20–22).
It has been reported that the expression and activity of proteasome
are increased during chronic pressure overload, thus leading to
ventricular hypertrophy (30). In
the present study, the expression of 20S proteasome and ubiquitin
was increased in the HF rats, accompanied by an increase in the
expression of Cx43, suggesting that an increase in the activity of
ubiquitin-proteasome system may contribute to the downregulation of
Cx43. Furthermore, the MG132 proteasome inhibitor inhibited the
expression of 20S proteasome and ubiquitin, and upregulated the
expression of Cx43 in the HF rats, further confirming the role of
the ubiquitin-proteasome system in regulating the expression of
Cx43 in HF.
In conclusion, the present study demonstrated that
the MG132 proteasome inhibitor inhibited the expression of 20S
proteasome and ubiquitin, and upregulated the expression of Cx43
and ZO-1 in rats with adriamycin-induced HF. These results suggest
that inhibition of the ubiquitin-proteasome system may effectively
prevent the degradation of Cx43 in HF, and thus may reduce the
occurrence of ventricular arrhythmia.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81300134), the
Research Fund for the Doctoral Program of Higher Education of China
(grant no. 20112307120014), and the Heilongjiang Province Natural
Science Foundation of China (grant no. H201443).
References
|
1
|
Packer M: Sudden unexpected death in
patients with congestive heart failure: A second frontier.
Circulation. 72:681–685. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jongsma HJ and Wilders R: Gap junctions in
cardiovascular disease. Circ Res. 86:1193–1197. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saffitz JE, Davis LM, Darrow BJ, Kanter
HL, Laing JG and Beyer EC: The molecular basis of anisotropy: Role
of gap junctions. J Cardiovasc Electrophysiol. 6:498–510. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dupont E, Matsushita T, Kaba RA, Vozzi C,
Coppen SR, Khan N, Kaprielian R, Yacoub MH and Severs NJ: Altered
connexin expression in human congestive heart failure. J Mol Cell
Cardiol. 33:359–371. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kostin S, Rieger M, Dammer S, Hein S,
Richter M, Klovekorn WP, Bauer EP and Schaper J: Gap junction
remodeling and altered connexin43 expression in the failing human
heart. Mol Cell Biochem. 242:135–144. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang X and Gerdes AM: Chronic pressure
overload cardiac hypertrophy and failure in guinea pigs: III.
Intercalated disc remodeling. J Mol Cell Cardiol. 31:333–343. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sepp R, Severs NJ and Gourdie RG: Altered
patterns of cardiac intercellular junction distribution in
hypertrophic cardiomyopathy. Heart. 76:412–417. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko
YS and Matsushita T: Gap junction alterations in human cardiac
disease. Cardiovasc Res. 62:368–377. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kitamura H, Ohnishi Y, Yoshida A, Okajima
K, Azumi H, Ishida A, Galeano EJ, Kubo S, Hayashi Y, Itoh H and
Yokoyama M: Heterogeneous loss of connexin43 protein in nonischemic
dilated cardiomyopathy with ventricular tachycardia. J Cardiovasc
Electrophysiol. 13:865–870. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hahn AF, Ainsworth PJ, Naus CC, Mao J and
Bolton CF: Clinical and pathological observations in men lacking
the gap junction protein connexin 32. Muscle Nerve Suppl. 9(Suppl):
S39–S48. 2000. View Article : Google Scholar
|
|
11
|
Gutstein DE, Liu FY, Meyers MB, Choo A and
Fishman GI: The organization of adherens junctions and desmosomes
at the cardiac intercalated disc is independent of gap junctions. J
Cell Sci. 116:875–885. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Patel VV, Kostetskii I, Xiong Y, Chu
AF, Jacobson JT, Yu C, Morley GE, Molkentin JD and Radice GL:
Cardiac-specific loss of N-cadherin leads to alteration in
connexins with conduction slowing and arrhythmogenesis. Circ Res.
97:474–481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sanford JL, Edwards JD, Mays TA, Gong B,
Merriam AP and Rafael-Fortney JA: Claudin-5 localizes to the
lateral membranes of cardiomyocytes and is altered in
utrophin/dystrophin-deficient cardiomyopathic mice. J Mol Cell
Cardiol. 38:323–332. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Toyofuku T, Yabuki M, Otsu K, Kuzuya T,
Hori M and Tada M: Direct association of the gap junction protein
connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem.
273:12725–12731. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hunter AW, Barker RJ, Zhu C and Gourdie
RG: Zonula occludens-1 alters connexin43 gap junction size and
organization by influencing channel accretion. Mol Biol Cell.
16:5686–5698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hunter AW, Jourdan J and Gourdie RG:
Fusion of GFP to the carboxyl terminus of connexin43 increases gap
junction size in HeLa cells. Cell Commun Adhes. 10:211–214. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin C, Martyn KD, Kurata WE, Warn-Cramer
BJ and Lau AF: Connexin43 PDZ2 binding domain mutants create
functional gap junctions and exhibit altered phosphorylation. Cell
Commun Adhes. 11:67–87. 2004. View Article : Google Scholar
|
|
18
|
Bruce AF, Rothery S, Dupont E and Severs
NJ: Gap junction remodelling in human heart failure is associated
with increased interaction of connexin43 with ZO-1. Cardiovasc Res.
77:757–765. 2008. View Article : Google Scholar
|
|
19
|
Laing JG, Saffitz JE, Steinberg TH and
Yamada KA: Diminished zonula occludens-1 expression in the failing
human heart. Cardiovasc Pathol. 16:159–164. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Laing JG, Tadros PN, Westphale EM and
Beyer EC: Degradation of connexin43 gap junctions involves both the
proteasome and the lysosome. Exp Cell Res. 236:482–492. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leithe E and Rivedal E: Epidermal growth
factor regulates ubiquitination, internalization and
proteasome-dependent degradation of connexin43. J Cell Sci.
117:1211–1220. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Musil LS, Le AC, VanSlyke JK and Roberts
LM: Regulation of connexin degradation as a mechanism to increase
gap junction assembly and function. J Biol Chem. 275:25207–25215.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berthoud VM, Tadros PN and Beyer EC:
Connexin and gap junction degradation. Methods. 20:180–187. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Girao H and Pereira P: The proteasome
regulates the interaction between Cx43 and ZO-1. J Cell Biochem.
102:719–728. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leithe E and Rivedal E: Ubiquitination and
down-regulation of gap junction protein connexin-43 in response to
12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem.
279:50089–50096. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Borer JS, Jason M, Devereux RB, Fisher J,
Green MV, Bacharach SL, Pickering T and Laragh JH: Function of the
hypertrophied left ventricle at rest and during exercise.
Hypertension and aortic stenosis. Am J Med. 75:34–39. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mettler FP, Young DM and Ward JM:
Adriamycin-induced cardiotoxicity (cardiomyopathy and congestive
heart failure) in rats. Cancer Res. 37:2705–2713. 1977.PubMed/NCBI
|
|
28
|
Rabelo E, De Angelis K, Bock P, Gatelli
Fernandes T, Cervo F, Bellό Klein A, Clausell N and Cláudia
Irigoyen M: Baroreflex sensitivity and oxidative stress in
adriamycin-induced heart failure. Hypertension. 38:576–580. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Singal PK, Iliskovic N, Li T and Kumar D:
Adriamycin cardiomyopathy: Pathophysiology and prevention. Faseb J.
11:931–936. 1997.PubMed/NCBI
|
|
30
|
Depre C, Wang Q, Yan L, Hedhli N, Peter P,
Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, et al:
Activation of the cardiac proteasome during pressure overload
promotes ventricular hypertrophy. Circulation. 114:1821–1828. 2006.
View Article : Google Scholar : PubMed/NCBI
|