Introduction
Prostate cancer is one of the most common types of
non-cutaneous malignancy in males. In the United States, the
incidence of prostate cancer is highest among male malignant tumors
(1). In China, increasing evidence
suggests that the age of onset of prostate cancer has been
decreasing in recent years (2).
Although early-stage prostate cancer can be cured by radical
prostatectomy, for patients with clinically detectable metastasis,
there remains a lack of effective treatment options (3).
Intratumoral hypoxia prevails in several types of
fast growing solid tumor and is a marker of poor clinical prognosis
in prostate cancer (4). The
reaction of tumor cells to hypoxia includes the generation of
signal transducing molecules, predominantly growth factors and
cytokines, and changes in tumor cell function, which affect cell
proliferation, de-differentiation, resistance to apoptosis and
metastatic potential (5).
Chemokines are small, chemoattractant proteins,
which have the ability to chemoattract cells expressing their
cognate G protein-coupled receptors. On the basis of structure,
chemokines are divided into four subfamilies: CXC, CC, C and CX3C
(6). Several studies have
suggested that chemokines and their receptors are involved in a
number of disease states, including cardiovascular disease
(7), systemic inflammation
(8), cancer (9) and infectious disorders (10). Fractalkine (FKN, also known as
CX3CL1), is a unique chemokine subclass and the only member of the
CX3C chemokine family, existing as a membrane-bound form and a
soluble form. The membrane-bound form is synthesized as a
transmembrane molecule with an extracellular N-terminal domain
attached by a mucin-like stalk to the cell surface (6). Soluble FKN is generated via cleavage
at the base of the mucin-like stalk by the metalloproteinases, A
disintegrin and metalloproteinase 10 and 17 (11). Several studies have identified that
FKN is expressed by a variety of tumors, including prostate cancer,
and there has been increasing interest in its involvement in
prostate cancer (12–15). FKN-CX3CR1 is reported to be
involved in the molecular events that regulate the adhesion,
migration and survival of human prostate cancer cells (15). In addition, FKN-CX3CR1 binding has
also been observed to be crucial in the progression of prostate
cancer and skeletal metastasis (12).
Hypoxia leads to tumor cell proliferation and tumor
growth. Emerging evidence has indicated that hypoxia can promote
tumor growth by upregulating specific chemokine receptors,
including CCR2 (16). Our previous
study demonstrated that hypoxia increases the expression of CX3CR1
via the hypoxia-inducible factor and nuclear factor-κB signaling
pathway in androgen-independent prostate cancer cells (17). However, the association between FKN
and hypoxia-induced prostate cancer cell proliferation remains to
be fully elucidated.
The present study aimed to determine the effect of a
hypoxic microenvironment on the expression of FKN and the role of
FKN in hypoxia-induced prostate cancer cell proliferation. The
results showed that FKN expression was upregulated under hypoxic
conditions, which resulted in enhanced proliferation of prostate
cancer cells.
Materials and methods
Cell lines and reagents
DU145 and PC-3 human prostate cancer cell lines were
purchased from American Type Culture Collection (Manassas, VA,
USA). Human recombinant FKN was purchased from R&D systems,
Inc. (Minneapolis, MN, USA). Anti-human FKN antibody was purchased
from Abcam (Cambridge, MA, USA). CDK2 primary antibody [mouse
monoclonal immunoglobulin (Ig)G; cat no. sc-6248] was obtained from
Santa Cruz Biotechnology Inc. (Dallas, TX, USA) and cyclin E
primary antibody (rabbit monoclonal IgG; cat no. 2978s) was
purchased from Cell Signaling Technology Inc. (Danvers, MA, USA).
The secondary antibodies for CDK2 (anti-mouse IgG; cat no. A16027)
and cyclin E (anti-rabbit IgG; cat no. A16104) were from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). All primers
used in the present study were synthesized by Sangon Biotech Co.,
Ltd (Shanghai, China).
Cell culture and hypoxia exposure
Under normoxic conditions the cells were maintained
in RPMI-1640 medium containing 10% fetal bovine serum (Gibco Life
Technologies, Carlsbad, CA, USA) and 1X penicillin/streptomycin
(Invitrogen Life Technologies, Carlsbad, CA, USA) at 37°C in a
humidified atmosphere containing 5% CO2 and 95% air. The
cells that were grown under hypoxic conditions were incubated in a
hypoxic chamber (Thermo Fisher Scientific, Inc.) containing 1%
O2, 5% CO2 and 94% N2 at 37°C.
Cell proliferation assay
The cells were seeded at a density of
3×103 cells/well into a 96-well plate. Cell
proliferation was measured following 12, 24, 36, 48 and 72 h of
normoxic or hypoxic culture using a Cell Counting kit-8 (CCK-8;
Dojindo Molecular Technologies, Inc., Kumamoto, Japan), according
to the manufacturer's instructions. Briefly, CCK-8 reagents were
added to a subset of wells and incubated for 2 h at 37°C, following
which the absorbance was measured at a test wave length of 450 nm
on an automated plate reader (SpectraMax M3; Molecular Devices,
Sunnyvale, CA, USA).
Colony formation assay
The cells were seeded into a 6-well plate at a
density of 200 cells/well and incubated for 10 days to allow colony
formation. The colonies were fixed with 3% formaldehyde (Tianjin
Fuyu Fine Chemical Co., Ltd., Tianjin, China) and stained with 0.1%
crystal violet (Shanghai Bogoo Biotechnology Co., Ltd., Shanghai,
China). The number of colonies containing >50 cells were
counted. The colonies were manually counted using a microscope
(Olympus TH4-200; Olympus, Tokyo, Japan) and images of the colonies
were captured.
Flow cytometric analysis of cell
cycle
Cell cycle analysis was performed using flow
cytometry. DU145 cells were pre-incubated with FKN, with 200 pg/ml
human recombinant FKN protein added to the medium when the cells
were 80% confluent. The cells were harvested and fixed with 70%
ethanol prior to being stored at 4°C overnight. The fixed cells
were incubated with RNase (25 µg/ml; Sigma-Aldrich, St
Louis, MO, USA) at 37°C for 30 min, and the DNA was subsequently
stained with propidium iodide (50 µg/ml; Sigma-Aldrich) for
30 min in the dark. The stained cells were then analyzed by
fluorescence-activated cell sorting using a FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells incubated
under normoxic or hypoxic conditions using TRIzol®
reagent (Invitrogen Life Technologies). RNA was reverse transcribed
into cDNA using Superscript II Reverse Transcriptase (Invitrogen
Life Technologies), according to the manufacturer's instructions.
The cDNA (1 µg in 4 µl buffer) was amplified by qPCR
in an ABI 7500 real-time PCR system (Applied Biosystems; Thermo
Fisher Scientific) using the SYBR Select Master Mix kit (Applied
Biosystems) and with gene-specific primers (1 µl). The
primers used in the present study were as follows: β-actin sense,
5′-TACCTCATGAAGATCCTCACC-3′ and antisense,
5′-TTTCGTGGATGCCACAGGAC-3′; CDK2 sense, 5′-CAGGATGTGACCAAGCCAGTA-3′
and antisense, 5′-CCAACCCTCTCCAGCAATAA-3′; cyclin E sense,
5′-ACGACGACGACGAAAAACTC-3′ and antisense,
5′-GTTGCGACGCTGAAGAGAAC-3′; and FKN sense,
5′-CTTTCTCATCCACTATCAACA-3′; and antisense, 5′-CTCCACTACTCTTTC-3′.
All the primers were purchased from Sangon Biotech Co. Ltd. The
thermocycling conditions were initial denaturation at 94°C for 2
min, followed by 30 cycles of 98°C for 10 sec, 60°C for 15 sec and
68°C for 2 min. Each reaction was repeated in triplicate to
minimize experimental variation, and the expression levels of
β-actin were used as an internal control. The results were
quantitated by scanning densitometry using a Bio-Rad 620 Video
Densitometer (Bio-Rad Laboratories, Hercules, CA, USA)
Western blot analysis
The total protein of cyclin E and CDK2 were
extracted prior to western blot analysis. Western blot analysis was
performed, as described previously (18). Briefly, the cells were washed with
PBS and re-suspended in cold lysis buffer with
phenylmethanesulfonylfluoride. The cell lysate was incubated on ice
for 30 min and centrifuged at 12,000 × g for 15 min at 4°C. The
protein concentration of the lysate was determined using a BCA-200
protein assay kit (Beyotime Institute of Biotechnology, Inc.,
Haimen, China). Equal quantities of protein (40 µg/lane)
were separated by 12% SDS-PAGE and transferred onto nitrocellulose
membranes (Pall Corp., Port Washington, NY, USA). Following
blocking with 5% fat-free milk for 2 h at room temperature, the
membranes were incubated with various primary antibodies (1:500) at
4°C overnight. Subsequently, the bound primary antibody was
detected by incubating with appropriate horseradish
peroxidase-conjugated secondary antibodies (1:1,000) for 2 h at
room temperature, followed by washing with Tris-buffered saline
containing Tween 20 three times. The immunoreactive bands were
visualized using blot analysis Super ECL Plus Detection Reagents
(Applygen Technologies Inc., Beijing, China). The volumes of the
protein bands were quantified using a Bio-Rad Chemi Doc™ EQ
densitometer and Bio-Rad Quantity One software (Bio-Rad
Laboratories). The expression levels of tubulin were assessed as an
internal control.
ELISA assay
The concentration of soluble FKN in the supernatant
was determined using an FKN ELISA kit (R&D Systems, Inc.,
Minneapolis, MN, USA) following the manufacturer's instructions and
in accordance with the procedure of a previous study (19).
Statistical analysis
The results of the present study are expressed as
the mean ± standard error of the mean. Each experiment was repeated
in triplicate. One-way analysis of variance was used for multiple
comparisons. Statistical analyses were performed using SAS software
version 9.1.3 (SAS Institute Inc., Cary, NC, USA). Statistical
differences between two groups were analyzed using an unpaired two
tailed Student's t-tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
Hypoxia induces prostate cancer cell
proliferation
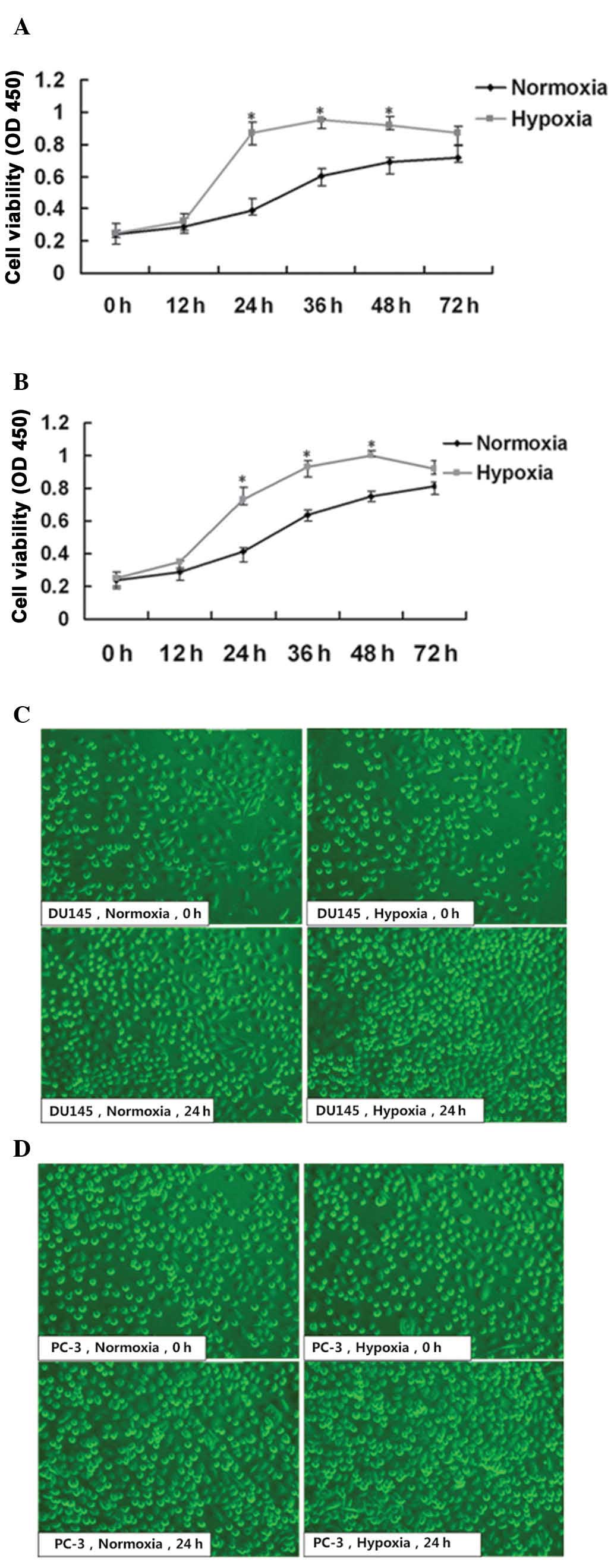
To examine the response of prostate cancer cell
proliferation to hypoxia, two types of androgen-independent
prostate cancer cell lines, DU145 and PC-3, were cultured under
normoxic or hypoxic conditions, respectively. Firstly, cell
viability was determined following 12, 24, 36, 48 and 72 h of
culture using a CCK-8 assay. Morphological changes were also
observed using an inverted microscope. DU145 and PC-3 cell
proliferation was continuous under the hypoxic and normoxic
conditions. The cell proliferation rates of the DU145 and PC-3
cells exposed to hypoxia were significantly higher, compared with
those under normoxia at 24-48 h (P<0.05; Fig. 1A and B). Furthermore, microscopic
observation revealed markedly increased cell density in the two
cell lines following 24 h culture under hypoxic conditions,
compared with normoxic conditions, and cell growth under hypoxic
and normoxic conditions remained normal as observed by inverted
microscopy (Fig. 1C and D).
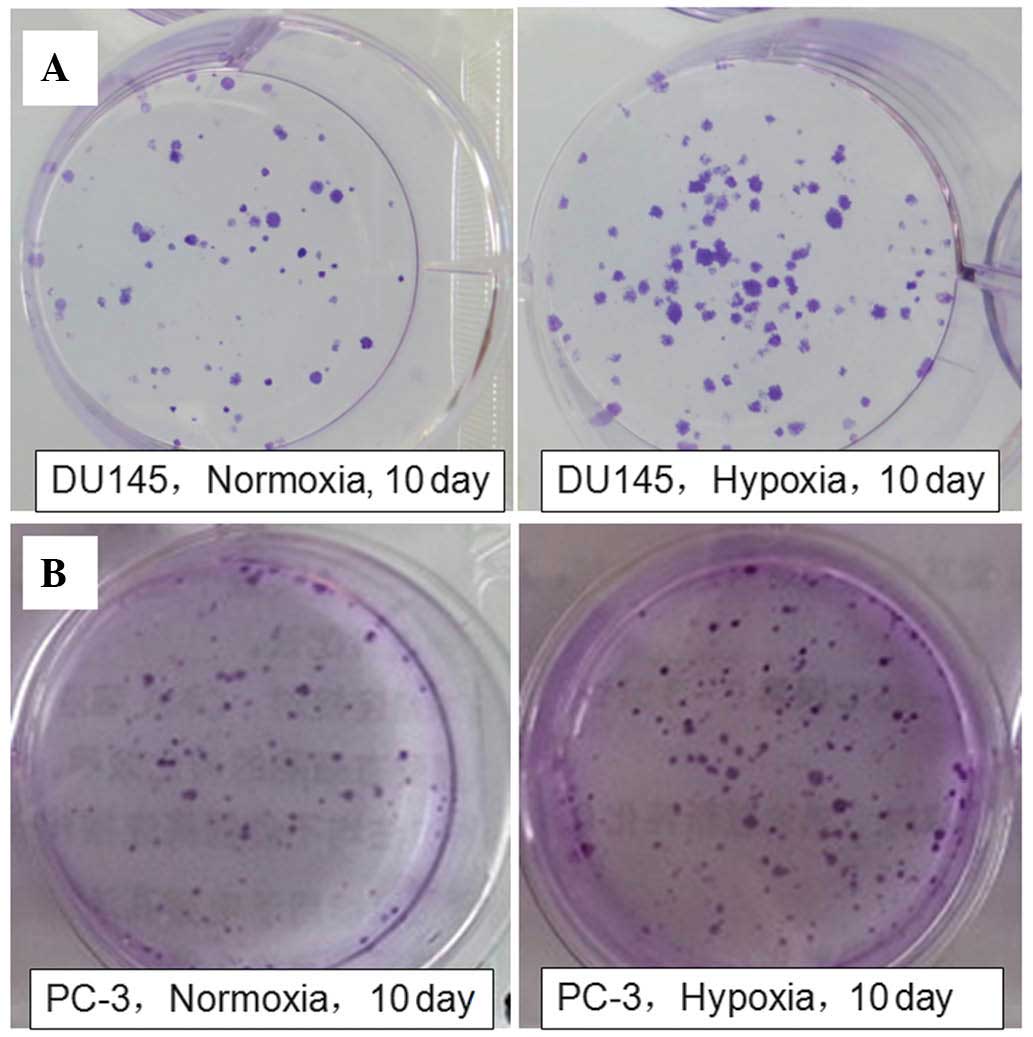
Subsequently, the colony-forming ability of the DU145 and PC-3
cells under the two conditions were determined in order to further
confirm the effect of hypoxia on prostate cancer cell
proliferation. The clone clusters of the two cell lines under
hypoxic conditions were larger and more numerous, compared with
those under normoxic conditions for 10 days (Fig. 2). The results suggested that
hypoxia induced DU145 and PC-3 cell proliferation. In addition, the
DU145 cell proliferative response to hypoxic treatment was more
marked, compared with that of the PC-3 cells. Therefore, DU145
cells were selected as the experimental cells for subsequent
investigation of the underlying molecular mechanisms.
Hypoxia upregulates the mRNA expression
and secretion of FKN
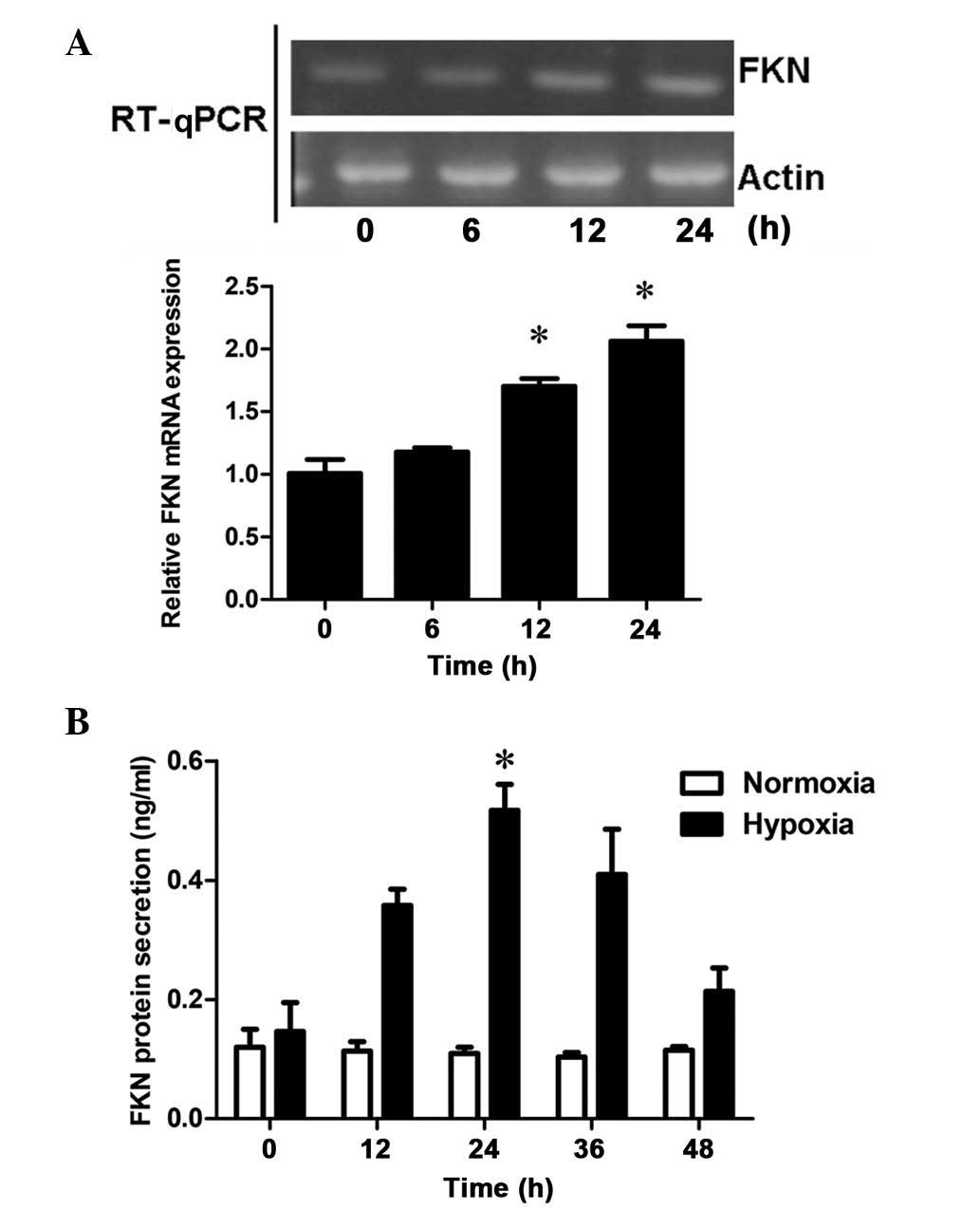
To assess the effects of hypoxia on mRNA expression,
the mRNA expression levels of FKN in DU145 cells was examined 0, 6,
12 and 24 h following hypoxic treatment. RT-qPCR demonstrated that
FKN was expressed at low levels in the DU145 cells. However, with
increasing duration of hypoxic treatment, the mRNA expression of
FKN increased in a time-dependent manner, with significant
differences at 12 and 24 h (P<0.05; Fig. 3A and B), which resembled the
response of cell proliferation to hypoxia. These results suggested
that hypoxia-upregulated the mRNA expression of FNK in a
time-dependent manner in the prostate cancer cells. In addition,
the concentration levels of soluble FKN in the supernatant,
determined using an ELISA kit, revealed that the protein secretion
levels of FKN in the DU145 cells increased significantly following
24 h of hypoxic treatment, compared with those following normoxic
treatment (P<0.05; Fig. 3C).
Therefore, the results suggested a positive correlation between FKN
and hypoxic prostate cell proliferation.
Hypoxia-induced prostate cancer cell
proliferation is mediated by FKN
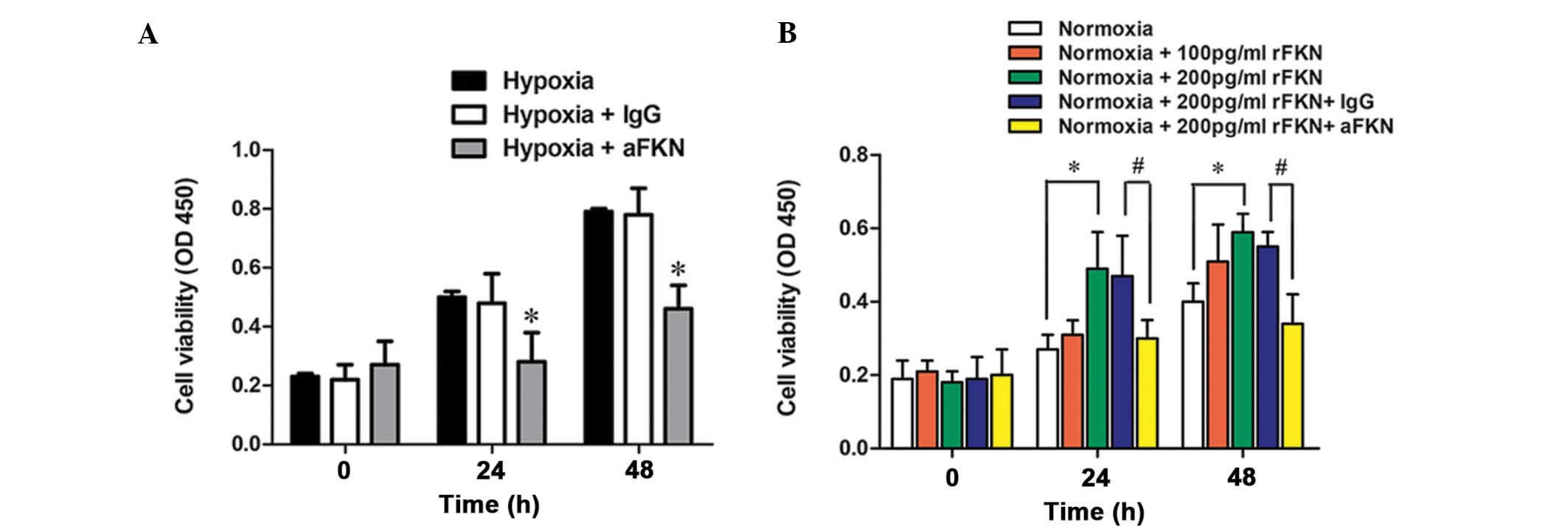
To investigate the role of FKN in hypoxia-induced
prostate cancer cell proliferation, FKN-specific antibody anti-FKN
or isotype IgG were added to the hypoxic DU145 cells, and cell
proliferation was determined following 24 and 48 h of incubation
using a CCK-8. As shown in Fig.
4A, DU145 cell proliferation was markedly inhibited by anti-FKN
pretreatment (P<0.05). To further verify the role of FKN in
prostate cancer cell proliferation, the proliferation of the cells
was also examined following exogenous administration of human
recombinant FKN protein (100 pg/ml or 200 pg/ml) under normoxic
conditions. As shown in Fig. 4B,
DU145 cell proliferation was markedly enhanced by treatment with
200 pg/ml exogenous FKN protein (P<0.05), and this increase was
significantly alleviated by anti-FKN (P<0.05). These data
suggested that FKN was involved in the regulation of hypoxic
prostate cancer cell proliferation.
FKN promotes G1/S phase
transition by upregulating the expression levels of cyclin E and
CDK2
Cell proliferation is closely associated with cell
cycle regulation. In order to determine whether hypoxia-induced
prostate cancer cell proliferation was associated with cell cycle
regulation by FKN, the cell cycle distribution in the DU145 cells
was measured under normoxic or hypoxic conditions using flow
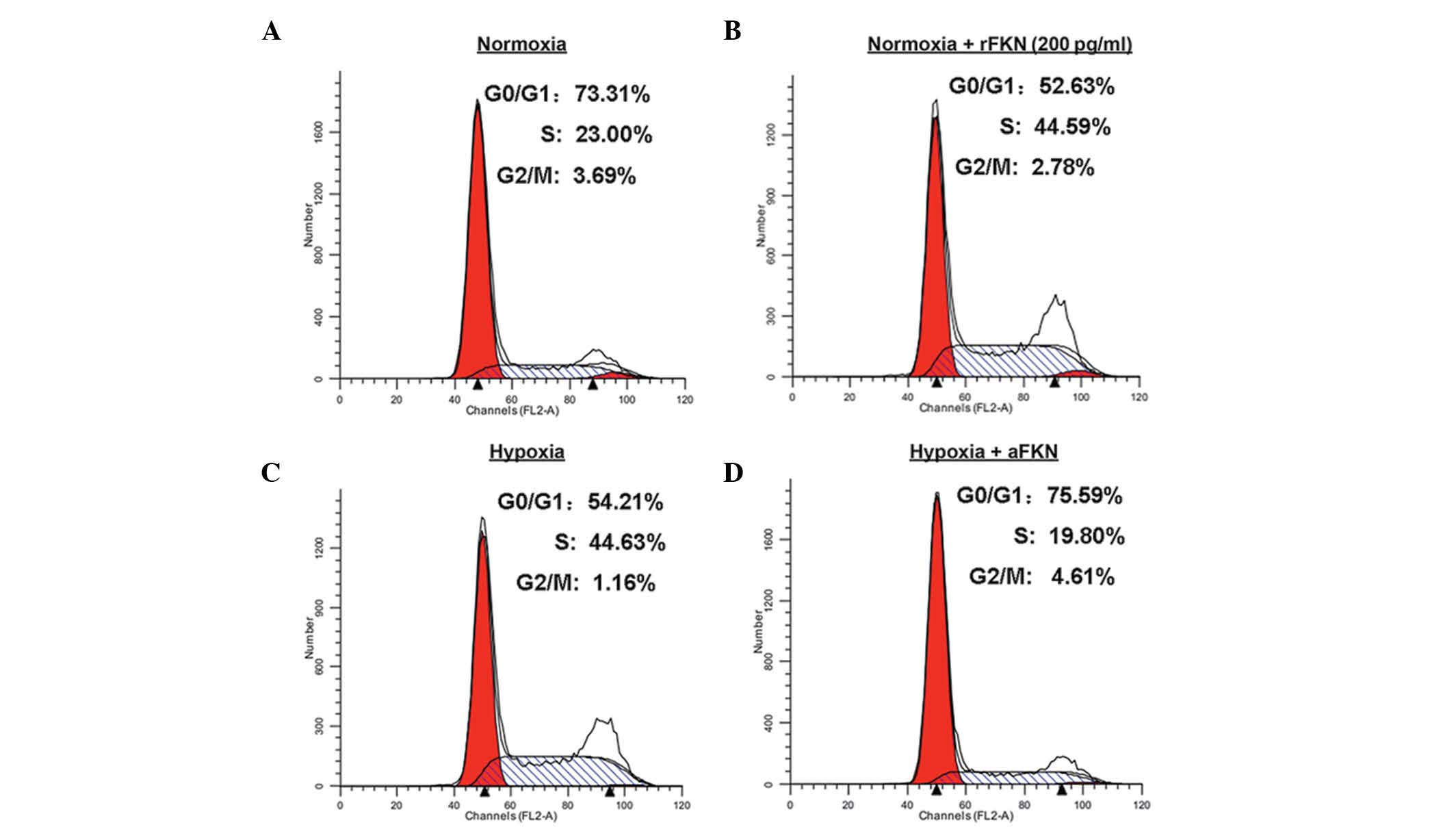
cytometric analysis (Fig. 5).
Compared with normoxic conditions (Fig. 5A and B), hypoxic treatment
decreased the proportion of cells in the G1phase,
between 73.31 and 54.21%, and increased the proportion of cells in
the S phase, between 23.00 and 44.63%. However, anti-FKN
pre-treatment increased the proportion of cells in the
G1 phase between 54.21 and 75.59%. and decreased the
proportion in the S phase, between 44.63 and 19.80%, which
suggested that antagonizing FKN inhibited the G1 to S
phase transition induced by hypoxia (Fig. 5C and D). To further elucidate the
role of FKN in cell cycle regulation, the cell cycle distribution
of DU145 cells pre-incubated with 200 pg/ml human recombinant FKN
protein under normoxic conditions was analyzed. As shown in
Fig. 5A and B, exogenous FKN
protein administration decreased the proportion of cells in the
G1 phase between 73.31 to 52.63%, and increased the
proportion of cells in the S phase between 23.00 and 44.59%. These
results suggested that FKN promoted the G1/S phase cell
cycle transition.
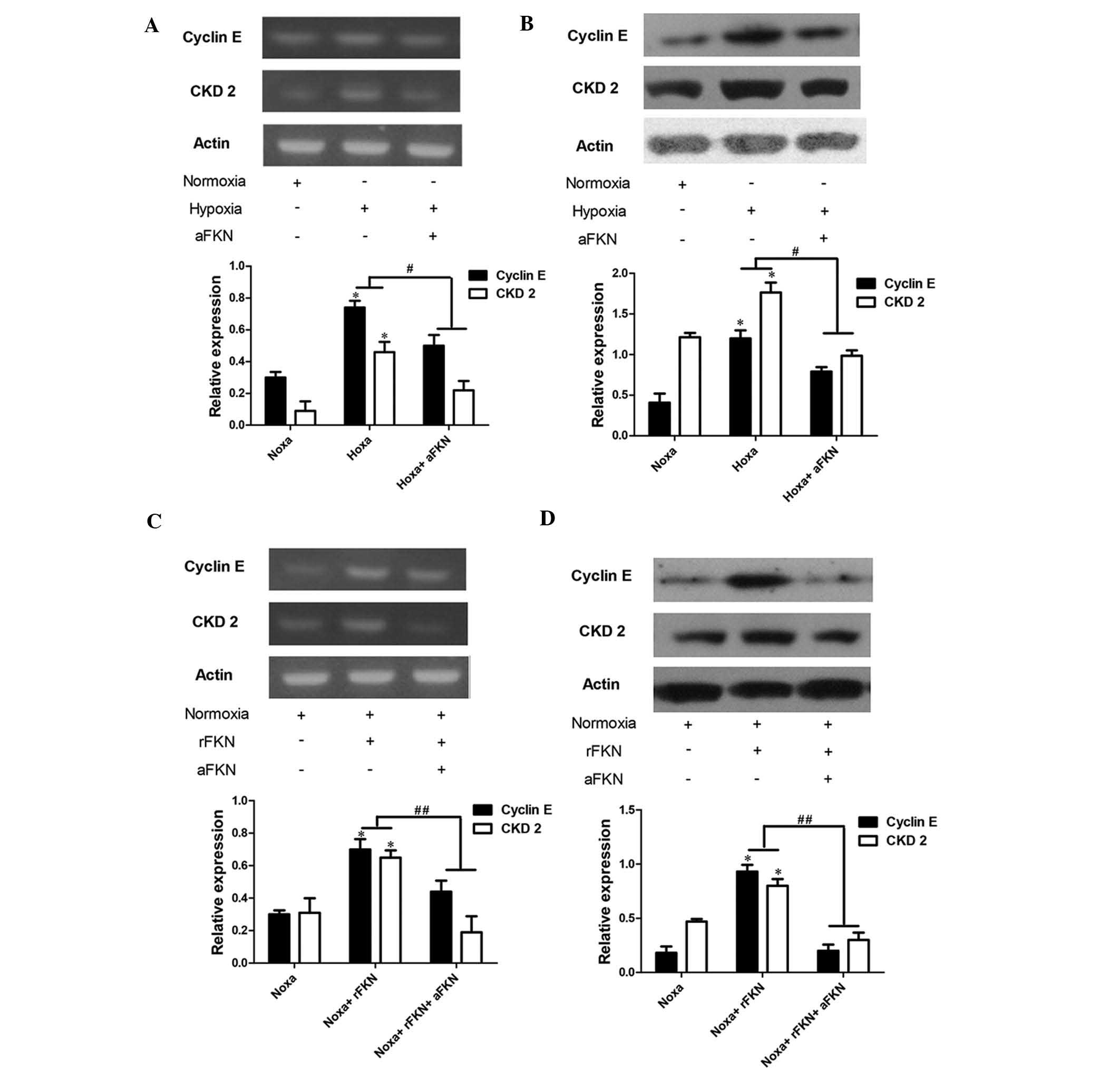
Cell cycle progression depends on the formation of
CDK/cyclin complexes, and cyclin E and CDK2 are G1/S
phase-specific regulators (20).
Therefore, the present study determined the mRNA and protein
expression levels of cyclin E and CDK2 using RT-qPCR and western
blot analysis. As shown in Fig.
6A–D, the mRNA and protein expression levels of cyclin E and
CDK2 in the DU145 cells exposed to hypoxia were significantly
higher than those in the cells exposed to normoxic conditions
(P<0.05), and anti-FKN pre-treatment markedly attenuated the
increased expression levels of cyclin E and CDK2 (P<0.05). To
further verify the role of FKN in the regulation of cyclin E and
CDK2, the expression levels of cyclin E and CDK2 in DU145 cells
exposed to normoxia with anti-FKN or exogenous FKN protein
pre-treatment were also examined. The mRNA and protein expression
levels of cyclin E and CDK2 were significantly elevated by 200
pg/ml exogenous FKN protein administration (P<0.05), and
anti-FKN pretreatment markedly alleviated this increase (P<0.05;
Fig. 6C and D).
Discussion
Prostate cancer is the most frequently diagnosed
type of malignancy and is the second leading cause of
cancer-associated mortality in males (21). Early-stage prostate cancer depends
on androgens for growth and survival, and androgen ablation therapy
causes them to regress (22).
However, cancer, which is not cured by surgical procedures
eventually become androgen-independent, and androgen ablation fails
to target androgen-independent cells (23). Therefore, examining the underlying
molecular mechanisms and potential therapeutic targets in
androgen-independent prostate cancer is essential for improving
clinical outcomes. The present study presented data, which
demonstrated that hypoxia, a common phenomenon in prostate cancer,
induced the expression and secretion of FKN, and that antagonizing
FKN inhibited hypoxic prostate cancer cell proliferation.
A hypoxic microenvironment contributes to the
progression of cancer (19). Under
certain conditions, the interaction between tumor cells and their
microenvironment is facilitated by a variety of soluble factors,
including growth factors and chemokines (24). Hypoxia has been suggested to
promote cell migration and invasion in various types of tumor by
regulating specific chemokine receptors (25–27).
FKN is the only member recognized so far, which belongs to the CX3C
chemokine subfamily. FKN exists as a membrane-bound form
functioning as an adhesion molecule, and as a soluble form acting
as a potent chemoattractant for monocytes, natural killer cells and
T cells (28). Therefore, FKN is
important in several disorders comprising imbalance of the immune
response, including arthritis (29), asthma (30), human immunodeficiency virus
(31) and Crohn's disease
(32). In addition, the
involvement of FKN/CX3CR1 in the pathogenesis and progression of
multiple malignant diseases has also been a focus of interest. For
certain tumor entities, FKN was demonstrated to be correlated with
a higher local recurrence risk and metastatic potential (33,34).
In epithelial ovarian cancer cells, FKN functions as an important
regulator of malignant cell proliferation via binding to CX3CR1 and
consequently activating AKT signaling (35). In prostate cancer, neutralizing
antibodies against FKN markedly repress the ability of prostate
cancer cells to adhere to the bone marrow endothelium (15). In the present study, the results
demonstrated that hypoxia promoted cell proliferation in two types
of androgen-independent prostate cancer cell lines, DU145 and PC-3.
Subsequently, the effects of hypoxia on FKN synthesis and secretion
were investigated in the prostate cancer cells. The results
demonstrated that hypoxia induced the secretion and mRNA expression
of FKN in a sustained manner. Based on these observations, it was
hypothesized that FKN was associated with prostate cancer cell
proliferation. The present study subsequently determined DU145 cell
proliferation rates following FKN inhibition with the anti-FKN
FKN-specific antibody under hypoxic conditions. The results
demonstrated that anti-FKN pre-treatment markedly inhibited DU145
cell proliferation. To further investigate the role of FKN under
normoxic conditions, DU145 cell proliferation was examined
following the administration of exogenous human recombinant FKN
protein. The results revealed that DU145 cell proliferation
markedly increased, and this increase was significantly alleviated
by treatment with anti-FKN, which indicated that DU145 cell
proliferation was mediated by FKN. These data suggested that
upregulation of the expression and secretion of FKN was involved in
hypoxia-induced prostate cancer cell proliferation.
Cellular proliferation follows an orderly
progression through the cell cycle. The traditional subdivisons of
the standard cell cycle include the G1, S, G2
and M phases (36). Accumulating
evidence has suggested the relevance of cell cycle deregulation in
human cancer. The majority of types of human cancer exhibit
deregulated control of G1 phase progression, a period
during which the cells either initiate proliferation or remain
quiescent (37). The present study
examined the role of FKN in prostate cancer cell cycle regulation
using flow cytometry, and the results revealed that either
exogenous FKN protein administration or hypoxic treatment notably
increased the proportion of cells in the S phase, and anti-FKN
pre-treatment attenuated hypoxia-induced cell accumulation in the S
phase. These data indicated that FKN regulated the G1/S
checkpoint, resulting in promotion of the G1 to S phase
transition. The transition between one cycle phase and another
occurs in order and is regulated by a variety of cell cycle
regulators, the most prominent of which are cyclins and their
associated CDKs. Of these, cyclin E is important for initiation of
the S phase, and its association with CDK2 regulates the
progression between the G1 and S phase (38). The results of the present study
demonstrated that antagonizing FKN attenuated the hypoxia-induced
mRNA and protein expression levels of cyclin E and CDK2 in the
prostate cancer cells. By contrast, prostate cancer cells, exposed
to exogenous FKN protein pre-treatment under normoxic conditions
exhibited elevated mRNA and protein expression levels of cyclin E
and CDK2, and anti-FKN pre-treatment markedly alleviated this
elevation. These findings suggested that FKN promoted
G1/S cell cycle progression through upregulating the
expression levels of cyclin E and CDK2, resulting in hypoxic
prostate cancer cell proliferation.
Aberrant epidermal growth factor receptor (EGFR)
signaling is associated with characteristics of aggressive
malignancies, including increased proliferative potential, nitric
oxide synthesis and accelerated G1/S cell cycle
progression. White et al (39) reported that EGFR activation is
involved in the proliferation of human vascular smooth muscle cells
in response to FKN treatment. Therefore, hypoxia may induce the
secretion of FKN and subsequent EGFR signaling activation, which
promotes cell cycle progression, resulting in increased cell
proliferation.
In conclusion, the results of the present study
provided evidence of a novel function for FKN, which enhances cell
proliferation by promoting cell cycle progression in hypoxic
prostate cancer cells. Further investigations are required to
further expand on these findings, with the goal of identifying a
potential therapeutic target.
Abbreviations:
|
CCK-8
|
cell counting kit-8
|
|
CDKs
|
cyclin-dependent kinases
|
|
EGFR
|
epidermal growth factor receptor
|
|
FBS
|
fetal bovine serum
|
|
FKN
|
fractalkine
|
|
NK
|
natural killer
|
|
CCK-8
|
cell counting kit-8
|
|
CDKs
|
cyclin-dependent kinases
|
|
EGFR
|
epidermal growth factor receptor
|
|
FBS
|
fetal bovine serum
|
|
FKN
|
fractalkine
|
|
NK
|
natural killer
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (no. 30973010), the Natural
Science Foundation of Heilongjiang Province of China (no.
QC2009C115) and the startup Fund of The Affiliated Third Hospital
of Harbin Medical University (no. JJ2011-12).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baade PD, Youlden DR, Cramb SM, Dunn J and
Gardiner RA: Epidemiology of prostate cancer in the Asia-Pacific
region. Prostate Int. 1:47–58. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Freedland SJ: Screening, risk assessment
and the approach to therapy in patients with prostate cancer.
Cancer. 117:1123–1135. 2011. View Article : Google Scholar
|
|
4
|
Taiakina D, Dal Pra A and Bristow RG:
Intratumoral hypoxia as the genesis of genetic instability and
clinical prognosis in prostate cancer. Adv Exp Med Biol.
772:189–204. 2014. View Article : Google Scholar
|
|
5
|
Voss MJ, Niggemann B, Zänker KS and
Entschladen F: Tumour reactions to hypoxia. Curr Mol Med.
10:381–386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jones BA, Beamer M and Ahmed S:
Fractalkine/CX3CL1: A potential new target for inflammatory
diseases. Mol Interv. 10:263–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lucas AD, Bursill C, Guzik TJ, Sadowski J,
Channon KM and Greaves DR: Smooth muscle cells in human
atherosclerotic plaques express the fractalkine receptor CX3CR1 and
undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1).
Circulation. 108:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blaschke S, Koziolek M, Schwarz A, Benöhr
P, Middel P, Schwarz G, Hummel KM and Müller GA: Proinflammatory
role of fractalkine (CX3CL1) in rheumatoid arthritis. J Rheumatol.
30:1918–1927. 2003.PubMed/NCBI
|
|
9
|
Tang L, Hu HD, Hu P, Lan YH, Peng ML, Chen
M and Ren H: Gene therapy with CX3CL1/Fractalkine induces antitumor
immunity to regress effectively mouse hepatocellular carcinoma.
Gene Ther. 14:1226–1234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Faure S, Meyer L, Costagliola D,
Vaneensberghe C, Genin E, Autran B, Delfraissy JF, McDermott DH,
Murphy PM, Debré P, et al: Rapid progression to AIDS in HIV+
individuals with a structural variant of the chemokine receptor
CX3CR1. Science. 287:2274–2277. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ludwig A, Hundhausen C, Lambert MH,
Broadway N, Andrews RC, Bickett DM, Leesnitzer MA and Becherer JD:
Metalloproteinase inhibitors for the disintegrin-like
metalloproteinases ADAM10 and ADAM17 that differentially block
constitutive and phorbol ester-inducible shedding of cell surface
molecules. Comb Chem High Throughput Screen. 8:161–171. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jamieson WL, Shimizu S, D'Ambrosio JA,
Meucci O and Fatatis A: CX3CR1 is expressed by prostate epithelial
cells and androgens regulate the levels of CX3CL1/fractalkine in
the bone marrow: Potential role in prostate cancer bone tropism.
Cancer Res. 68:1715–1722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nevo I, Sagi-Assif O, Meshel T, Ben-Baruch
A, Jöhrer K, Greil R, Trejo LE, Kharenko O, Feinmesser M, Yron I
and Witz IP: The involvement of the fractalkine receptor in the
transmigration of neuroblastoma cells through bone-marrow
endothelial cells. Cancer Lett. 273:127–139. 2009. View Article : Google Scholar
|
|
14
|
Erreni M, Solinas G, Brescia P, Osti D,
Zunino F, Colombo P, Destro A, Roncalli M, Mantovani A, Draghi R,
et al: Human glioblastoma tumours and neural cancer stem cells
express the chemokine CX3CL1 and its receptor CX3CR1. Eur J Cancer.
46:3383–3392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shulby SA, Dolloff NG, Stearns ME, Meucci
O and Fatatis A: CX3CR1-fractalkine expression regulates cellular
mechanisms involved in adhesion, migration and survival of human
prostate cancer cells. Cancer Res. 64:4693–4698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu Y, Cai Z, Galson DL, Xiao G, Liu Y,
George DE, Melhem MF, Yao Z and Zhang J: Monocyte chemotactic
protein-1 (MCP-1) acts as a paracrine and autocrine factor for
prostate cancer growth and invasion. Prostate. 66:1311–1318. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xiao LJ, Chen YY, Lin P, Zou HF, Lin F,
Zhao LN, Li D, Guo L, Tang JB, Zheng XL and Yu XG: Hypoxia
increases CX3CR1 expression via HIF-1 and NFκB in
androgen-independent prostate cancer cells. Int J Oncol.
41:1827–1836. 2012.PubMed/NCBI
|
|
18
|
Xiao LJ, Lin P, Lin F, Liu X, Qin W, Zou
HF, Guo L, Liu W, Wang SJ and Yu XG: ADAM17 targets MMP-2 and MMP-9
via EGFR-MEK-ERK pathway activation to promote prostate cancer cell
invasion. Int J Oncol. 40:1714–1724. 2012.
|
|
19
|
Hasegawa M, Sato S, Echigo T, Hamaguchi Y,
Yasui M and Takehara K: Up regulated expression of
fractalkine/CX3CL1 and CX3CR1 in patients with systemic sclerosis.
Ann Rheum Dis. 64:21–28. 2005. View Article : Google Scholar
|
|
20
|
Harbour JW, Luo RX, Dei Santi A, Postigo
AA and Dean DC: Cdk phosphorylation triggers sequential
intramolecular interactions that progressively block Rb functions
as cells move through G1. Cell. 98:859–869. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
22
|
Nishimura S, Hato M, Hyugaji S, Feng F and
Amano M: Glycomics for drug discovery: Metabolic perturbation in
androgen-independent prostate cancer cells induced by unnatural
hexosamine mimics. Angew Chem Int Ed Engl. 51:3386–3390. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feldman BJ and Feldman D: The development
of androgen-independent prostate cancer. Nat Rev Cancer. 1:34–45.
2001. View
Article : Google Scholar
|
|
24
|
Borsig L, Wolf MJ, Roblek M, Lorentzen A
and Heikenwalder M: Inflammatory chemokines and metastasis-tracing
the accessory. Oncogene. 33:3217–3224. 2013. View Article : Google Scholar
|
|
25
|
Waugh DJ, Wilson C, Seaton A and Maxwell
PJ: Multi-faceted roles for CXC-chemokines in prostate cancer
progression. Front Biosci. 13:4595–4604. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang S, Qi L, Li M, Zhang D, Xu S, Wang N
and Sun B: Chemokine CXCL12 and its receptor CXCR4 expression are
associated with perineural invasion of prostate cancer. J Exp Clin
Cancer Res. 27(62)2008. View Article : Google Scholar
|
|
27
|
Wang J, Lu Y, Wang J, Koch AE, Zhang J and
Taichman RS: CXCR6 induces prostate cancer progression by the
AKT/mammalian target of rapamycin signaling pathway. Cancer Res.
68:10367–10376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bazan JF, Bacon KB, Hardiman G, Wang W,
Soo K, Rossi D, Greaves DR, Zlotnik A and Schall T: A new class of
membrane-bound chemokine with a CX3C motif. Nature. 385:640–644.
1997. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koch AE: Chemokines and their receptors in
rheumatoid arthritis: Future targets? Arthritis Rheum. 52:710–721.
2005. View Article : Google Scholar
|
|
30
|
Rimaniol AC, Till SJ, Garcia G, Capel F,
Godot V, Balabanian K, Durand-Gasselin I, Varga EM, Simonneau G,
Emilie D, et al: The CX3C chemokine fractalkine in allergic asthma
and rhinitis. J Allergy Clin Immunol. 112:1139–1146. 2003.
View Article : Google Scholar
|
|
31
|
Foussat A, Bouchet-Delbos L, Berrebi D,
Durand-Gasselin I, Coulomb-L'Hermine A, Krzysiek R, Galanaud P,
Levy Y and Emilie D: Deregulation of the expression of the
fractalkine/frac-talkine receptor complex in HIV-1-infected
patients. Blood. 98:1678–1686. 2001. View Article : Google Scholar
|
|
32
|
Muehlhoefer A, Saubermann LJ, Gu X,
Luedtke-Heckenkamp K, Xavier R, Blumberg RS, Podolsky DK,
MacDermott RP and Reinecker HC: Fractalkine is an epithelial and
endothelial cell-derived chemoattractant for intraepithelial
lymphocytes in the small intestinal mucosa. J Immunol.
164:3368–3376. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Blum DL, Koyama T, M'Koma AE, Iturregui
JM, Martinez-Ferrer M, Uwamariya C, Smith JA Jr, Clark PE and
Bhowmick NA: Chemokine markers predict biochemical recurrence of
prostate cancer following prostatectomy. Clin Cancer Res.
14:7790–7797. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu X, Wang Y, Chen J, Ma H, Shao Z, Chen H
and Jin G: High expression of CX3CL1/CX3CR1 axis predicts a poor
prognosis of pancreatic ductal adenocarcinoma. J Gastrointest Surg.
16:1493–1498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gaudin F, Nasreddine S, Donnadieu AC,
Emilie D, Combadière C, Prévot S, Machelon V and Balabanian K:
Identification of the chemokine CX3CL1 as a new regulator of
malignant cell proliferation in epithelial ovarian cancer. PloS
one. 6:e215462011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Malumbres M and Carnero A: Cell cycle
deregulation: A common motif in cancer. Prog Cell Cycle Res.
5:5–18. 2003.PubMed/NCBI
|
|
38
|
Golias CH, Charalabopoulos A and
Charalabopoulos K: Cell proliferation and cell cycle control: A
mini review. Int J Clin Pract. 58:1134–1141. 2004. View Article : Google Scholar
|
|
39
|
White GE, Tan TC, John AE, Whatling C,
McPheat WL and Greaves DR: Fractalkine has anti-apoptotic and
proliferative effects on human vascular smooth muscle cells via
epidermal growth factor receptor signalling. Cardiovasc Res.
85:825–835. 2010. View Article : Google Scholar :
|