Introduction
Hepatocellular carcinoma (HCC), a primary hepatic
tumor with aggressive malignancy and high prevalence, is the third
leading cause of cancer-associated mortality worldwide (1,2).
Although progress in HCC treatment has been achieved during the
last three decades, patients with advanced and terminal stages of
HCC continue to have a poor prognosis, owing to the paucity of
effective and tolerable systemic chemotherapy strategies (3,4). Due
to the multi-fold molecular pathogenesis and high chemoresistance
of HCC, as well as the low selectivity of chemotherapeutic drugs,
it is essential to develop multi-target compounds, which can target
several aberrant signaling pathways concurrently, which may not
only improve medication efficacy, but also minimize drug toxicity
(5,6).
Multiple pathophysiological processes, including
viral replication, protein overloading, oxidative stress and the
inflammatory reaction, are involved in the oncogenesis of HCC
caused by chronic liver diseases, including hepatitis B and C viral
infection, alcoholic cirrhosis and obesity-associated non-alcoholic
fatty liver disease (7). Notably,
these events in hepatocytes induce a state of stress in the
endoplasmic reticulum (ER), termed ER stress (8,9).
There has been increasing awareness with regards to the role of ER
stress in the homeostasis of cancer cells (5,9). ER
stress occurs when ER homeostasis is lost due to an overload of
protein folding in the ER. Abnormalities in ER function can cause
ER stress, which results in an unfolded protein response (UPR),
including three key signaling proteins: Inositol-requiring enzyme
1α (IRE1α)-apoptosis signal-regulating kinase (ASK)-p38/c-Jun
N-terminal kinase (JNK), protein kinase R-like endoplasmic
reticulum kinase (PERK)-eukaryotic translation initiation factor 2α
(eIF2α)-activating transcription factor (ATF)4 and ATF6, which act
as the sensors of ER stresses. Furthermore, several mechanisms have
been suggested to link ER stress with apoptosis, including the B
cell lymphoma 2 (Bcl-2) and caspase family (10,11).
Tetramethylpyrazine (TMP) is one of the major
bioactive components purified from the Chinese herb, Chuanxiong
(Ligusticum wallichi Franchat), and has been widely used in
the treatment of various types of human cancer, including lung
cancer (12), osteosarcoma
(13), gliomas (14,15),
ovarian carcinoma (16), breast
cancer (17), gastric cancer
(18) and HCC (19). CXC195 is a TMP analogue, which can
protect human umbilical vein endothelial cells from
H2O2-induced apoptosis through inhibition of
the mitochondria- and caspase 3-dependent signaling pathways
(20,21). In addition, CXC195 exhibits
antioxidant activity and anti-apoptotic effects in transient focal
ischemia by inhibiting the expression levels of NADPH oxidase and
nitric oxide synthase (22), and
by regulating the PI3K/Akt/glycogen synthase kinase 3β signaling
pathway (23). Despite evidence
indicating the anticancer effects of TMP, there is a lack of data
describing the anticancer activity of its analogue, CXC195. The
present study examined whether CXC195 induced apoptosis and ER
stress in human HCC HepG2 cells, and investigated the possible
mechanisms underlying these effects.
Materials and methods
Cell lines and culture
Human HCC HepG2 cells were purchased from the
American Type Culture Collection (Manassas, VA, USA) and maintained
in Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher
Scientific., Inc., Waltham, MA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Invitrogen; Thermo Fisher
Scientific., Inc.), 100 U/ml penicillin (Invitrogen) and 10
µg/ml streptomycin (Invitrogen) at 37°C in an atmosphere
containing 5% CO2. The media were replaced every 2–3
days and sub-cultured when the cell population density reached
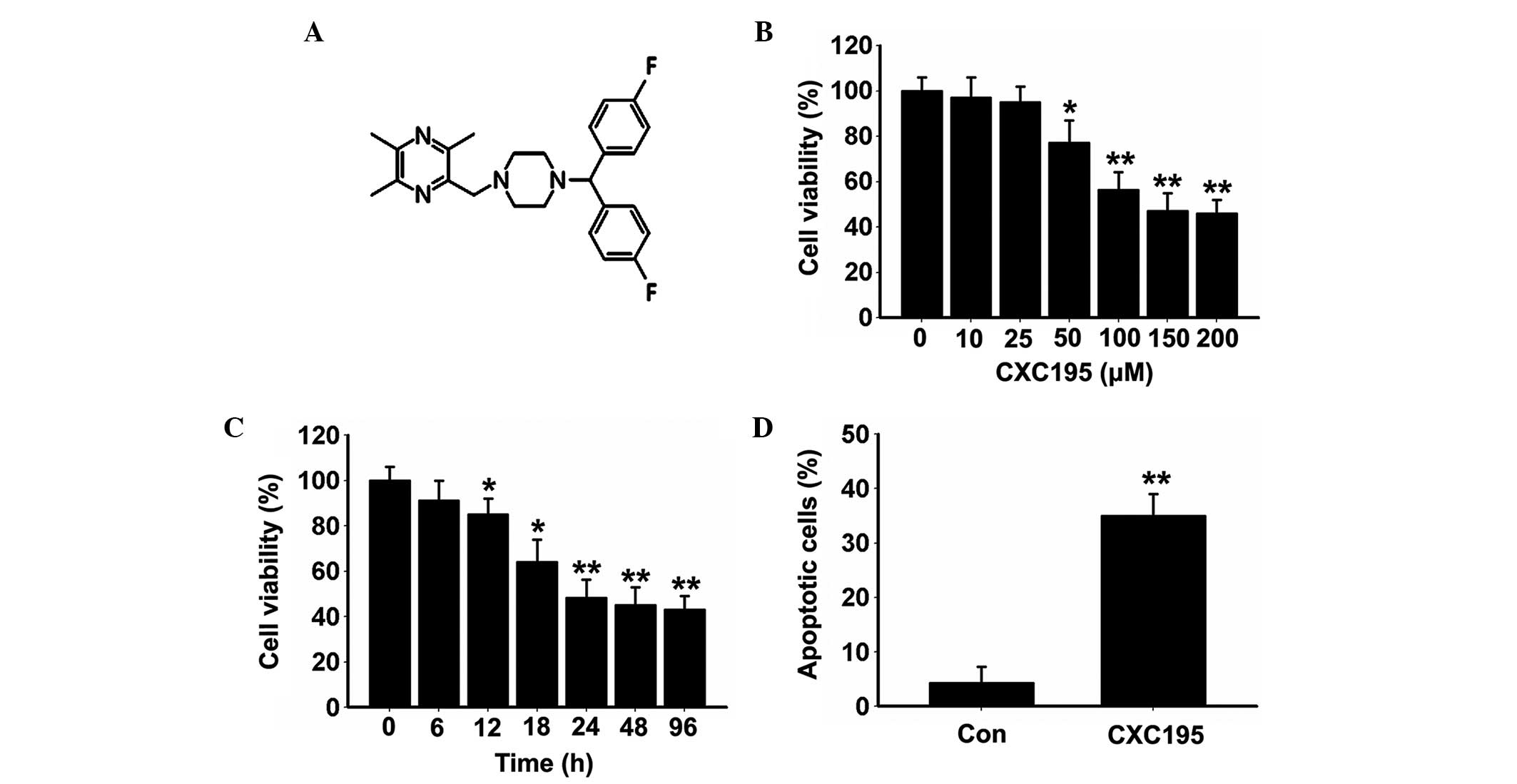
70–80% confluence. CXC195 (Fig.
1A) was synthesized by the direct reaction of
2-chloromethyl-3,5,6-trimethylpyrazine hydrochloride
(Sigma-Aldrich, St. Louis, MO, USA) with
4,4′-difluorobenzhydrylpiperazine (Sigma-Aldrich). The purity of
CXC195 (>98%) was determined using high-performance liquid
chromatography (Waters, Milford, MA, USA). The examined compounds
and positive control were dissolved in 0.1% dimethylsulfoxide
(DMSO; Sigma-Aldrich)). LY294002, SH-6 and rapamycin were purchased
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Cell viability assay
Cell viability was assessed using an MTT assay
(Santa Cruz Biotechnology, Inc.). The spent medium was removed and
10 µl MTT solution (5 mg/ml) was added to 100 µl
respective growth medium without phenol red, and the plates
containing 1×106 cells/well were incubated at 37°C for 4
h in a humidified atmosphere of 5% CO2. The formazan
crystals formed by the mitochondrial reduction of MTT were
solubilized in DMSO (100 µl/well) and the absorbance was
read at 540 nm using a microplate reader (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Double staining for Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI)
Briefly, the HepG2 cells were detached with 0.25%
trypsin (Invitrogen) and washed twice with phosphate-buffered
saline (PBS), followed by centrifugation at 12,000 × g for 5 min. A
total of 1×106 cells were then suspended in 1.25 mM
extracellular calcium (131 mM NaCl, 5 mM KCl, 1.3 mM
MgSO4, 1.3 mM CaCl2, 0.4 mM
KH2PO4, 6 mM glucose, 20 mM HEPES; pH 7.4)
and double-stained with Annexin V-FITC and PI (1:100; BD
Biosciences, San Jose, CA, USA) at 4°C for 30 min at room
temperature. The fluorescence of each sample was subsequently
quantitatively analyzed using a FACSCalibur flow cytometer (BD
Biosciences) and CellQuest software (version 5.1; BD
Biosciences).
Measurement of the cell cycle
The cells were treated with 20 µg/ml RNase A
(Invitrogen), followed by 25 µg/ml PI. The cell population
at each stage of the cell cycle was then determined by examining
the intensity of PI fluorescence on a flow cytometer using an argon
laser and a 570 nm bandpass filter (FACSort; BD Biosciences).
Caspase 3, 8, and 9 activity assays
HepG2 cells were gently lysed in ice-cold lysis
buffer [250 mM sucrose, 1 mM EDTA, 0.05% digitonin, 25 mM Tris (pH
6.8), 1 mM dithiothreitol, 1 µg/ml leupeptin, 1 μg/ml
pepstatin, 1 µg/ml aprotinin, 1 mM benzamidine and 0.1 mM
phenylmethylsulphonyl fluoride] for 30 min, and centrifuged at
12,000 ×g at 4°C. The cell lysates (30 µg) from the HepG2
cells were determined spectrophotometrically at 405 nm using a
microtiter plate reader (model M100L; Microfluidics, Newton, MA,
USA). The assays to determine the activities of caspase 3, 8 and 9
were performed by incubating the cell lysates with 0.2 mM of one of
the following caspase-specific colorimetric tetrapeptide
substrates: Ac-DEVD-pNA for caspase 3, Ac-IETD-pNA for caspase 8
and Ac-LEHD-p-nitroaniline (pNA) for caspase 9, for 1 h at 37°C, as
previously described (24). The
increase in the absorbance at 405 nm, which corresponds to the
quantity of pNA released from the peptide substrates was then
converted into units of enzyme activity using a standard curve
generated with free pNA. One unit of caspase 3, 8, or 9 activity
corresponded to the quantity of enzyme that releases 1 pmol/min pNA
from 0.2 mM DEVD-pNA, IETD-pNA or Ac-LEHD-pNA, respectively.
Lysates from HepG2 cells treated with DMSO were also used in these
assays, which served as a control group.
Determining the release of cytochrome c
(Cyt-c) and apoptosis-inducing factor (AIF)
The HepG2 cells were collected by
centrifugation at 12,000 ×g at 4°C for 5 min at 48°C, and washed
with ice-cold PBS. Subsequent fractionation of the mitochondrial
and cytosolic proteins were performed using a Mitochondrial Protein
Extraction kit (Santa Cruz Biotechnology, Inc.), according to the
manufacturer's protocol. The cell nuclear and cytosolic fractions
were prepared using a Nuclear/Cytosol Fractionation kit, purchased
from BioVision Inc. (Milpitas, CA, USA), according to the
manufacturer's protocol. Following centrifugation, the supernatants
were obtained for western blot analysis.
Western blot analysis
HepG2 cells were gently lysed in ice-cold lysis
buffer [250 mM sucrose, 1 mM EDTA, 0.05% digitonin, 25 mM Tris (pH
6.8), 1 mM dithiothreitol, 1 µg/ml leupeptin, 1 µg/ml
pepstatin, 1 µg/ml aprotinin, 1 mM benzam-idine and 0.1 mM
phenylmethylsulphonyl fluoride] for 30 min and centrifuged at
12,000 ×g at 4°C. Protein samples from the HepG2 cells extracts
were separated using 8% SDS-PAGE (high molecular weight) or 10%
SDS-PAGE (regular molecular weight) (Sigma-Aldrich) and transferred
onto a nitrocellulose membrane (GE Healthcare Life Sciences, Little
Chalfont, UK). The membrane was blocked with 5% skimmed milk and
incubated with PBS containing Tween 20 and the following primary
antibodies at 1:500 dilution: Monoclonal/polyclonal mouse/rabbit
anti-human proliferating cell nuclear antigen (PCNA) (cat no.
sc-25280), p21 (sc-397), p27 (sc-393380), p53 (sc-126), Bcl-2
(sc-7382), Bcl-2-associated X (Bax) (sc-7480), AIF (sc-13116),
Cyt-c (sc-13561), cyclooxygenase (COX)4 (sc-292052), tubulin
(sc-9104), histone (sc-10806), glucose-regulated protein (GRP)94
(sc-11402), GRP78 (sc-1050), CCAAT-enhancer-binding protein
homologous protein (CHOP) (sc-575) and GAPDH (sc-365062) (Santa
Cruz Biotechnology, Dallas, TX, USA) and cyclin-dependend kinase
(CDK)4 (cat no. 12790), cyclin D1 (1044), phosphorylated (p)-PERK
(3179), PERK (3192), eIF2α (9722), p-eIF2α (3597), ATF4 (11815),
IRE1α (3294), p-ASK (3765), ASK (37626S), p-p38 (4511), p38 (9213),
ATF6 (11815S), p-PI3K (4288S), PI3K (4249S), p-AKT (4060P), AKT
(#2920S), mammalian target of rapamycin (mTOR) (2983S) and p-mTOR
(5536S) (Cell Signaling Technology, Inc., Danvers, MA, USA)
overnight at 4°C. After washing with Tris-buffered saline
containing 1% Tween 20 (Sigma-Aldrich), membranes were incubated
with the horseradish peroxidase-conjugated: Goat anti-rabbit
immunoglobulin (Ig)G (cat. no. sc-34661) or donkey anti-goat IgG
(cat. no. sc-362265; Santa Cruz Biotechnology, Inc.) for 2 h at
4°C. The blots were developed using enhanced chemiluminescence
western blotting detection reagents (Santa Cruz Biotechnology,
Inc.). Densitometric analysis of the bands was performed using
Image Master™ 2D Elite 3.1 software (GE Healthcare Life
Sciences).
Statistical analysis
Values are expressed as the mean ± standard
deviation. Differences were analyzed using an unpaired Student's
t-test and one-way analysis of variance. All data were analyzed
using Graph Prism Software version 5 (GraphPad Inc., La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of CXC195 on the proliferation of
HepG2 cells
The present study first investigated the
antiproliferative effects of CXC195 on HepG2 cells at various
concentrations (10, 25, 50, 100, 150 and 200 µM) and time
points (6, 12, 18, 24, 48 and 96 h) using an MTT assay. As shown in
Fig. 1B, HepG2 cell viability
decreased gradually following treatment with 50 µM CXC195
for 24 h, and was maintained at its lowest levels when treated with
150–200 µM CXC195 for 24 h. Treatment with 150 µM
CXC195 induced a marked downregulation in cell viability, in a
time-dependent manner, with maximum inhibition of cell viability
detected between 24 and 96 h (Fig.
1C). Therefore, treatment with 150 µM CXC195 for 24 h
was selected for the subsequent experiments.
Annexin V/PI staining was used to evaluate the
effects of CXC195 on the type of cell death. As shown in Fig. 1D, CXC195 significantly increased
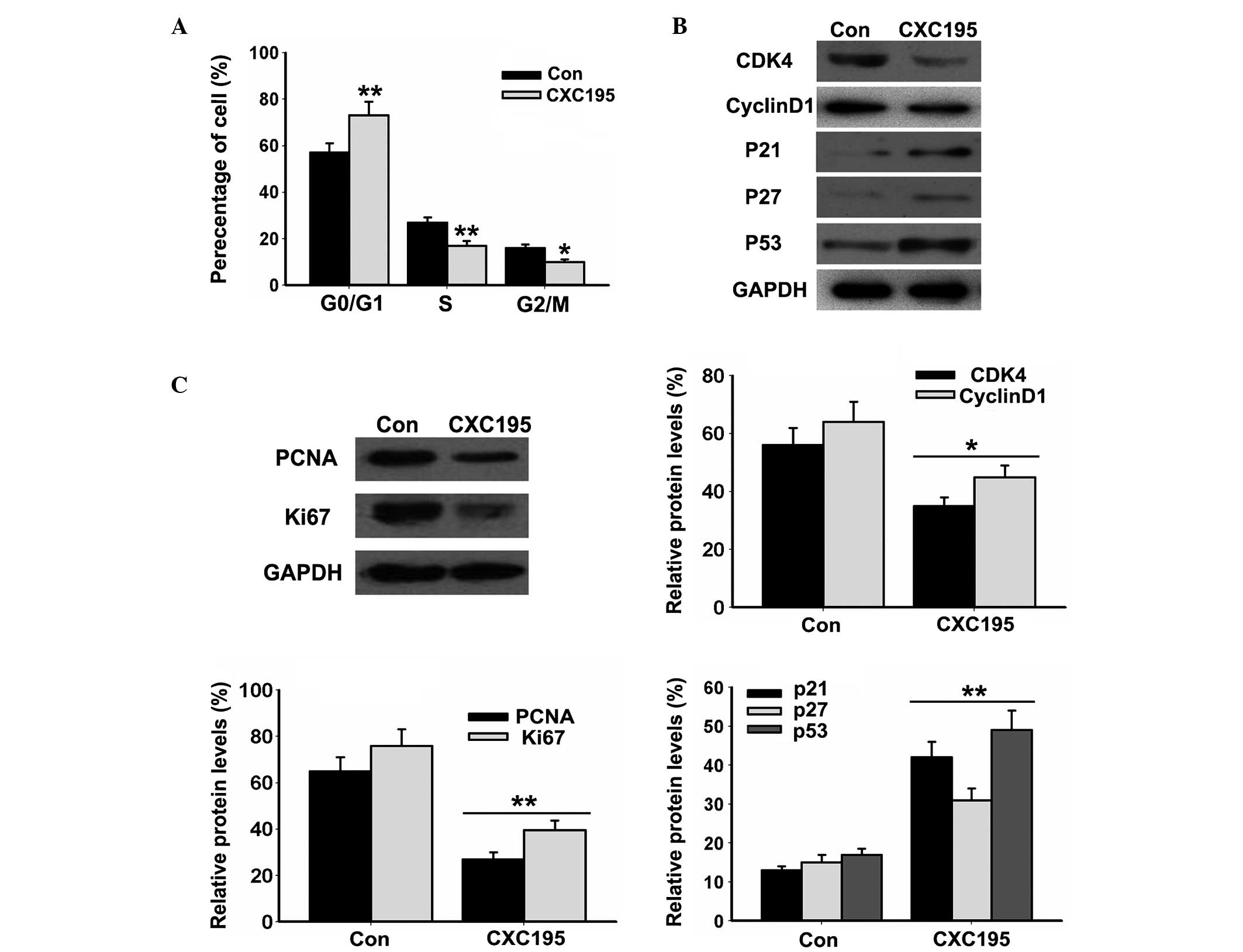
the rate of apoptosis in the HepG2 cells. Cell cycle analysis
demonstrated that CXC195 inhibited cell cycle progression of the
HepG2 cells, by increasing the proportion of cells in the
G0/G1 phase and decreasing the proportion of
cells in the S and G2/M phases (Fig. 2A). The results also demonstrated
that CXC195 significantly inhibited the expression levels of the
CDK4 and cyclin D1 cell cycle-associated proteins (Fig. 2B), and the PCNA and Ki67 markers of
cell proliferation (Fig. 2C). By
contrast, treatment with CXC195 elevated the expression levels of
the p21, p27 and p53 CDK inhibitors (Fig. 2C) in the HepG2 cells. These results
suggested that CXC195 inhibited the proliferation and induced the
apoptosis of the HepG2 cells.
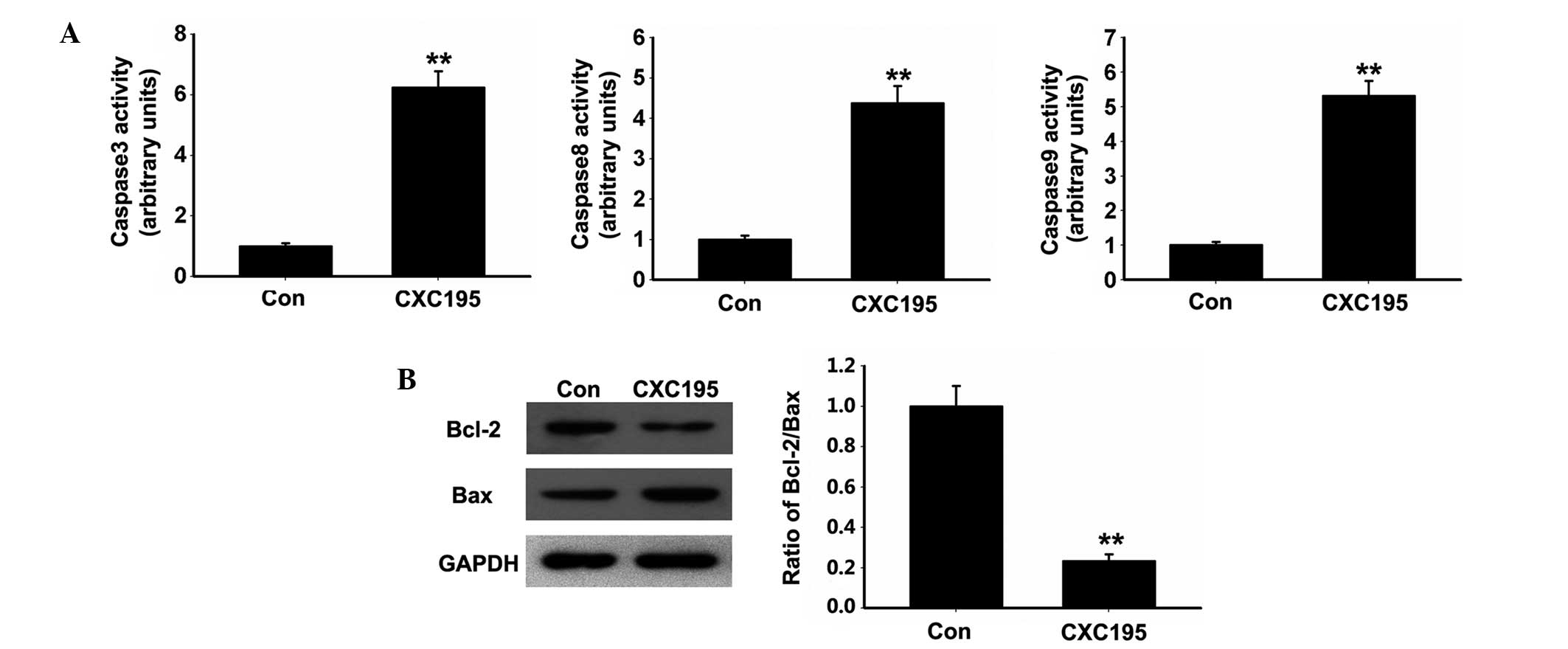
Effects of CXC195 on the caspase and
mitochondria-dependent signaling pathway in HepG2 cells
Apoptosis induced by various cytotoxic agents is
dependent on the activation of caspases, which are important in
cleaving specific target proteins (25). Therefore, the present study
assessed whether CXC195 activated caspase signaling pathways in the
HepG2 cells. As shown in Fig. 3A,
CXC195 was found to increase the activities of caspase 3, 8 and 9
following treatment of the HepG2 cells with 150 µM CXC195
for 24 h.
As Bcl-2 family proteins are reported to regulate
the mitochondria-mediated apoptosis signaling pathway by
maintaining a balance between pro- and anti-apoptotic members
(26), the present study examined
the effects of CXC195 on the expression levels of Bcl-2 family
proteins in the HepG2 cells. CXC195 increased the protein
expression of pro-apoptotic Bax, but decreased the protein
expression of Bcl-2 in the HepG2 cells. In addition, the Bcl-2/Bax
ratio was decreased in the HepG2 cells following treatment with
CXC195 (Fig. 3B).
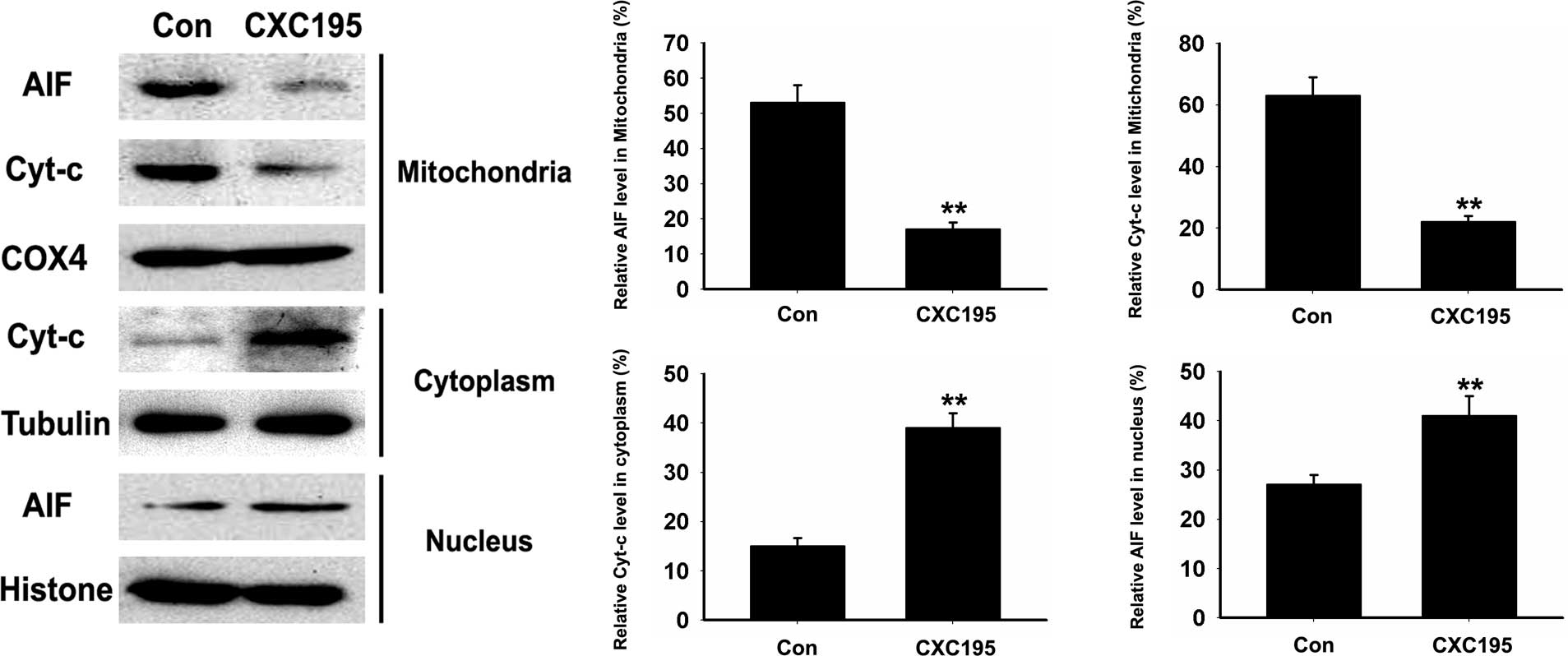
The release of pro-apoptotic proteins, including
Cyt-c and AIF, from the intermembrane space into the cytosol
is required for caspase activation, which initiates the apoptotic
program (27). As shown in
Fig. 4, following treatment with
CXC195, the expression levels of Cyt-c and AIF in the HepG2
cells significantly decreased in the mitochondria, whereas the
expression of Cyt-c increased significantly in the cytosol.
AIF can translocate from the cytosol to the nucleus, where it is
involved in chromatinolysis (27);
therefore, the present study investigated whether CXC195 induces
the relocation of AIF to the nucleus. As shown in Fig. 4, the expression of AIF was detected
in the nuclei of the CXC195-treated HepG2 cells, whereas minimal
AIF was detected in the untreated HepG2 cells. These data suggested
that CXC195 induced HepG2 cell apoptosis through caspase and
mitochondria-dependent mechanisms.
Effects of CXC195 on ER stress in HepG2
cells
There is increasing evidence that ER stress is
crucial in the regulation of apoptosis (8,9). To
confirm the hypothesis that ER stress is involved in mediating the
apoptotic effects of CXC195, the effect of CXC195 on the expression
levels of the ER stress-associated proteins, GRP78, GRP94 and CHOP,
were examined in the HepG2 cells. Western blotting demonstrated
that the expression levels of GRP78, GRP94 and CHOP increased
following treatment with 150 µM CXC195 for 24 h (Fig. 5A). ER stress induced three key
signaling pathways: IRE1α-ASK-P38/JNK, PERK-eIF2α-ATF4 and ATF6. As
shown in Fig. 5B–D, CXC195
increased the expression levels of p-PERK, p-eIF2α, p-ASK, p-p38
and p-JNK, and induced the activation of ATF4, IRE1α and ATF6 in
the HepG2 cells. These results suggested that ER stress is involved
in the CXC195-induced apoptosis of HepG2 cells.
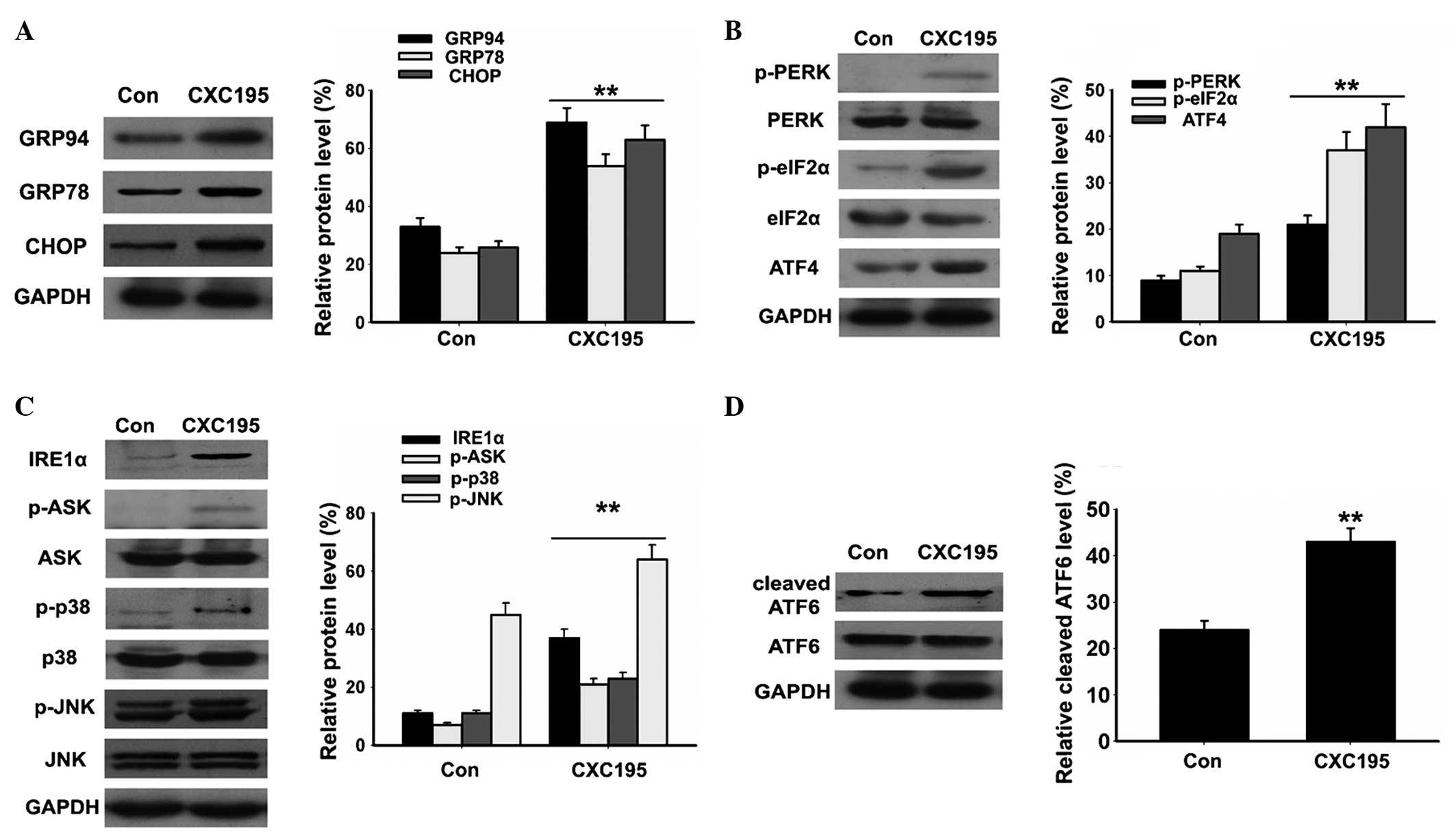 | Figure 5Effects of CXC195 on endoplasmic
reticulum stress in the HepG2 cells. The HepG2 cells were treated
with 150 µM CXC195 for 24 h. (A) Expression levels of GRP78,
GRP94 and CHOP were measured in the HepG2 cells using western blot
analysis. (B) Expression levels of p-PERK, PERK, p-eIF2α, eIF2α and
ATF4 were measured in the HepG2 cells using western blot analysis.
(C) Expression levels of IRE1α, p-ASK, ASK, p-p38, p38, p-JNK and
JNK were measured in the HepG2 cells using western blot analysis.
(D) Expression of cleaved ATF6 was measured in the HepG2 cells
using western blot analysis. GAPDH was used to confirm equal
protein loading. Data are presented as the mean ± standard
deviation (n=6). **P<0.01, vs. control. GRP,
glucose-regulated protein; CHOP, CCAAT-enhancer-binding protein
homologous protein; p-, phosphorylated; PERK, protein kinase R-like
endoplasmic reticulum kinase; eIF2α, eukaryotic translation
initiation factor 2α; AIF4, apoptosis-inducing factor 4; IRE1α,
inositol-requiring enzyme 1α; ASK, apoptosis signal-regulating
kinase; JNK, c-Jun N-terminal kinase; ATF6, activating
transcription factor 6; Con, control. |
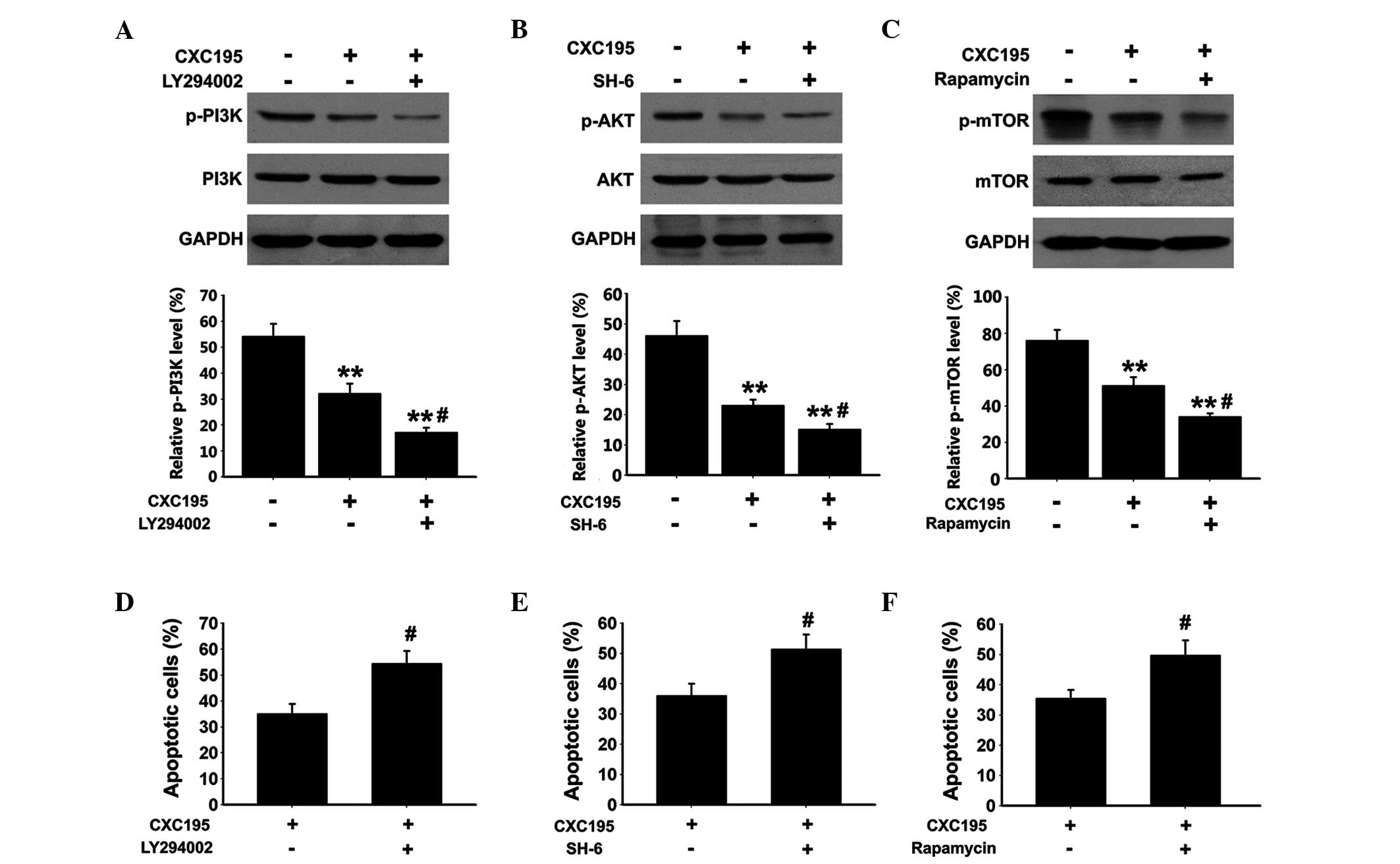
Effects of CXC195 on PI3K/Akt/mTOR
activation in HepG2 cells
To further understand the molecular mechanisms
underlying the induction of apoptosis in HepG2 cells following
treatment with CXC195, the expression of PI3K, Akt and mTOR, which
are critical signaling proteins associated with cell apoptosis,
were examined. As shown in Fig.
6A–C, the protein expression levels of p-PI3K, p-Akt and p-mTOR
were determined using western blot analysis, which demonstrated
that CXC195 significantly inhibited the phosphorylation of PI3K at
Tyr458, Akt at Ser473 and mTOR at Ser2448. The HepG2 cells were
also treated with either the selective PI3K inhibitor, LY294002,
the selective Akt inhibitor, SH-6, or the selective mTOR inhibitor,
rapamycin. The selective inhibitors were observed to further
inhibit the phosphorylation of PI3K, Akt and mTOR in the
CXC195-treated HepG2 cells. Furthermore, the HepG2 cells treated
with LY294002/SH-6/rapamycin and CXC195 exhibited significant
increases in apoptosis, compared with the cells treated with CXC195
only (Fig. 6D–F). These results
indicated that the pro-apoptotic effects of CXC195 in HepG2 cells
were associated with inhibition of the PI3K/Akt/mTOR signaling
cascade.
Discussion
Apoptosis is defined as programmed cell death and
has been suggested as an efficient antitumor mechanism (25). Malignant tumor cells can be
eliminated following treatment with anticancer chemotherapies
though apoptosis (28), and
apoptosis can be controlled by extrinsic and intrinsic signaling
pathways (29). The extrinsic
signaling pathway involves the death receptor, in which the death
domains target caspase 8. The activation of caspase 8 subsequently
activates caspase 3, which induces apoptosis (29). Caspase 3 is a member of the caspase
family of enzymes, which are major inducers of apoptosis, and the
levels of caspase 3 activity are often measured in investigations
into antitumor drugs targeting apoptosis (29). In the present study, the activation
of caspases 3 and 8 were induced following treatment with 150
µM CXC195 for 24 h. The results indicated that CXC195
induced HepG2 cell apoptosis through the regulation of the
extrinsic signaling pathway.
Members of the Bcl-2 family (intrinsic or
mitochondrial signaling pathway) are also key regulators of
apoptosis and the intrinsic pathway of apoptosis is associated with
DNA damage. Oligomerization of Bax or Bak, which are pro-apoptotic
proteins of the Bcl-2 family, promotes the release of mitochondrial
Cyt-c into the cytoplasm, which combines with the caspase 9
precursor to form an apoptosis complex, and the activation of
caspase 9 activates caspase 3 to induce apoptosis (30). The results of the present study
demonstrated that decreased expression of anti-apoptotic Bcl-2 and
increased expression of pro-apoptotic Bax were observed in the
CXC195-treated HepG2 cells. In addition, the activation of caspase
9 was induced following treatment with CXC195. Furthermore, the
levels of Cyt-c and AIF significantly decreased in the
mitochondria, whereas the level of Cyt-c increased in the
cytosol, and that of AIF increased in the nucleus. Therefore,
CXC195 may have initiated the intrinsic signaling pathway, which
induced caspase-dependent apoptotic signaling in the HepG2
cells.
The Bcl-2 family proteins also localise to the ER
during stress, where their suggested functions include the
regulation of apoptosis and the UPR (10,11).
ER stress occurs when ER homeostasis is lost due to an overload of
protein folding in the ER, and the UPR induces the upregulation of
ER-resident chaperones, including GRP78 and GRP94 (8,9). The
results of the present study demonstrated that CXC195 increased the
expression levels of GRP78 and GRP94 in the HepG2 cells.
Abnormalities in ER function can cause ER stress, which results in
the UPR and includes three key signaling pathways:
IRE1α-ASK-P38/JNK, PERK-eIF2α-ATF4 and ATF6, which act as the
sensors of ER stresses (8,9). In the present study, CXC195 increased
the activation of these signaling pathways in the HepG2 cells. The
GRP78 ER stress sensor and three branches of the UPR pathway,
together with the CHOP mediator constitute an essential and
dominant arm, and the interaction between this arm and TRB3 has
been demonstrated in wild-type HepG2 cells. Previous studies have
reported that the UPR pathway was the candidate pathway owing to
its ability to initiate CHOP-mediated programmed cell death, which
is independent of the activation of the intrinsic and extrinsic
apoptotic signaling pathways (31,32).
In agreement with the effect of CXC195 on ER stress in the present
study, CXC195 induced increased expression levels of CHOP in the
HepG2 cells. These results indicated that ER stress contributed to
the pro-apoptotic effect of CXC195 in HepG2 cells.
The PI3K-Akt-mTOR signaling pathway is one of the
most important signaling pathways in the regulation of cell
proliferation, growth and apoptosis in various types of cancer, as
it increases the activity of anti-apoptotic Akt (33,34).
The phosphorylation of PI3K, Akt and mTOR is required for the
suppression of cancer cell apoptosis and tumor progression
(35). The PI3K/Akt/mTOR signaling
pathway inhibits apoptosis by inactivating important members of the
apoptotic cascade, including caspase 3 and 9, Bax and Bad (35,36).
UPR initiates programmed cell death through the transcriptional
regulation of genes involved in cell death, including Bcl-2, Bim,
DR5 and TRB3. TRB3 is a pseudokinase, which inhibits the activation
of Akt by directly binding to Akt and preventing the
phosphorylation at Ser473 of Akt (36). Therefore, there is cross-talk
between the UPR and Akt signaling pathways. The data obtained in
the present study revealed that CXC195 downregulated the
phosphorylation of PI3K, Akt and mTOR, whereas the inhibitors of
these proteins (LY294002, SH-6 and rapamycin, respectively)
enhanced the inhibitory effects of CXC195 in the HepG2 cells. In
addition, these inhibitors increased the number of apoptotic HepG2
cells induced by CXC195. Overall, these results t indicated that
the pro-apoptotic effects of CXC195 in HepG2 cells were associated
with inhibiting the activation of the PI3K-Akt-mTOR signaling
cascade.
In conclusion, the present study reported for the
first time, to the best of our knowledge, that CXC195 inhibited the
proliferation and induced the apoptosis of HepG2 cells, which was
mediated through the caspase-, mitochondrial- and ER
stress-dependent signaling pathways. In addition, CXC195 caused
inactivation of the PI3K-Akt-mTOR signaling pathway, which may
exhibit a critical function in CXC195-induced apoptosis in HepG2
cells.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruix J and Llovet JM: Major achievements
in hepatocellular carcinoma. Lancet. 373:614–616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Whittaker S, Marais R and Zhu AX: The role
of signaling pathways in the development and treatment of
hepatocellular carcinoma. Oncogene. 29:4989–5005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wei L, Lu N, Dai Q, Rong J, Chen Y, Li Z,
You Q and Guo Q: Different apoptotic effects of wogonin via
induction of H(2) O(2) generation and Ca(2+) overload in malignant
hepatoma and normal hepatic cells. J Cell Biochem. 111:1629–1641.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dara L, Ji C and Kaplowitz N: The
contribution of endoplasmic reticulum stress to liver diseases.
Hepatology. 53:1752–1763. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malhi H and Kaufman RJ: Endoplasmic
reticulum stress in liver disease. J Hepatol. 54:795–809. 2011.
View Article : Google Scholar
|
|
9
|
Xu M, Lu N, Zhang H, Dai Q, Wei L, Li Z,
You Q and Guo Q: Wogonin induced cytotoxicity in human
hepatocellular carcinoma cells by activation of unfolded protein
response and inactivation of Akt. Hepatol Res. 43:890–905. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oakes SA, Lin SS and Bassik MC: The
control of endoplasmic reticulum-initiated apoptosis by the BCL-2
family of proteins. Curr Mol Med. 6:99–109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rong Y and Distelhorst CW: Bcl-2 protein
family members: Versatile regulators of calcium signaling in cell
survival and apoptosis. Annu Rev Physiol. 70:73–91. 2008.
View Article : Google Scholar
|
|
12
|
Zheng CY, Xiao W, Zhu MX, Pan XJ, Yang ZH
and Zhou SY: Inhibition of cyclooxygenase-2 by tetramethylpyrazine
and its effects on A549 cell invasion and metastasis. Int J Oncol.
40:2029–2037. 2012.PubMed/NCBI
|
|
13
|
Wang Y, Fu Q and Zhao W:
Tetramethylpyrazine inhibits osteosarcoma cell proliferation via
downregulation of NF-κB in vitro and in vivo. Mol Med Rep.
8:984–988. 2013.PubMed/NCBI
|
|
14
|
Yu K, Chen Z, Pan X, Yang Y, Tian S, Zhang
J, Ge J, Ambati B and Zhuang J: Tetramethylpyrazine-mediated
suppression of C6 gliomas involves inhibition of chemokine receptor
CXCR4 expression. Oncol Rep. 28:955–960. 2012.PubMed/NCBI
|
|
15
|
Fu YS, Lin YY, Chou SC, Tsai TH, Kao LS,
Hsu SY, Cheng FC, Shih YH, Cheng H, Fu YY and Wang JY:
Tetramethylpyrazine inhibits activities of glioma cells and
glutamate neuro-excitotoxicity: Potential therapeutic application
for treatment of gliomas. Neuro Oncol. 10:139–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yin J, Yu C, Yang Z, He JL, Chen WJ, Liu
HZ, Li WM, Liu HT and Wang YX: Tetramethylpyrazine inhibits
migration of SKOV3 human ovarian carcinoma cells and decreases the
expression of interleukin-8 via the ERK1/2, p38 and AP-1 signaling
pathways. Oncol Rep. 26:671–679. 2011.PubMed/NCBI
|
|
17
|
Zhang Y, Liu X, Zuo T, Liu Y and Zhang JH:
Tetramethylpyrazine reverses multidrug resistance in breast cancer
cells through regulating the expression and function of
P-glycoprotein. Med Oncol. 29:534–538. 2012. View Article : Google Scholar
|
|
18
|
Yi B, Liu D, He M, Li Q, Liu T and Shao J:
Role of the ROS/AMPK signaling pathway in
tetramethylpyrazine-induced apoptosis in gastric cancer cells.
Oncol Lett. 6:583–589. 2013.PubMed/NCBI
|
|
19
|
Wang XB, Wang SS, Zhang QF, Liu M, Li HL,
Liu Y, Wang JN, Zheng F, Guo LY and Xiang JZ: Inhibition of
tetramethyl-pyrazine on P-gp, MRP2, MRP3 and MRP5 in multidrug
resistant human hepatocellular carcinoma cells. Oncol Rep.
23:211–215. 2010.
|
|
20
|
Cheng XC, Liu XY, Xu WF, Guo XL and Ou Y:
Design, synthesis and biological activities of novel ligustrazine
derivatives. Bioorg Med Chem. 15:3315–3320. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ou Y, Dong X, Liu XY, Cheng XC, Cheng YN,
Yu LG and Guo XL: Mechanism of tetramethylpyrazine analogue CXC195
inhibition of hydrogen peroxide-induced apoptosis in human
endothelial cells. Biol Pharm Bull. 33:432–438. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu H, Wei X, Chen L, Liu X, Li S, Liu X
and Zhang X: Tetramethylpyrazine analogue CXC195 protects against
cerebral ischemia/reperfusion injury in the rat by an antioxidant
action via inhibition of NADPH oxidase and iNOS expression.
Pharmacology. 92:198–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen L, Wei X, Hou Y, Liu X, Li S, Sun B,
Liu X and Liu H: Tetramethylpyrazine analogue CXC195 protects
against cerebral ischemia/reperfusion-induced apoptosis through
PI3K/Akt/GSK3β pathway in rats. Neurochem Int. 66:27–32. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Masoud L, Vijayasarathy C,
Fernandez-Cabezudo M, Petroianu G and Saleh AM: Effect of malathion
on apoptosis of murine L929 fibroblasts: A possible mechanism for
toxicity in low dose exposure. Toxicology. 185:89–102. 2003.
View Article : Google Scholar
|
|
25
|
Kuwana T and Newmeyer DD: Bcl-2-family
proteins and the role of mitochondria in apoptosis. Curr Opin Cell
Biol. 15:691–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Loeffler M and Kroemer G: The
mitochondrion in cell death control: Certainties and incognita. Exp
Cell Res. 256:19–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Palanca JM, Aguirre-Rueda D, Granell MV,
Aldasoro M, Garcia A, Iradi A, Obrador E, Mauricio MD, Vila J,
Gil-Bisquert A and Valles SL: Sugammadex, a neuromuscular blockade
reversal agent, causes neuronal apoptosis in primary cultures. Int
J Med Sci. 10:1278–1285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hannun YA: Apoptosis and the dilemma of
cancer chemotherapy. Blood. 89:1845–1853. 1997.PubMed/NCBI
|
|
29
|
Chiantore MV, Vannucchi S, Mangino G,
Percario ZA, Affabris E, Fiorucci G and Romeo G: Senescence and
cell death pathways and their role in cancer therapeutic outcome.
Curr Med Chem. 16:287–300. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He K, Si P, Wang H, Tahir U, Chen K, Xiao
J, Duan X, Huang R and Xiang G: Crocetin induces apoptosis of
BGC-823 human gastric cancer cells. Mol Med Rep. 9:521–526.
2014.
|
|
31
|
Rao RV, Ellerby HM and Bredesen DE:
Coupling endoplasmic reticulum stress to the cell death program.
Cell Death Differ. 11:372–380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
33
|
Huang KF, Zhang GD, Huang YQ and Diao Y:
Wogonin induces apoptosis and down-regulates survivin in human
breast cancer MCF-7 cells by modulating PI3K-Akt pathway. Int
Immunopharmacol. 12:334–341. 2012. View Article : Google Scholar
|
|
34
|
Sui T, Ma L, Bai X, Li Q and Xu X:
Resveratrol inhibits the phosphatidylinositide 3-kinase/protein
kinase B/mammalian target of rapamycin signaling pathway in the
human chronic myeloid leukemia K562 cell line. Oncol Lett.
7:2093–2098. 2014.PubMed/NCBI
|
|
35
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: Rationale and
promise. Cancer cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamaguchi H and Wang HG: CHOP is involved
in endoplasmic reticulum stress-induced apoptosis by enhancing DR5
expression in human carcinoma cells. J Biol Chem. 279:45495–45502.
2004. View Article : Google Scholar : PubMed/NCBI
|