Introduction
Primary spontaneous pneumothorax [PSP; Online
Mendelian Inheritance in Man (OMIM)#173600] occurs in patients
without clinically apparent underlying lung disease. Of PSP cases,
~10% are caused by germline mutations in the FLCN gene
(1,2), which is known to cause Birt-Hogg-Dube
syndrome (BHD; OMIM#135150), an autosomal dominant condition
characterized by skin fibrofolliculomas, pulmonary
cysts/spontaneous pneumothorax and renal cancers (3,4).
FLCN mutation may result in isolated PSP with no skin or
renal manifestations, presumably due to an incomplete penetration,
as has been reported in multiple previous case studies (1,2,5–7).
Lung cysts are the common pathogenic ground of PSP
(8,9). Multiple lung cysts in patients with
FLCN mutations are often observed randomly and bilaterally
distributed in the lung, particularly in its lower portion
(10–13). By contrast, apical bullae are often
observed in patients with sporadic PSP without FLCN
mutations (14–16). Notably, FLCN mutation-like
lung cysts are also observed in a significant portion of
non-FLCN mutant sporadic PSP cases (6). Whether these cases are in any way
associated with FLCN disruption remains to be
elucidated.
The FLCN gene (OMIM#607273) encodes an
evolutionarily conserved protein, folliculin, with no apparent
functional motif currently recognized. The majority of the
pathogenic FLCN mutations, identified to date, resulted in
premature truncation of the protein (http://www.skingene-database.com/) (17). FLCN missense mutations and
small in-frame deletions were reported to predominantly disrupt
protein stability and lead to significant reductions in the
expression of FLCN (18).
No evidence for a dominant negative effect of FLCN mutants
was observed in the transfected cells. Additionally, large
intragenic deletions spanning the putative FLCN promoter
region have been reported in families with BHD. Luciferase reporter
assays demonstrated that a deletion of the putative promoter
dramatically reduced the gene expression in vitro (19). In addition, FLCN
inactivation caused by promoter methylation has also been detected
in types of renal tumor (20,21).
This raised the possibility that epigenetic regulation of
FLCN may contribute to the pathology of BHD.
The present study selected 71 patients with PSP, who
harbored a FLCN mutation-like lung phenotype, however,
exhibited no germline and somatic mutations in the FLCN
coding regions. Significant variations in the expression of
FLCN were observed in the lung cysts of these patients, when
compared with those of the patients with BHD and the controls. It
was hypothesized that transcriptional irregulation of FLCN
may be an important mechanism contributing to the development of
lung cysts and, subsequently, PSP. The present study aimed to
search for epigenetic variations in the putative promoter of
FLCN in the patients with PSP.
Materials and methods
Patients
The present study was approved by the ethics
committees of Nanjing University Medical School, Nanjing Chest
Hospital and Taizhou Hospital of Zhejiang Province. The 71 selected
patients with PSP included 69 sporadic patients and 2 patients with
a family history. The patients were clinically diagnosed with PSP
on the basis of a thorax computed tomography scan and underwent
surgeries for the treatment of pneumothorax at two tertiary
hospitals, Taizhou Hospital of Zhejiang Province and Nanjing Chest
Hospital. The patients were enrolled for the present study since
they exhibited FLCN-like multiple lung cysts, however,
exhibited no mutations in the FLCN coding region. A thorough
screen for skin and renal abnormalities was performed by cutaneous
examination and abdominal ultrasonography, respectively. Peripheral
blood samples and tissue samples derived from the clinically
resected lung lesions were collected. The control group included 11
morphologically normal lung tissues, which were obtained from
patients with stage I non-small cell lung carcinomas that underwent
lobectomy. These samples were obtained at least 5 cm from the tumor
locus. Written informed consent was obtained from all patients
involved in the present study.
DNA sequencing
Genomic DNA was extracted using the DNeasy Blood and
Tissue kit (Qiagen, Hilden, Germany). The FLCN exons and
flanking intron regions were sequenced, as previously reported
(1). The PCR products were
amplified using the BigDye Terminator kit (Applied Biosystems Life
Technologies, Foster City, CA, USA) and sequenced on an ABI 3130
Genetic Analyzer (Applied Biosystems Life Technologies). The data
were analyzed by referring to the reference sequence (NM_144997.5)
obtained from the NCBI database (http://www.ncbi.nlm.nih.gov/gene/201163#reference-sequences).
Haplotype analysis
Haplotype analysis was performed in the family of
patient F260. A total of 16 individuals were geno-typed, among
which 7 were affected and 9 were unaffected. Eight microsatellite
markers were used, which spanned an 11.4-cM distance flanking the
FLCN locus on chromosome 17, including D17S799, D17S921,
D17S122, D17S1857, D17S740, D17S2196, D17S2187 and D17S798.
Haplotypes were determined from the genotype order in which the
least number of recombinants occurred.
Multiplex ligation-dependent probe
amplification assay (MLPA)
A total of two sets of MLPA assays were used to
detect deletions/duplications in FLCN and its up/downstream
regions. The commercial kit, P256-B1 FLCN (MRC-Holland, Amsterdam,
Netherlands), contained probes targeting all 14 exons. Another MLPA
kit was synthesized (available on request) with 15 probes targeting
9 kb upstream and 5 kb downstream of the gene. The MLPA reactions
were performed, according to the manufacturer's instructions. The
data were analyzed using the Coffalyser, NET software (version 9.0;
MRC-Holland). The patient data was normalized against those of the
control individuals. Threshold values were set at 0.7–1.4 for
normal, <0.4 for deletion and >1.5 for duplication.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was extracted from the lung tissues
using an RNeasy Mini kit (Qiagen). The RNA was reverse transcribed
into cDNA using PrimeScript RT reagent kit (Takara Bio, Inc.,
Dalian, China). The mRNA expression of FLCN was determined
using FastStart TaqMan Probe Master (Roche, Basel, Switzerland) on
a StepOne Real-Time PCR system (Applied Biosystems Life
Technologies). Probe #63 (cat. no. 4688627001; Roche) and the
intron-spanning primer pair, Forward: 5′-GGA CCA GTG CCT CGT CTG-3′
and reverse: 5′-GGT GAA CTT AAA AAG CAC CTTT CA-3′, were selected
for FLCN. GAPDH was selected as a control gene for
normalization. The data were analyzed using the 2−ΔΔCt
method (22). Each sample was run
in triplicate and all reactions were performed twice.
Western blot analysis
Lung cyst samples were homogenized using a dounce
homogenizer (Corning, Shanghai, China) and suspended in ice cold
lysis buffer containing protease inhibitors (Beyotime Institute of
Biotechnology, Shanghai, China). The homogenates were centrifuged
at 13,000 × g for 20 min at 4°C. The protein concentrations of the
supernatants were quantified using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology). A total of 100 mg protein
was separated by 12% SDS-PAGE and transferred onto immobilon
membranes (Millipore, Bedford, MA, USA). The membranes were blocked
with 5% non-fat milk in Tris-buffered saline with 0.05% Tween20
(TBST; Millipore) at room temperature and then washed with TBST for
15 min three times. The membranes were incubated overnight at 4°C
with the primary antibodies rabbit anti-FLCN (D14G9) monoclonal
antibody (mAb) (cat no. 3697; Cell Signaling Technology, Beverly,
MA, USA; 1:1,000 dilution) and rabbit β-actin (13E5) mAb (cat no.
4970; Cell Signaling Technology; 1:1,000 dilution). Blots were
washed in TBST for 15 min three times and subsequently incubated
with horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G (cat no. ab6721; Abcam, Cambridge, UK; 1:3,000
dilution). Immunoreactive bands were detected using an eECL western
blot kit (CWBio, Beijing, China). The images of the bands were
captured using a digital G-box chemiluminescent imaging system
(Syngene, Frederick, MD, USA). Each sample was analyzed at least
twice.
Methylation analysis
The CpG island region in the FLCN gene was
predicted using the University of California Santa Cruz (UCSC)
genome browser (http://genome.ucsc.edu) and CpGplot software (version
EMBOSS 6.6.0.0; http://www.ebi.ac.uk). A panel of 13
pyrosequencing assays were performed to quantify the methylation
value of 69 CpG islands for each sample. The primers for
amplification and pyrosequencing were designed using PyroMark assay
design software v.2.0 (Qiagen) (Table
I). A total of 500 ng DNA sample was treated with EZ DNA
methylation-Gold kit (Zymo Research, Orange, CA, USA), and the
converted DNA was amplified using a PyroMark PCR kit (Qiagen),
according to the manufacturer's instruction. The PCR products were
pyrosequenced and the data were analyzed on a PyroMark Q96 ID
(Qiagen). A value of 6% was set as a convincing absolute threshold
for methylation, according to manufacturer's instructions and
suggestions from a previous study (23).
 | Table IPrimers for pyrosequencing analysis
of FLCN. |
Table I
Primers for pyrosequencing analysis
of FLCN.
| Primer ID | Forward
(5′-3′) | Reverse
(5′-3′)a | Pyrosequencing
primer |
|---|
| 1 |
AAAAAGATTTATTGAGGGAGGAAGA |
TCCCTACATTAAAAATATAAAAAATAC |
AAAAAGATTTATTGAGGGAGGAAGA |
| 2 |
AGGGATGTAATTTTTATATTTTTTAT |
TCTTCCTCCCTCAATAAATCTTTTT |
AGGGATGTAATTTTTATATTTTTTAT |
| 3 |
GTTTTTTTTAGTATTTTTAGTTGGTG |
CAAACCCAAAAACACAATCC |
GTTTTTTTTAGTATTTTTAGTTGGTG |
| 4 |
GTTTTTTTTAGTATTTTTAGTTGGTG |
CAAACCCAAAAACACAATCC |
GGTGGAAAAGGGTAATTGT |
| 5 |
GGTTTTTGTTTTTTAGTTAGATTT |
TACCACCACCCAACACT |
GGTTTTTGTTTTTTAGTTAGATTT |
| 6 |
GGTTTTTGTTTTTTAGTTAGATTT |
TACCACCACCCAACACT |
GAGAGTTAGGGAGGTT |
| 7 |
GTAGTGTTGGGTGGTGGTA |
ACCCAAAACCCCCAAACC |
GTAGTGTTGGGTGGTGGTA |
| 8 |
GTAGTGTTGGGTGGTGGTA |
ACCCAAAACCCCCAAACC |
CCAAACCCCCAAAACCCA |
| 9 |
GTAGTGTTGGGTGGTGGTA |
ACCCAAAACCCCCAAACC |
ACCAAAACAATCAAAAAAACTCTC |
| 10 |
GGGGTTTTGGGTGTGATTT |
CCTAAACCCTCCCCCATTACT |
GGGGTTTTGGGTGTGATTT |
| 11 |
GGGGTTTTGGGTGTGATTT |
CCTAAACCCTCCCCCATTACT |
GGGGTTGGGATTTAGAG |
| 12 |
GGGGTTTTGGGTGTGATTT |
CCTAAACCCTCCCCCATTACT |
GATTTGGGGAGTAGGTAGG |
| 13 |
GATTTGGGGAGTAGGTAGG |
ATCCTCATCCCCATAAACTT |
GAGGGTTTAGGGTTAGAGTTTAGG |
Combined bisulphite restriction analysis (COBRA) was
further performed, as described previously (24). A 281 bp sequence within the CpG
islands, including 26 CpGs (CpG28-53), were amplified by
semi-nested PCRs with no bias towards methylated or unmethylated
templates. The PCR products were treated with the restriction
endonucleases, TaqI (Promega, Madison, WI, USA) and
BstUI (BioLabs, Ipswich, MA, USA), and were subsequently
separated by gel electrophoresis to detect the digestion products.
The universal methylated human DNA standard (Zymo Research) was
used as a positive control.
Statistical analysis
All statistical analyses were performed using the
SPSS statistics 17.0 software (SPSS, Inc., Chicago, IL, USA). For
mRNA expression, the statistical significance was determined using
unpaired, two-tailed Student's t-test. For the DNA methylation
status, the statistical significance was evaluated using the
Mann-Whitney U-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
FLCN irregulation is observed in lung
cysts of PSP
To determine whether the FLCN mutation-like
lung phenotype was associated with an alteration in the expression
of FLCN, the mRNA expression levels of FLCN in lung
cyst lesions from the 71 patients with PSP were determined. A
wide-spread distribution of the mRNA expression levels of
FLCN was observed, when compared with those of the BHD and
control groups (Table II;
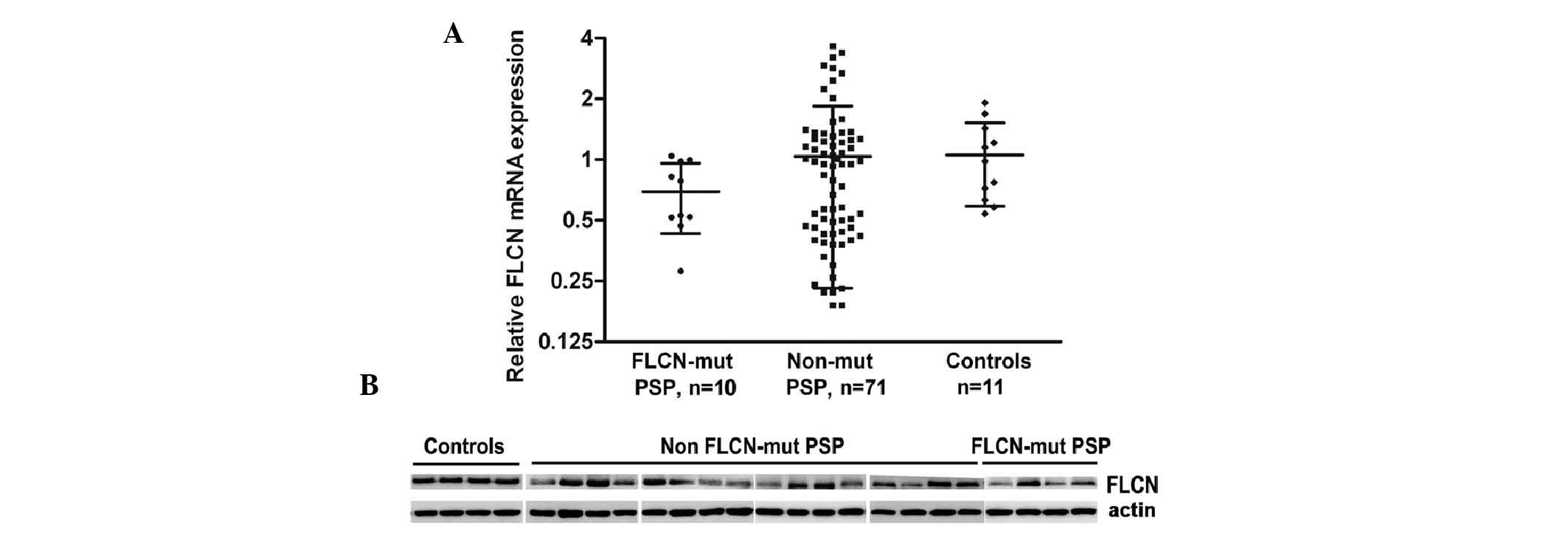
Fig. 1A). As seen in Fig. 1A, the mRNA expression levels of
FLCN in lung cyst lesions from the patients with BHD were
reduced (0.69±0.26), suggesting that certain mutant transcripts may
be unstable. Notably, the levels of the FLCN transcript from
the 71 patients with PSP may be divided in to three groups: Low,
medium and high. About one third of the patients with PSP (35.2%,
25/71) exhibited a 2–5 fold decrease in the mRNA expression of
FLCN, with the group average of 0.37±0.11, when compared
with that of the control group (1.06±0.47). Additionally, ~12.7%
patients (9/71) revealed an increase in the mRNA expression of
FLCN in the cyst lesions (2.83±0.54). Similar changes were
observed on the protein expression of FLCN (Table II; Fig. 1B). In ~80% of the patients, the
tendency of the changes in the mRNA and protein expression levels
of FLCN was consistent. This data argued that FLCN
irregulation may be associated with the pathogenesis of lung cysts
in the patients with sporadic PSP.
| Table IImRNA and protein expression levels of
LCN in 71 patients with PSP without FLCN coding
mutations. |
Table II
mRNA and protein expression levels of
LCN in 71 patients with PSP without FLCN coding
mutations.
| Group | n | mRNA level | Protein level |
|---|
| FLCN-low
PSP | 25 | 0.37±0.11 | 0.39±0.40 |
| FLCN-normal
PSP | 37 | 1.05±0.29 | 0.92±0.57 |
| FLCN-high
PSP | 9 | 2.83±0.54 | 2.02±1.12 |
| Control lung
tissue | 11 | 1.06±0.47 | 1.01±0.25 |
Hypoid insufficiency of FLCN is
pathogenic
To investigate the mechanism of FLCN
irregulation in the patients with PSP, the sequence variations in
the cis-regulatory region of FLCN were initially identified.
A 1.6 kb upstream fragment, including the 5′-flank (650 bp),
non-coding exon 1 (228 bp) and partial intron 1 (700 bp) of
FLCN, was sequenced. No sequence mutation was identified in
any of the 71 patients. A total of three common single nucleotide
polymorphisms were confirmed, including rs1708629, rs1736209 and
rs1736208.
The samples were subsequently investigated by MLPA
analysis for large deletions/duplications. An FLCN
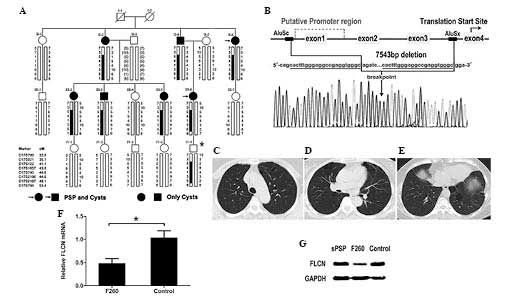
intragenic deletion was detected in patient F260, with a positive
family history. Pedigree and haplotype analysis revealed that the
deletion co-segregated among the affected individuals in the family
(Fig. 2A). PCR amplification and
bidirectional sequencing of the junction fragment revealed a 7,543
bp deletion, including the non-coding exons 1–3 and a 1.3 kb
upstream sequence (Fig. 2B). The
deletion resulted in the removal of the putative FLCN
promoter region. All family members harboring the deletion
exhibited multiple lung cysts, basally located or randomly
distributed on the lung (Fig.
2C–E), and two experienced pneumothorax. These phenotypic
features were similar with the clinical manifestations of patients
with pathogenic FLCN mutations.
To confirm that this heterozygous deletion disrupted
the transcription of the mutant allele of FLCN, the
expression of FLCN in patient F260 was determined. About a
50% reduction in the level of FLCN was observed in lung cyst
lesions from patient F260 at the mRNA (47.4%) and protein (43.5%)
expression levels, as compared with those of the controls (Fig. 2F and G). These results suggested
that FLCN insufficiency caused by the heterozygous FLCN
promoter deletion is pathogenic in the development of lung
cysts.
Smallest region of overlap (SRO) for the
FLCN promoter deletions contains CpG islands
The FLCN promoter appeared to be a
recombination hotspot, in which several large deletions were
identified in families with BHD (19). Within the SRO (1,893 bp) for all
deletions, a CpG-enriched sequence was predicted by the UCSC genome
browser and CpGPlot software (Fig.
3A). The CpG islands, located −628 to +335 of the transcription
starting site, encompass the 5′-flank, exon 1 and partial intron 1,
with 72 CpG loci (Fig. 3B). This
region is predicted to contain multiple putative regulatory
elements, including Sp1, AP-2, GCF and Early-Seq1 (Promoter Scan
software, http://www-bimas.cit.nih.gov). It was speculated that
the methylation status of the CpG islands may potentially affect
the binding of transcription factors and consequently result in the
alteration of the expression of FLCN.
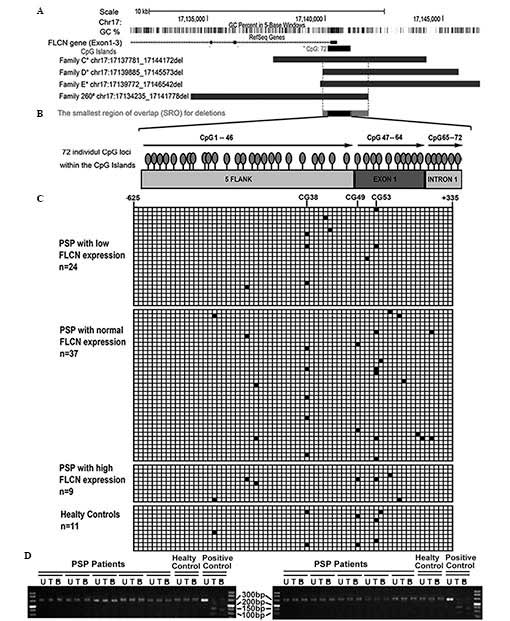 | Figure 3Methylation status of the FLCN
promoter in patients with PSP. (A) The genomic location of the
FLCN exons 1–3, CpG Islands, deletions identified and the
smallest region of overlap on chromosome 17p11.2. The information
refers to the GRCh37/hg19 annotation in the UCSC and GenBank
database. *Families C, D and E were previously reported
(19). #Family 260 was
identified in the present study. (B) The CpG Islands encompass the
5′ flank, exon 1 and partial intron 1 of FLCN, and includes
72 CpG loci. (C) The methylation status of 70 patients and 11
controls was assessed. Each line represents a subject analyzed by
pyrosequencing, and each row represents a CpG locus. The black
boxes indicate a methylated CpG island (value, ≥6%) and white boxes
indicate unmethylated CpG islands. No significant difference in the
methylation of the FLCN promoter was identified between the
patients and the controls. (D) Representative data of TaqI
and BstUI restriction analysis of the COBRA PCR products in
the patients and the controls. The absence of digestion products
indicated that the originally unmethylated restriction sites of
TaqI (tcga) and BstUI (cgcg) were lost due to
bisulphite conversion. All samples tested were, therefore,
unmethylated. The positive control was the universal methylated
human DNA standard. PCR, polymerase chain reaction; PSP, PSP,
primary spontaneous pneumothorax; U, PCR products without
digestion; T, PCR products digested by Taql; B: PCR products
digested by BstUI. |
Promoter methylation is not associated
with FLCN irregulation
The methylation status of the FLCN promoter
in 70 patients and 11 controls was quantitatively analyzed by
pyrosequencing. No statistical difference in the methylation
pattern of the FLCN CpG islands was observed between
different groups of patients with PSP and the controls. Dense
methylation across the CpG islands was not observed in any sample
analyzed, neither patients nor the controls (Fig. 3C). For each sample, >95% of the
CpG loci were unmethylated. Low methylation values, often <15%,
at seldom individual loci (CpG38,49,53) were detected in a few
samples, however, none were demonstrated to be associated with the
expression of FLCN. The methylated CpGs were often separated
by neighboring unmethylated CpGs. In short, no correlation between
methylation levels and the expression of FLCN was
established.
To further confirm the pyrosequencing results, COBRA
methylation analysis of the CpG islands was performed in all
patients and controls. The 281 bp amplified region (CpG28-53)
included two TaqI sites (tcga) and BstUI (cgcg)
sites, however, following bisulphite treatment, no digestion
products were observed in any sample. The absence of the digestion
products of restriction endonucleases demonstrated that the
originally unmethylated sites were lost due to bisulphite
conversion. All the CpG loci analyzed were unmethylated.
Discussion
PSP is a clinical hallmark of BHD syndrome, since
lung cysts/spontaneous pneumothorax are the most frequent and the
initial presenting manifestation compared with the skin or renal
features (10–12). Up to 80% of patients with BHD were
identified to exhibit multiple lung cysts, and 24% experienced
pneumothorax. Our previous study demonstrated that ~10% of PSP
cases are caused by germline mutations in the FLCN gene
(2). However, more sporadic PSP
cases with FLCN-like lung cysts, however, no FLCN
mutations, were observed. Whether an FLCN-associated
mechanism was involved in these cases, and in addition, whether a
mechanism other than genetic defect, including epigenetic
alteration, is responsible for the development of FLCN-like
lung cysts are interesting and worthy of further
investigations.
Previous studies of BHD are predominantly focused on
the germline mutations in the FLCN coding region. In the
present study, a heterozygous FLCN promoter deletion was
identified in a family with PSP exhibiting characteristic
FLCN-like lung cysts. A reduced expression of FLCN
was observed in the lesion tissues, consistent with a previous
study, which assessed the FLCN promoter function in
vitro (19). The present study
provided further evidence to support the notion that the
downregulation of FLCN is pathogenic in lung cyst formation.
Epigenetic mechanisms have long been associated with gene
irregulation and human disease (25). In renal tumors, the involvement of
FLCN promoter methylation has been hypothesized (20,21),
however, inconsistent observations were reported (24,26).
No previous studies have investigated the association between
FLCN promoter methylation and the development of lung cysts.
The present study observed significant variability in the
expression of FLCN in the lung lesions of patients with
non-FLCN mutant sporadic PSP, however, demonstrated no
evidence for the association of FLCN promoter methylation
with these cases. The lack of FLCN promoter methylation in
patients with PSP with FLCN-like lung cysts is a valuable
observation, which provided evidence to refocus our future
research. Future studies targeting both genetic and epigenetic
mechanisms are required to elucidate the molecular nature of
FLCN irregulation in PSP.
Acknowledgments
The authors would like to thank the patients and
family members involved in the present study. This study was
supported by grants from the National Basic Research Program of
China (nos. 2010CB945103 and 2009CB918704), the National Natural
Science Foundation of China (nos. 81030013, 81170002 and 81471095),
the Science and Technology Project of Jiangsu Province (no.
BL2014053), the Ph.D. Programs Foundation of Ministry of Education
of China (no. 20110091120032), the Jiangsu Province Health
Department of Medicine leading talent and innovation team project
(no. LJ201109) and the Foundation of Nanjing Municipal Public
Health Bureau (no. ZKX11030).
References
|
1
|
Graham RB, Nolasco M, Peterlin B and
Garcia CK: Nonsense mutations in folliculin presenting as isolated
familial spontaneous pneumothorax in adults. Am J Respir Crit Care
Med. 172:39–44. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ren HZ, Zhu CC, Yang C, Chen SL, Xie J,
Hou YY, Xu ZF, Wang DJ, Mu DK, Ma DH, et al: Mutation analysis of
the FLCN gene in Chinese patients with sporadic and familial
isolated primary spontaneous pneumothorax. Clin Genet. 74:178–183.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nickerson ML, Warren MB, Toro JR,
Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P,
Pavlovich CP, et al: Mutations in a novel gene lead to kidney
tumors, lung wall defects and benign tumors of the hair follicle in
patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2:157–164.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Menko FH, van Steensel MA, Giraud S,
Friis-Hansen L, Richard S, Ungari S, Nordenskjöld M, Hansen TV,
Solly J, Maher ER, et al: Birt-Hogg-Dubé syndrome: Diagnosis and
management. Lancet Oncol. 10:1199–1206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Painter JN, Tapanainen H, Somer M,
Tukiainen P and Aittomäki K: A 4-bp deletion in the Birt-Hogg-Dubé
gene (FLCN) causes dominantly inherited spontaneous pneumothorax.
Am J Hum Genet. 76:522–527. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gunji Y, Akiyoshi T, Sato T, Kurihara M,
Tominaga S, Takahashi K and Seyama K: Mutations of the
Birt-Hogg-Dube gene in patients with multiple lung cysts and
recurrent pneumothorax. J Med Genet. 44:588–593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kunogi M, Kurihara M, Ikegami TS,
Kobayashi T, Shindo N, Kumasaka T, Gunji Y, Kikkawa M, Iwakami S,
Hino O, et al: Clinical and genetic spectrum of Birt-Hogg-Dube
syndrome patients in whom pneumothorax and/or multiple lung cysts
are the presenting feature. J Med Genet. 47:281–287. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abdala OA, Levy RR, Bibiloni RH, Viso HD,
De Souza M and Satler VH: Advantages of video assisted thoracic
surgery in the treatment of spontaneous pneumothorax. Medicina (B
Aires). 61:157–160. 2001.In Spanish.
|
|
9
|
Chen YJ, Luh SP, Hsu KY, Chen CR, Tsao TC
and Chen JY: Video-assisted thoracoscopic surgery (VATS) for
bilateral primary spontaneous pneumothorax. J Zhejiang Univ Sci B.
9:335–340. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Toro JR, Pautler SE, Stewart L, Glenn GM,
Weinreich M, Toure O, Wei MH, Schmidt LS, Davis L, Zbar B, et al:
Lung cysts, spontaneous pneumothorax and genetic associations in 89
families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med.
175:1044–1053. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Toro JR, Wei MH, Glenn GM, Weinreich M,
Toure O, Vocke C, Turner M, Choyke P, Merino MJ, Pinto PA, et al:
BHD mutations, clinical and molecular genetic investigations of
Birt-Hogg-Dubé syndrome: A new series of 50 families and a review
of published reports. J Med Genet. 45:321–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sahn SA and Heffner JE: Spontaneous
pneumothorax. N Engl J Med. 342:868–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Grant LA, Babar J and Griffin N: Cysts,
cavities and honeycombing inmultisystem disorders: Differential
diagnosis and findings on thin-section CT. Clin Radiol. 64:439–448.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guimaraes CV, Donnelly LF and Warner BW:
CT findings for blebs and bullae in children with spontaneous
pneumothorax and comparison with findings in normal age-matched
controls. Pediatr Radiol. 37:879–884. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jordan KG, Kwong JS, Flint J and Müller
NL: Surgically treated pneumothorax. Radiologic, pathologic
findings. Chest. 111:280–285. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shih CH, Yu HW, Tseng YC, Chang YT, Liu CM
and Hsu JW: Clinical manifestations of primary spontaneous
pneumothorax in pediatric patients: An analysis of 78 patients.
Pediatr Neonatol. 52:150–154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lim DH, Rehal PK, Nahorski MS, Macdonald
F, Claessens T, Van Geel M, Gijezen L, Gille JJ, Giraud S, Richard
S, et al: A new locus-specific database (LSDB) for mutations in the
folliculin (FLCN) gene. Human Mutat. 31:E1043–E1051. 2010.
View Article : Google Scholar
|
|
18
|
Nahorski MS, Reiman A, Lim DH, Nookala RK,
Seabra L, Lu X, Fenton J, Boora U, Nordenskjöld M, Latif F, et al:
Birt Hogg-Dubé syndrome-associated FLCN mutations disrupt protein
stability. Hum Mutat. 32:921–929. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Benhammou JN, Vocke CD, Santani A, Schmidt
LS, Baba M, Seyama K, Wu X, Korolevich S, Nathanson KL, Stolle CA
and Linehan WM: Identification of intragenic deletions and
duplication in the FLCN gene in Birt-Hogg-Dubé syndrome. Genes
chromosomes cancer. 50:466–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khoo SK, Kahnoski K, Sugimura J, Petillo
D, Chen J, Shockley K, Ludlow J, Knapp R, Giraud S, Richard S, et
al: Inactivation of BHD in sporadic renal tumors. Cancer Res.
63:4583–4587. 2003.PubMed/NCBI
|
|
21
|
Gatalica Z, Lilleberg SL, Vranic S,
Eyzaguirre E, Orihuela E and Velagaleti G: Novel intronic germline
FLCN gene mutation in a patient with multiple ipsilateral renal
neoplasms. Human pathol. 40:1813–1819. 2009. View Article : Google Scholar
|
|
22
|
Hasumi Y, Baba M, Ajima R, Hasumi H,
Valera VA, Klein ME, Haines DC, Merino MJ, Hong SB, Yamaguchi TP,
et al: Homozygous loss of BHD causes early embryonic lethality and
kidney tumor development with activation of mTORC1 and mTORC2. Proc
Natl Acad Sci USA. 106:18722–18727. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hansmann T, Pliushch G, Leubner M, Kroll
P, Endt D, Gehrig A, Preisler-Adams S, Wieacker P and Haaf T:
Constitutive promoter methylation of BRCA1 and RAD51C in patients
with familial ovarian cancer and early-onset sporadic breast
cancer. Hum Mol Genet. 21:4669–4679. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
da Silva NF, Gentle D, Hesson LB, Morton
DG, Latif F and Maher ER: Analysis of the Birt-Hogg-Dubé (BHD)
tumour suppressor gene in sporadic renal cell carcinoma and
colorectal cancer. J Med Genet. 40:820–824. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
El-Osta A and Wolffe AP: DNA methylation
and histone deacetylation in the control of gene expression: Basic
biochemistry to human development and disease. Gene Expr. 9:63–75.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kahnoski K, Khoo SK, Nassif NT, Chen J,
Lobo GP, Segelov E and Teh BT: Alterations of the Birt-Hogg-Dubé
gene (BHD) in sporadic colorectal tumours. J Med Genet. 40:511–515.
2003. View Article : Google Scholar : PubMed/NCBI
|