Introduction
Cardiomyocyte hypertrophy is a compensatory heart
response towards harmful stimuli, and patients with cardiomyocyte
hypertrophy usually have a poor prognosis, with the onset of heart
systolic and diastolic dysfunction, and ultimately sudden heart
failure-induced mortality (1). In
order to develop novel therapeutic strategies, the molecular
pathways underlying cardiomyocyte hypertrophy require further
elucidation.
MicroRNAs (miRNAs; miRs) belong to a group of 20–22
nucleotide-long non-coding RNAs, which suppress the expression of
target mRNAs by binding their 3′-untranslated region (UTR) in a
miRNA recognition element (MRE)-dependent mechanism (2). Accumulated evidence has demonstrated
that miRNA is closely associated with the initiation and
progression of cardiomyocyte hypertrophy (3). miR-1 has been demonstrated to
suppress cardiac hypertrophy by decreasing the expression levels of
calmodulin and Mef2a (4). The
anti-hypertrophic effect of miR-1 may also be associated
with the insulin-like growth factor signaling pathway (5), the twinfilin-1 cytoskeleton
regulatory protein (6), NCX1 and
AnxA5 proteins (7). The
introduction of miR-1 was demonstrated to reverse the
pathological remodeling induced by pro-hypertrophic stimuli
(8). Isoproterenol (ISO) is a
β-adrenoceptor activator, which has been confirmed to exert
cardioprotective effects by promoting the expression of
miR-1 (9). Notably, cardiac
miR-1 abundance can be indirectly detected by evaluating the
serum expression levels of fatty acid binding protein 3 in patients
with cardiomyocyte hypertrophy, suggesting that miR-1 is a
promising diagnostic biomarker for susceptibility to cardiomyocyte
hypertrophy (10). These findings
indicate that miR-1 is an attractive epigenetic factor
regulating cardiac hypertrophy. However, the downstream effectors
of miR-1 have not been examined in full.
Nuclear factor of activated T cells cytoplasmic 3
(NFATC3) is important in the initiation and progression of
cardiomyocyte hypertrophy. Calcineurin is a serine/threonine
protein phosphatase, which dephosphorylates NFATC3, resulting in
its nuclear localization, and these events lead to the pathological
remodeling of hypertrophic cardiomyocytes (11,12).
However, the regulatory pathways located upstream of NFATC3,
particularly miRNAs targeting this pro-hypertrophic factor, remain
to be fully elucidated.
The present study aimed to investigate the possible
molecular associations between miR-1 and NFATC3 during the
response of cardiomyocytes in the presence of pro-hypertrophic
stimuli. The present study may enhance the current understanding of
the molecular mechanisms associated with cardiac hypertrophy.
Materials and methods
Patients and samples
Heart tissue samples were collected from patients
succumbed to cardiac hypertrophy (n=15; age, 40–86 years; males,
n=7; females, n=8) and patients who succumbed to conditions other
than cardiac hypertrophy as controls (n=15; age, 32–77 years;
males, n=9; females, n=6) at the First Affiliated Hospital of
Xinxiang Medical University (Weihui, China). Fresh tissues were
obtained with written informed consent from all patients according
to protocols approved by Ethical Review Board in the First
Affiliated Hospital of Xinxiang Medical University (Weihui,
China).
Cardiomyocyte isolation and culture
The experimental animal protocol of the present
study was approved by the ethics committee of the First Affiliated
Hospital of Xinxiang Medical University (Weihui, China).
Cardiomyocytes were obtained from 8-week-old Wistar rats (n=9;
male; weight, 4–6 g; purchased from Jinfeng Experimental Animal
Company, Jinan, China) kept in ventilated housing with 50–60%
humidity at 18–22° C following previously described procedures
(13). Briefly, the hearts were
harvested and homogenized with HEPES (Sigma-Aldrich)-buffered
saline following sacrification of the animals by CO2
aspiration. The heart tissue samples were subsequently treated with
1.2 mg/ml pancreatin and 0.14 mg/ml collagenase (Worthington
Biochemical Corporation, Lakewood, NJ, USA) in HEPES-buffered
saline. The cells were then resuspended in Dulbecco's modified
Eagle's medium/F-12 (Invitrogen Life Technologies, Carlsbad, CA,
USA) supplemented with 5% heat-inactivated horse serum (Invitrogen
Life Technologies), 0.1 mM ascorbate (Sigma-Aldrich),
insulin-transferring-sodium selenite medium (Sigma-Aldrich), 100
U/ml penicillin, 100 µg/ml streptomycin and 0.1 mM
bromodeoxyuridine (all from Sigma-Aldrich) at 1×106
cells/ml prior to being plated at 5×105 cells/well and
cultured at 37° C for 1 h. Cells were treated with ISO (10
µM; Sigma-Aldrich) and Aldomet (Aldo; 1 µM;
Sigma-Aldrich) for the indicated durations (0, 1, 2, 4 or 8 h).
Analysis of miR-1 expression using
RT-qPCR
Total RNA was extracted from the coronary tissue
samples using TRIzol™ reagent (Sigma-Aldrich, St Louis, MO, USA).
The RT reaction was performed using the TaqMan® MicroRNA
Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Waltham, MA, USA) following the manufacturer's
instructions. qPCR was performed using TaqMan® 2X
Universal PCR Master mix (Applied Biosystems) on a CFX96™ Real-Time
PCR Detection system (Bio-Rad Laboratories, Hercules, CA, USA)
supplied with analytical software. The reactions were incubated in
an 9700 Thermocycler (Applied Biosystems) in a 96-well plate for 30
min at 16° C, 30 min at 42° C, 5 min at 85° C and then held at 4°
C. The primers are as follows: miR-1, GAGTTGCCTGACTTCT, and
TTCAACAGGCCTAGTA (Thermo Fisher, Grand Island, NY, USA); β-myosin
heavy chain (β-MHC), CTGGCACCGTGGACTACAAC, and
CGCACAAAGTGAGGATAGGGT (Sangon Biotech, Shanghai, China). The ∆∆CT
method was used to quantify the abundance of miRNA or mRNA.
Adenoviral construction and
infection
A recombinant adenovirus expressing the
constitutively active form of NFATC3 (Ad-NFATC3) was obtained from
Dr Michael C. Naski (Department of Pathology, University of Texas
Health Science Center, San Antonio, TX, USA) (14). An adenoviral vector containing
enhanced green fluorescent protein (Ad-EGFP), was provided by Dr
Liu (Medical College, Qingdao University, Qingdao, China) (15) using a TCID50 Quick Determination
kit (Jimei Biotech, Shanghai, China), and used as a negative
control. The propagation and titration of the adenoviruses was
performed as previously described (16). The adenoviruses were added to the
cultures 48 h prior to the experiments at a multiplicity of
infection of 10 and incubated at 37° C for 3 h, following which the
adenoviruses were removed by replacing with fresh culture
medium.
Measurement of cell surface area
The cell surface area of the experimental cells was
determined based on the fluorescent staining of F-actin in the
cardiomyocytes, as previously described (17). Briefly, following the indicated
treatments, the cardiomyocytes were fixed with 4% paraformaldehyde
and treated with 0.1% Triton X-100. The cells were then incubated
overnight with fluorescent phalloidin-tetramethylrhodamine
conjugate (Sigma-Aldrich) at room temperature, prior to being
visualized using a Zeiss LSM 510 META laser confocal microscope
(Zeiss, Oberkochen, Germany). A total of 100 cells were counted in
30–50 randomly-selected fields. The size of the area outlined by
F-actin was determined using ImageJ software (version 1.48u;
National Institutes of Health, Bethesda, MD, USA). The data are
expressed as the mean ± standard deviation.
Analysis of the protein/DNA ratio
The evaluation of the protein/DNA ratio in the
experimental cardiomyocytes was performed, as described in a
previous study (13). Salmon
sperm, perchloric acid and KOH were purchased from Shanghai Sangon
Biotech. Hoechst dye and human serum albumin were purchased from
Sigma-Aldrich.
Western blot analysis
The procedures used to perform western blotting
assays were those previously described (15). Briefly, the total protein was
extracted from the cells using PER Mammalian Protein Extraction
reagent (200 µl for 1×106 cells; Thermo Fisher
Scientific) and the protein concentration was determined using the
bicinchoninic acid assay (Invitrogen Life Technologies). A total of
20 µg protein per lane was separated by 10% SDS-PAGE and
subsequent transfer onto 0.45-µm nitrocellulose membranes
(Millipore, Billerica, MA, USA). The membranes were then blocked
with 5% fat-free dry milk and incubated with the indicated primary
antibodies (Abs), including GAPDH (D16H11) XP® rabbit
monoclonal Ab (cat no. 5174; 1:1,000; Cell Signaling Technology,
Inc., Danvers, MA, USA) and NFATc3 mouse monoclonal Ab (cat no.
sc-8405; 1:500, Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
overnight at 4° C. After three washes with phosphate-buffered
saline containing Tween 20, membranes were incubated with goat
anti-rabbit and anti-mouse antibodies (cat. nos. ZDR-5306 and
ZDR-5307, respectively; 1:5,000; ZSGB-BIO, Beijing, China) for 1 h.
Finally, the blots were visualized using SuperSignal West Dura
Extended Duration substrate (Thermo Fisher Scientific, Inc.).
ImageJ software was used for quantification of the blots.
Treatment of the miRNA mimics
The miR-1 and control mimics were synthesized
by Shanghai GenePharma Co., Ltd. (Shanghai, China). The cells were
transfected with either 300 nM control mimics or 300 nM
miR-1 mimics. The experiments described above were performed
48 h post-transfection.
Luciferase assay
To investigate whether NFATC3 is a target of
miR-1, two recombinant vectors were constructed to express
luciferase under the regulation of the 3′-UTR of NFATC3 mRNA,
containing either wild-type miR-1 MREs or mutant MREs
(pMIR-REPORT-NFATC3-wt and pMIR-REPORT-NFATC3-mut, respectively).
At 48 h post-transfection with Lipofectamine® 2000
(Invitrogen Life Technologies), lysis buffer (Promega Corporation,
Madison, WI, USA) was added to the cell culture, and the expression
of luciferase was detected using a Dual-Luciferase Reporter system
(Promega Corp.), according to the manufacturers' instructions.
Statistical analysis
Statistical significance was calculated using
Students' t-test. All statistical analyses were performed
using SPSS software version 19.0 (International Business Machines,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
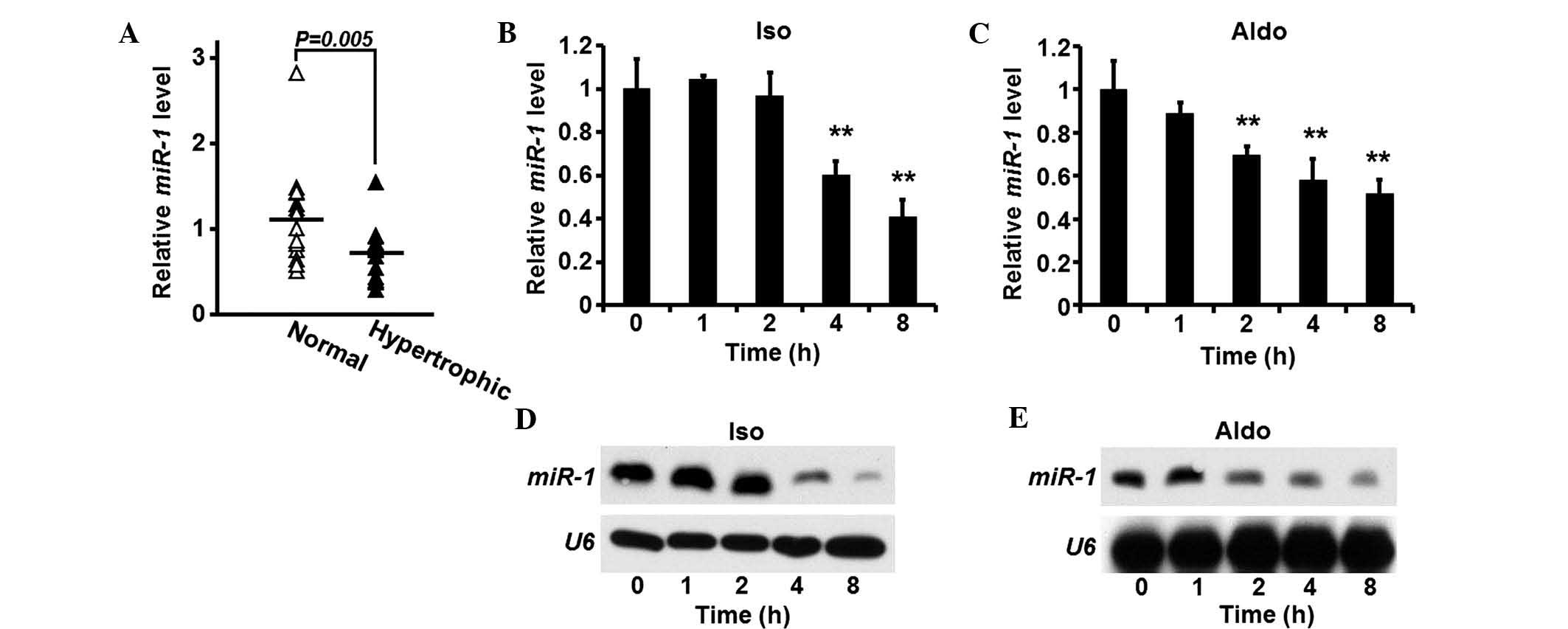
miR-1 is suppressed in heart tissue
samples of patients with cardiac hypertrophia
Heart tissue samples were obtained from patients
suffering from cardiomyocyte hypertrophia (n=15). In addition,
cardiac tissue samples obtained from donors without heart diseases,
which served as controls (n=15). The expression levels of
miR-1 were quantified in the tissue samples, and compared
between the two groups. The expression of miR-1 was
significantly suppressed in the hypertrophic heart tissue samples,
compared with the control group (Fig.
1A).
Expression of miR-1 is reduced in rat
cardiomyocytes following pro-hypertrophic treatment
As miR-1 is aberrantly expressed in human
hypertrophic heart tissue samples, the expression levels of
miR-1 were also investigated in rat cardiomyocytes. Isolated
rat cardiomyocytes were treated with ISO or Aldo, and the
expression levels of miR-1 were quantified. The expression
levels of miR-1 gradually decreased following each treatment
(Fig. 1B and C). Western blotting
was used to confirm the reduction in the expression levels of
miR-1 induced by ISO or Aldo (Fig. 1D and E).
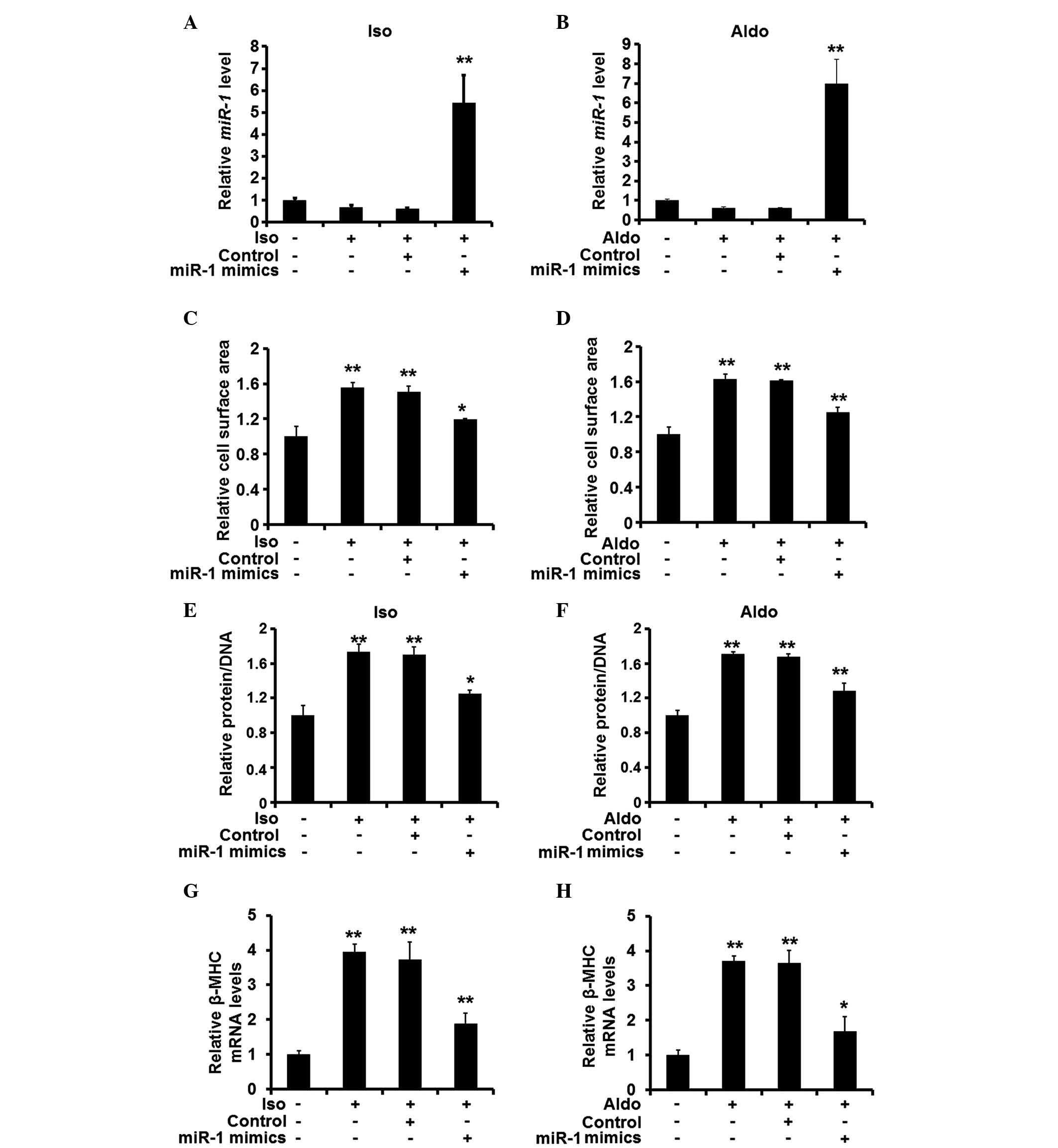
miR-1 attenuates hypertrophic responses
in rat cardiomyocytes in vitro
As the expression levels of miR-1 were found
to be reduced in the hypertrophic heart tissue samples, the
potential changes in the expression levels of miR-1 were
investigated, in order to determine whether the changes in
expression levels affected the hypertrophic response of the
cardiomyocytes. Synthetic miRNA mimics were then used to restore
the expression of miR-1 in the cardiomyocytes pre-treated
with ISO or Aldo (Fig. 2A and B).
Restoration of the expression of miR-1 was demonstrated to
reduce the cell surface area of the cardiomyocytes (Fig. 2C and D). In addition, the ratio of
protein to DNA was decreased when miR-1 was overexpressed
(Fig. 2E and F), and the mRNA
expression levels of β-MHC were also reduced (Fig. 2G and H). These results suggested
that miR-1 suppression is required for the pro-hypertrophic
activity of ISO and Aldo.
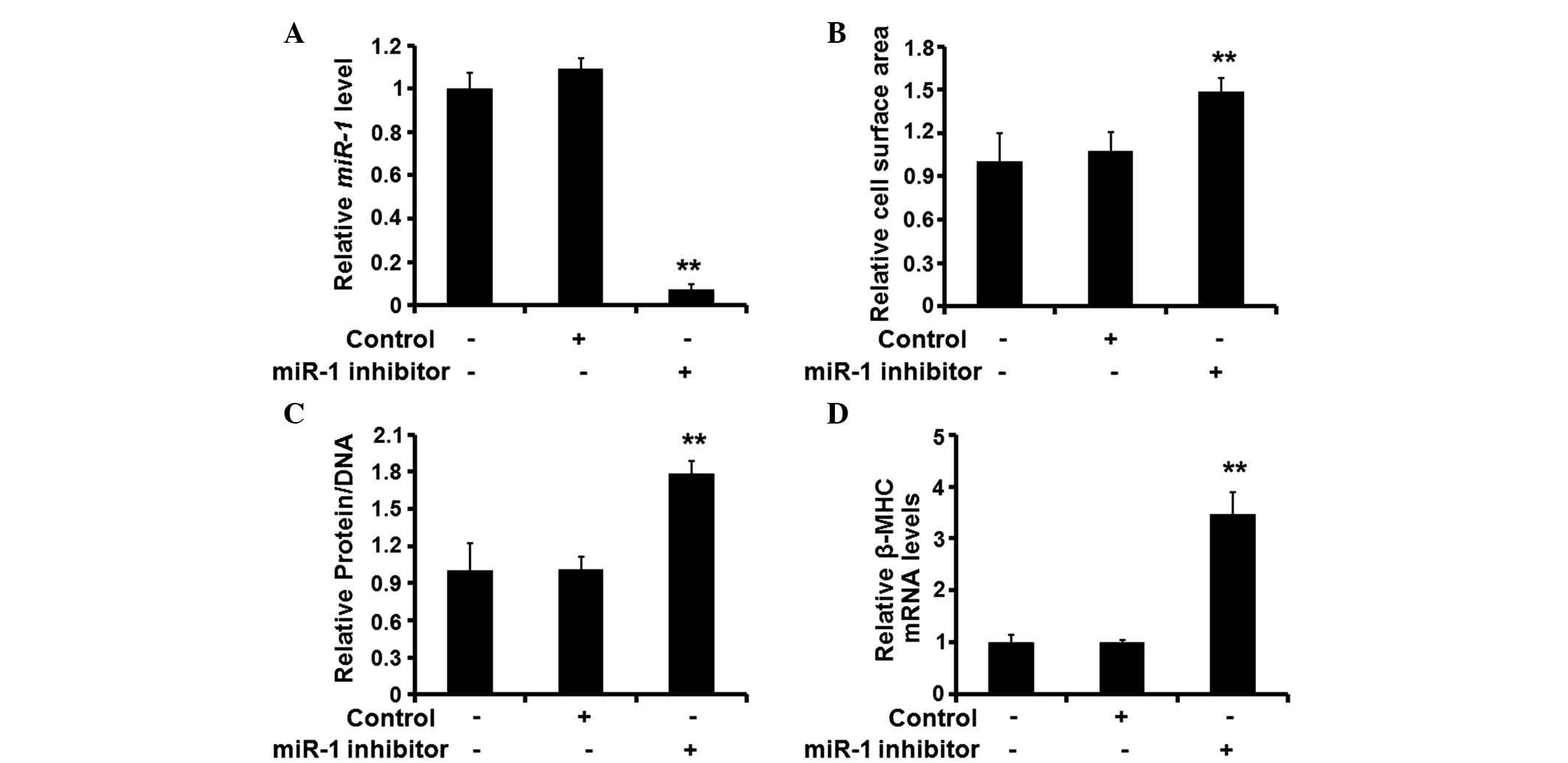
Silencing of miR-1 is sufficient to
initiate cardiomyocyte hypertrophy
Following establishment of the role of miR-1
in the hypertrophic response of rat cardiomyocytes, the present
study investigated the effects of miR-1 silencing on the
cardiomyocytes. A synthetic inhibitor specific for miR-1 was
used to reduce the expression levels of endogenous miR-1
(Fig. 3A). Following treatment
with the inhibitor, the surface area of the cardiomyocytes
increased significantly (Fig. 3B),
and the protein/DNA ratio (Fig.
3C) and expression of β-MHC also increased (Fig. 3D). These results suggested that
silencing of the expression of miR-1 induced cardiomyocyte
hypertrophy.
miR-1 targets NFATC3 in rat
cardiomyocytes
As miR-1 is important in suppressing
cardiomyocyte hypertrophy, the present study investigated its
underlying molecular mechanisms. Potential targets of miR-1
were screened using the online database, TargetScan (http://www.targetscan.org/). NFATC3, a putative
transcription factor that enhances hypertrophic response by
promoting the expression of myocardin (18), was among the predicted targets of
miR-1. One copy of miR-1 MRE was located within the
3′-UTR of NFATC3 mRNA (Fig. 4A).
There was an inverse association between the expression levels of
miR-1 and NFATC3 in the human heart tissue samples (Fig. 4B). The expression levels of NFATC3
increased and decreased in the rat cardiomyocytes following
miR-1 silencing and overexpression, respectively (Fig. 4C and D). A luciferase reporter
assay demonstrated that the suppression of endogenous miR-1
increased the expression levels of luciferase by
pMIR-REPORT-NFATC3-wt (Fig. 4E),
whereas increased expression of miR-1 reduced the expression
levels of luciferase by pMIR-REPORT-NFATC3-wt, but not
pMIR-REPORT-NFATC3-mut (Fig. 4F).
These results suggested that miR-1 targeted and suppressed
the expression of NFATC3.
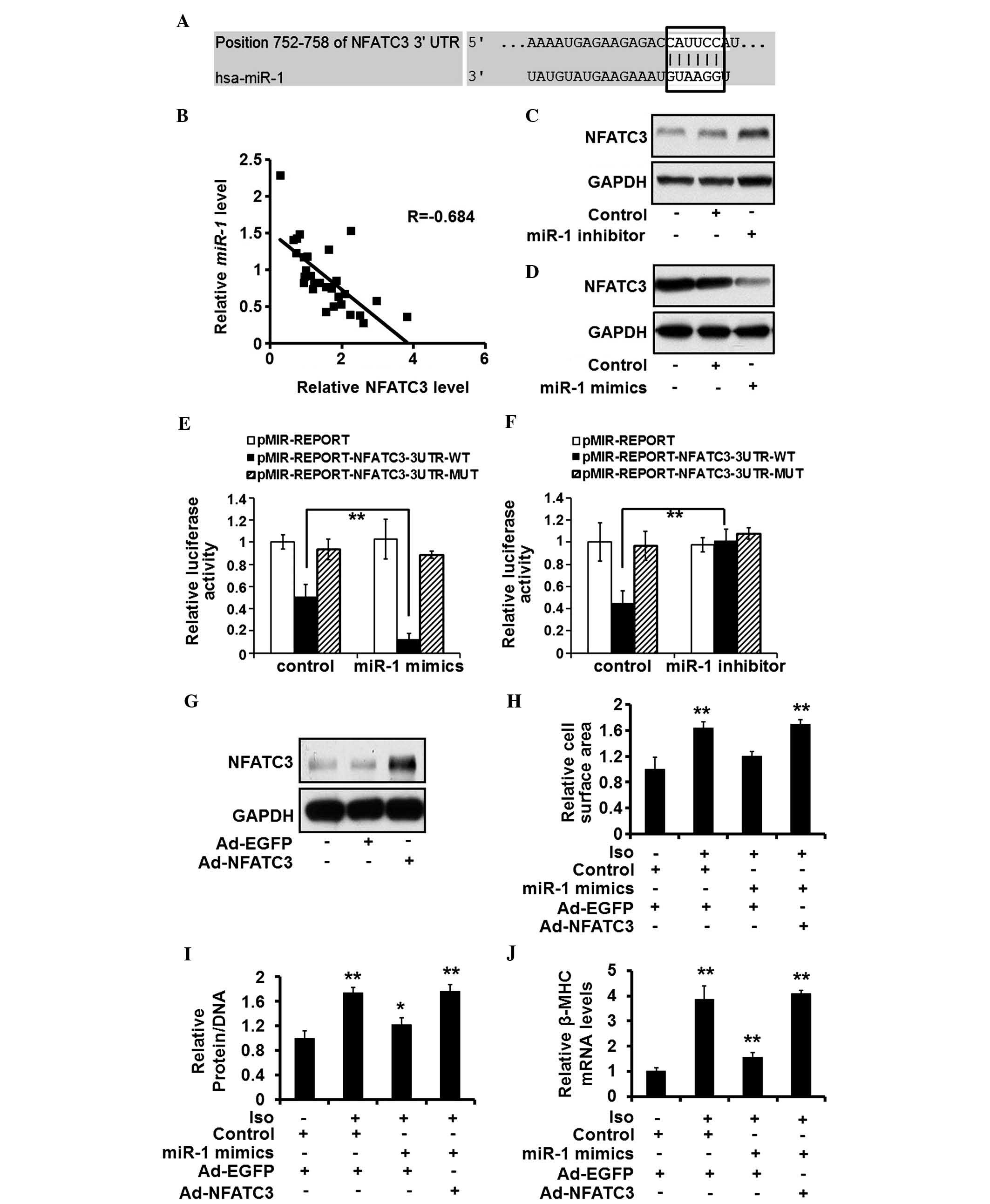 | Figure 4miR-1 targets NFATC3 and
NFATC3 suppression is required for the anti-hypertrophic effects of
miR-1. (A) Putative miR-1 seed sequence within the
3′-UTR of NFATC3 mRNA is highlighted. (B) Correlation between the
expression of miR-1 and NFATC3, determined using Pearson's
analysis. (C and D) Protein expression levels of NFATC3, quantified
using western blotting in rat cardiomyocytes treated with control,
miR-1 mimics (30 nM) or inhibitor (30 nM). (E and F)
Expression levels of luciferase induced by pMIR-REPORT,
pMIR-REPORT-NFATC3-3UTR-WT, or pMIR-REPORT-NFATC3-3UTR-MUT were
estimated in the rat cardiomyocytes treated with control,
miR-1 mimics (30 nM) or inhibitor (30 nM). (G) ISO (10
µM)-stimulated rat cardiomyocytes were treated with control
or miR-1 mimics (10 nM), and Ad-EGFP or Ad-NFATC3
(multiplicity of infection, 10). Protein expression of NFATC3 was
detected by western blotting. (H) Cell surface area, (I)
protein/DNA ratio and (J) mRNA expression of β-MHC were evaluated
in the rat cardiomyocytes exposed to the same treatment. Values are
expressed as the mean ± standard deviation of three independent
experiments. *P<0.05; **P<0.01 vs.
Ad-EGFP and Control-transfected group. miR-1, microRNA; Iso,
isoproterenol; NFATC3, nuclear factor of activated T cells
cytoplasmic 3; EGFP, enhanced green fluorescent protein enhanced
green fluorescent protein; UTR, untranslated region; Ad, adenoviral
vector. |
Suppression of NFATC3 is required for the
anti-hypertrophic effects of miR-1
As NFATC3 was identified as a novel target of
miR-1, the present study aimed to establish the role of
NFATC3 in the hypertrophic process of miR-1. An adenoviral
vector expressing NFATC3 was used to increase the expression levels
of NFATC3 in the rat cardiomyocytes (Fig. 4G). The overexpression of NFATC3
eradicated the effects of miR-1 restoration in
cardiomyocytes treated with ISO, evidenced by an increased cell
surface area (Fig. 4H),
protein/DNA ratio (Fig. 4I) and
expression of β-MHC (Fig. 4J).
These results suggested that the suppression of NFATC3 is required
for the anti-hypertrophic effects of miR-1.
Discussion
Although miR-1 is a closely associated with
the progression of cardiomyocyte hypertrophy, the molecular
mechanisms underlying its effects in heart disease remains to be
fully elucidated. The present study identified NFATC3 as a novel
target of cardiac hypertrophy-associated miRNA. The regulation of
NFATC3 by miR-1 was associated with the putative recognition
of binding sites within the 3′-UTR region of target mRNA molecules.
However, miR-1 may exert its regulatory effects on NFATC3 through
other mechanisms, including the suppression of NFATC3 inhibitory
modulators.
To the best of our knowledge, the present study
established for the first time a direct association between two
well-known cardiac hypertrophy-associated genes, miR-1 and NFATC3.
However, the molecular events, which underlie the aberrant
expression of NFATC3 in cardiomyocyte hypertrophy remain to be
fully elucidated, although its pro-hypertrophic function has been
well-established in previous studies (18,19).
In addition, the present study provided further clarification of
the molecular signaling pathway responsible for the high expression
levels of NFATC3 during the hypertrophic process.
Although miR-1 was identified as a negative
regulator of the expression of NFATC3 in the cardiomyocytes in the
present study, other miRNAs have also been found among the
predicted regulators of NFATC3. miR-122 has been demonstrated to be
an important factor for the development of hepatic tissues, and in
the physiological and pathological processes of the liver (20,21),
which is a potential regulator of NFATC3. Therefore, it is
important to investigate the association between these potential
epigenetic regulators and NFATC3. The mapping of a complete
regulatory network of NFATC3 is likely to contribute to an improved
understanding of the molecular signaling pathways, which are
associated with the formation and progression of cardiomyocyte
hypertrophy.
In addition to the examination of the molecular
mechanisms underlying the effects of miR-1, the present
study examined the clinical expression profile of miR-1 in
patients with cardiac hypertrophy. The results revealed that miR-1
was overexpressed in the cardiac tissue samples of patients with
cardiac hypertrophy. Previously, elevated serum miRNA levels have
been demonstrated to be of useful diagnostic and prognostic value
in patients suffering from various diseases, including cancer and
heart disease (22,23). Further investigations are required
in order to determine whether the serum levels of miR-1 can
be used for diagnosis and prognosis of patients with cardiac
hypertrophy.
In conclusion, the present study demonstrated that
miR-1 suppressed the expression of NFATC3 by targeting its
MRE within its 3′-UTR region, and the overexpression of
miR-1 may result in the aberrant expression of NFATC3 in
hypertrophic cardiomyocytes. These data may contribute to a better
understanding of the molecular mechanisms underlying cardiac
hypertrophy, and provide evidence supporting the targeting of
miR-1/NFATC3 as a promising therapeutic strategy for heart
diseases.
References
|
1
|
Huang J, Shelton JM, Richardson JA, Kamm
KE and Stull JT: Myosin regulatory light chain phosphorylation
attenuates cardiac hypertrophy. J Biol Chem. 283:19748–19756. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valencia-Sanchez MA, Liu J, Hannon GJ and
Parker R: Control of translation and mRNA degradation by miRNAs and
siRNAs. Genes Dev. 20:515–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Melman YF, Shah R and Das S: MicroRNAs in
heart failure: Is the picture becoming less miRky? Circ Heart Fail.
7:203–214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ikeda S, He A, Kong SW, Lu J, Bejar R,
Bodyak N, Lee KH, Ma Q, Kang PM, Golub TR and Pu WT: MicroRNA-1
negatively regulates expression of the hypertrophy-associated
calmodulin and Mef2a genes. Mol Cell Biol. 29:2193–2204. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elia L, Contu R, Quintavalle M, Varrone F,
Chimenti C, Russo MA, Cimino V, De Marinis L, Frustaci A, Catalucci
D and Condorelli G: Reciprocal regulation of microRNA-1 and
insulin-like growth factor-1 signal transduction cascade in cardiac
and skeletal muscle in physiological and pathological conditions.
Circulation. 120:2377–2385. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Q, Song XW, Zou J, Wang GK, Kremneva E,
Li XQ, Zhu N, Sun T, Lappalainen P, Yuan WJ, et al: Attenuation of
microRNA-1 derepresses the cytoskeleton regulatory protein
twinfilin-1 to provoke cardiac hypertrophy. J Cell Sci.
123:2444–2452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tritsch E, Mallat Y, Lefebvre F, Diguet N,
Escoubet B, Blanc J, De Windt LJ, Catalucci D, Vandecasteele G, Li
Z and Mericskay M: An SRF/miR-1 axis regulates NCX1 and annexin A5
protein levels in the normal and failing heart. Cardiovasc Res.
98:372–380. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karakikes I, Chaanine AH, Kang S, Mukete
BN, Jeong D, Zhang S, Hajjar RJ and Lebeche D: Therapeutic
cardiac-targeted delivery of miR-1 reverses pressure
overload-induced cardiac hypertrophy and attenuates pathological
remodeling. J Am Heart Assoc. 2:e0000782013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hou Y, Sun Y, Shan H, Li X, Zhang M, Zhou
X, Xing S, Sun H, Chu W, Qiao G and Lu Y: β-adrenoceptor regulates
miRNA expression in rat heart. Med Sci Monit. 18:BR309–BR314. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Varrone F, Gargano B, Carullo P, Di
Silvestre D, De Palma A, Grasso L, Di Somma C, Mauri P, Benazzi L,
Franzone A, et al: The circulating level of FABP3 is an indirect
biomarker of microRNA-1. J Am Coll Cardiol. 61:88–95. 2013.
View Article : Google Scholar
|
|
11
|
Wilkins BJ, De Windt LJ, Bueno OF, Braz
JC, Glascock BJ, Kimball TF and Molkentin JD: Targeted disruption
of NFATc3, but not NFATc4, reveals an intrinsic defect in
calci-neurin-mediated cardiac hypertrophic growth. Mol Cell Biol.
22:7603–7613. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wilkins BJ, Dai YS, Bueno OF, Parsons SA,
Xu J, Plank DM, Jones F, Kimball TR and Molkentin JD:
Calcineurin/NFAT coupling participates in pathological, but not
physiological, cardiac hypertrophy. Circ Res. 94:110–118. 2004.
View Article : Google Scholar
|
|
13
|
Tan WQ, Wang K, Lv DY and Li PF: Foxo3a
inhibits cardiomyocyte hypertrophy through transactivating
catalase. J Biol Chem. 283:29730–29739. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reinhold MI, Abe M, Kapadia RM, Liao Z and
Naski MC: FGF18 represses noggin expression and is induced by
calcineurin. J Biol Chem. 279:38209–38219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu J, Ma L, Li C, Zhang Z, Yang G and
Zhang W: Tumor-targeting TRAIL expression mediated by miRNA
response elements suppressed growth of uveal melanoma cells. Mol
Oncol. 7:1043–1055. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma L, Liu J, Shen J, Liu L, Wu J, Li W, et
al: Expression of miR-122 mediated by adenoviral vector induces
apoptosis and cell cycle arrest of cancer cells. Cancer Biol Ther.
9:554–561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murtaza I, Wang HX, Feng X, Alenina N,
Bader M, Prabhakar BS and Li PF: Down-regulation of catalase and
oxidative modification of protein kinase CK2 lead to the failure of
apoptosis repressor with caspase recruitment domain to inhibit
cardiomyocyte hypertrophy. J Biol Chem. 283:5996–6004. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang K, Long B, Zhou J and Li PF: MiR-9
and NFATc3 regulate myocardin in cardiac hypertrophy. J Biol Chem.
285:11903–11912. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai H, Jia G, Liu X, Liu Z and Wang H:
Astragalus polysac-charide inhibits isoprenaline-induced cardiac
hypertrophy via suppressing Ca2+-mediated
calcineurin/NFATc3 and CaMKII signaling cascades. Environ Toxicol
Pharmacol. 38:263–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma L, Liu J, Shen J, Liu L, Wu J, Li W,
Luo J, Chen Q and Qian C: Expression of miR-122 mediated by
adenoviral vector induces apoptosis and cell cycle arrest of cancer
cells. Cancer Biol Ther. 9:554–561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bandiera S, Pfeffer S, Baumert TF and
Zeisel MB: MiR-122-a key factor and therapeutic target in liver
disease. J Hepatol. 62:448–457. 2015. View Article : Google Scholar
|
|
22
|
Oliveira-Carvalho V, da Silva MM,
Guimarães GV, Bacal F and Bocchi EA: MicroRNAs: New players in
heart failure. Mol Biol Rep. 40:2663–2670. 2013. View Article : Google Scholar
|
|
23
|
Li J, Liu Y, Wang C, Deng T, Liang H, Wang
Y, Huang D, Fan Q, Wang X, Ning T, et al: Serum miRNA expression
profile as a prognostic biomarker of stage II/III colorectal
adenocarcinoma. Sci Rep. 5:129212015. View Article : Google Scholar : PubMed/NCBI
|