Introduction
Methanol intoxication resulting from the consumption
of fake wine has led to numerous incidents of disability, blindness
and mortality as a consequence of selective neurotoxic actions
(1). Methanol is metabolized
primarily in the liver by sequential oxidative steps to form formic
acid, formaldehyde and carbon dioxide (2,3).
Previous studies hypothesized that the retinal pathophysiology of
methanol intoxication is a consequence of formate-induced
mitochondrial dysfunction (4).
Formate disrupts mitochondrial electron transport and energy
production by inhibiting cytochrome oxidase activity and the
terminal electron acceptor of the electron transport chain
(4). Cell death resulting from
cytochrome oxidase inhibition by formate is considered to result
partly from the depletion of ATP, which reduces energy levels and
leads to the disruption of cell functions (5). Furthermore, concentrations of formate
in the retina and vitreous humor closely correspond to the
concentration of formate in the blood (6,7).
It was previously reported that formate may inhibit
cytochrome oxidase activity in the concentration range of 5–30 mM
in vitro and in vivo (8). Similar formate levels have been
measured in the blood, vitreous humor and cerebrospinal fluid of
methanol-poisoned humans and monkeys (9,10).
However, since tissues differ in their sensitivity to the toxic
effects of methanol poisoning, and the retina is highly susceptible
to depletion of retinal ATP due to its constant exposure to
irradiation and a high metabolic activity, it remains to be
elucidated whether patterns of cell death in the retina may be
attributable to induction by methanol poisoning.
The present study aimed to investigate the effects
of sodium formate on 661W cells in order to understand the
molecular events of cell death caused by methanol poisoning. An
improved understanding of this mechanism is required in order to
identify more effective treatments for patients with methanol
intoxication.
Materials and methods
Ethical approval
All human and animal experiments performed in the
present study were approved by the Human and Animal Research Ethics
Committees of The Second Hospital of Jilin University, (Changchun,
China).
Antibodies and reagents
Polyclonal rabbit anti-mouse antibodies raised
against Bax and Bcl-2 were obtained from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA; cat. nos. sc-6236 and sc-492). Antibodies
against cleaved caspase-9 (cat. no. 7237), cleaved caspase-3 (cat.
no. 9579), c-Jun N-terminal kinase (JNK; cat. no. 9251),
phosphorylated (p)-JNK (cat. no. 9252) and microtubule-associated
protein 1A/1B-light chain 3 (LC3; cat. no. 2775) were purchased
from Cell Signaling Technology, Inc. (Beverly, MA, USA). A mouse
monoclonal antibody against glyceraldehyde-3-phosphate
dehydrogenase was purchased from Kangchen Bio-Tech Co., Ltd.
(Shanghai, China; cat. no. kc-564). All primary antibodies were
diluted 1:1,000. The secondary antibodies (goat anti-rabbit; cat.
no. 31210; 1:5,000) were obtained from Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). An enhanced chemiluminescence (ECL)-Plus
kit was purchased from Beyotime Institute of Biotechnology
(Nantong, China). An annexin V-FLUOS Staining kit was purchased
from Roche Diagnostics (Mannheim, Germany). Sodium formate was
purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai,
China). Hoechst 33342 stain, propidium iodide (PI),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT), monodansylcadaverine (MDC), SP600125, Z-VAD-fmk and
2′,7′-dichlorofluorescein diacetate (DCFH-DA) were obtained from
Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The 661W cells were obtained from American Tissue
Culture Collection (Manassas, VA, USA) and were maintained in
Dulbecco's modified Eagle's medium, supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 g/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.). The cells were grown in a humidified incubator
with 95% air and 5% CO2 at 37°C. Subsequently, the cells
were passaged until they reached 80% confluence.
Cell viability assays
The 661W cells were plated at a density of
5×103 cells/well in 96-well plates. Following incubation
for 24 h, culture medium containing various final formate
concentrations (0, 15, 30, 60 or 120 mM) was added to each well.
Paired control cultures were prepared with medium containing
comparable levels of sodium chloride (sodium control). The parallel
cell cultures were incubated for 6, 12 or 24 h. Subsequently, 661W
cells were pretreated with a pan-caspase inhibitor (Z-VAD-fmk) and
a JNK inhibitor (SP600125) for 0.5 h prior to treatment with 30 mM
sodium formate for 24 h. MTT (20 µl) was added to each well
and the samples were incubated for an additional 4 h followed by
the addition of 100 µl dimethylsulfoxide to each well. The
absorbance of blue formazan at 570 nm was measured using a
microplate reader (Model 680; Bio-Tek Instruments, Inc., Winooski,
VT, USA).
Hoechst 33342 and PI staining
The level of apoptosis of the 661W cells was
determined using double staining with Hoechst 33342 and PI. The
cells were stained with 10 µg/ml Hoechst 33342 and 10
µg/ml PI for 30 min at 37°C. Following two successive washes
with phosphate-buffered saline (PBS), images of the cells were
captured with a digital camera attached to a fluorescence
microscope (IX70; Olympus Corporation, Tokyo, Japan).
Annexin V-fluorescein isothiocyanate
(FITC) assay
The percentage of cells actively undergoing
apoptosis was determined using flow cytometric analysis with the
annexin V-FITC Apoptosis Detection kit (BD Biosciences, San Jose,
CA, USA), according to the manufacturer's protocol. Briefly, the
cells were harvested and resuspended in binding buffer
(106 cells/ml). The cells at a density of 103
cells/well were mixed with 5 µl annexin V-FITC and 5
µl PI. Following incubation of the cells at room temperature
for 15 min in the dark, the flow cytometric analysis was performed
using the FACSCalibur™ system (BD Biosciences, Franklin Lakes, NJ,
USA).
ROS measurement
Intracellular ROS production was assessed using a
fluorescent probe, DCFH-DA, using flow cytometric analysis, as
previously described (10).
Detection of ROS was based on the fact that intracellular ROS are
able to oxidize DCFH, yielding the fluorescent product,
2′,7′-dichlorofluorescein (DCF). Following an incubation of the
cells with 0, 15, or 30 mM formate for 24 h, the supernatant was
removed, and the cells were washed with PBS three times. The cells
were subsequently harvested and suspended in PBS. DCFH-DA (10
µM final concentration) was added and the mixture was
incubated at 37°C for 15 min. Finally, ROS generation was measured,
according to the fluorescence intensity (FL-1; 530 nm) of
104 cells using a flow cytometer (FACSCalibur; BD
Biosciences).
MDC staining
MDC (Sigma-Aldrich) was used to visualize autophagic
vacuoles. The 661W cells were plated into 6-well plates and treated
with two different concentrations of formate (15 or 30 mM) for 6 h.
Following treatment, the cells were incubated with 0.1 mM MDC for
30 min. Fluorescent images were captured under a fluorescence
microscope (Olympus).
Western blotting
The proteins (2.6 µg/µl) were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), and western blotting was subsequently
performed, as previously described (11). Briefly, the cells were harvested,
resuspended in cell lysis buffer (Beyotime Institute of
Biotechnology) and incubated on ice for 30 min. The cell lysates
were centrifuged at 12,000 x g for 10 min at 4°C. The supernatants
were mixed with one-quarter volumes of 4X SDS sample buffer and
boiled for 10 min. The proteins were subsequently separated by
SDS-PAGE in a 10–15% gel. Following electrophoresis, the proteins
were transferred onto polyvinylidene fluoride membranes and blocked
with 5% non-fat milk powder in Tris-buffered saline (Thermo Fisher
Scientific, Inc.) with Tween-20 (TBST; Beyotime Institute of
Biotechnology) for 1 h at room temperature. The membrane was
subsequently incubated with a diluted primary antibody in blocking
buffer overnight at 4°C. The membrane was washed three times with
TBST and incubated with a horseradish peroxidase-conjugated
secondary antibody (Beyotime Institute of Biotechnology) for 30 min
at room temperature. Following extensive washing, the proteins were
visualized using an ECL-Plus kit and the blots were exposed to
Kodak radiographic film (Kodak. Corp., Rochester, NY, USA).
Statistical analysis
The data were obtained from >3 independent
experiments. Statistically significant differences between the
groups were assessed by one-way analysis of variance, followed by
the Bonferroni post-hoc test, using SPSS 13.0 software for
statistical analysis (SPSS Inc., Chicago, IL, USA). The data are
expressed as the standard deviation, unless otherwise stated.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of sodium formate on the
viability of 661W cells
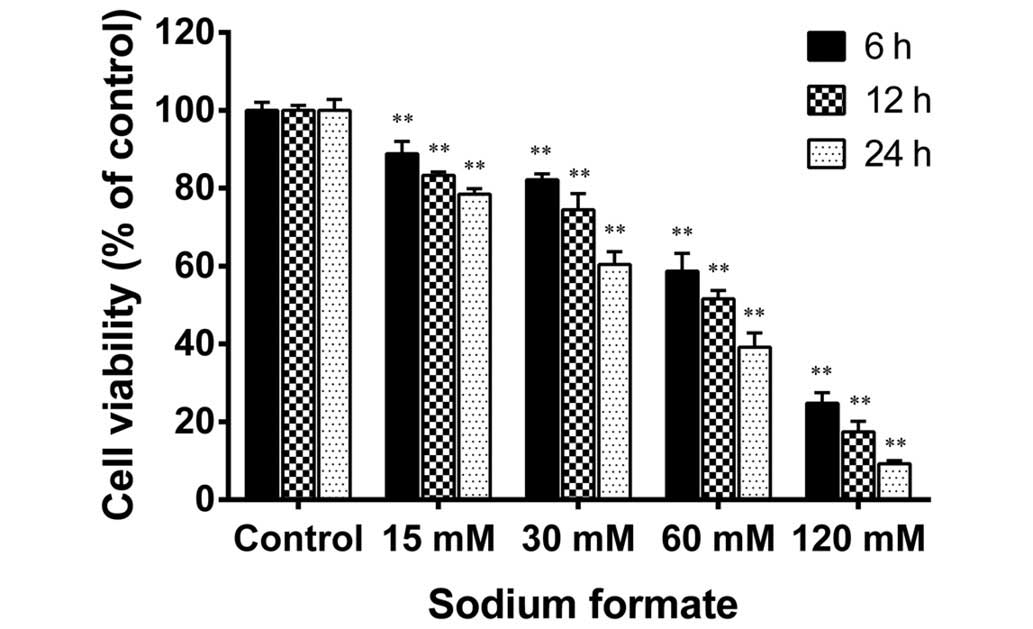
The cytotoxicity of sodium formate in 661W cells was
examined using an MTT assay. As shown in Fig. 1, sodium formate treatment reduced
the cell viability in a time- and a dose-dependent manner. The
viability of the 661W cells treated with 120 mM sodium formate for
24 h was decreased by a greater extent compared with the other
concentrations tested (15, 30, and 60 mM). The viability of the
661W cells treated with 15 or 30 mM sodium formate for 24 h was
78.5 and 60.4%, respectively, compared with the control group. The
661W cells were also treated with 5 mM sodium formate (data not
shown), and the results revealed no clear differences compared with
the control cultures. Therefore, the concentrations of sodium
formate used in the subsequent experiments were 15 and 30 mM. No
discrimination was made between the paired control cultures in the
viability assays and those in further experiments. Therefore, the
paired control results (15 mM sodium chloride) were used in the
following experiments. These results suggested that treatment of
the 661W cells with sodium formate led to a marked decline in their
viability.
Sodium formate induces apoptosis in 661W
cells
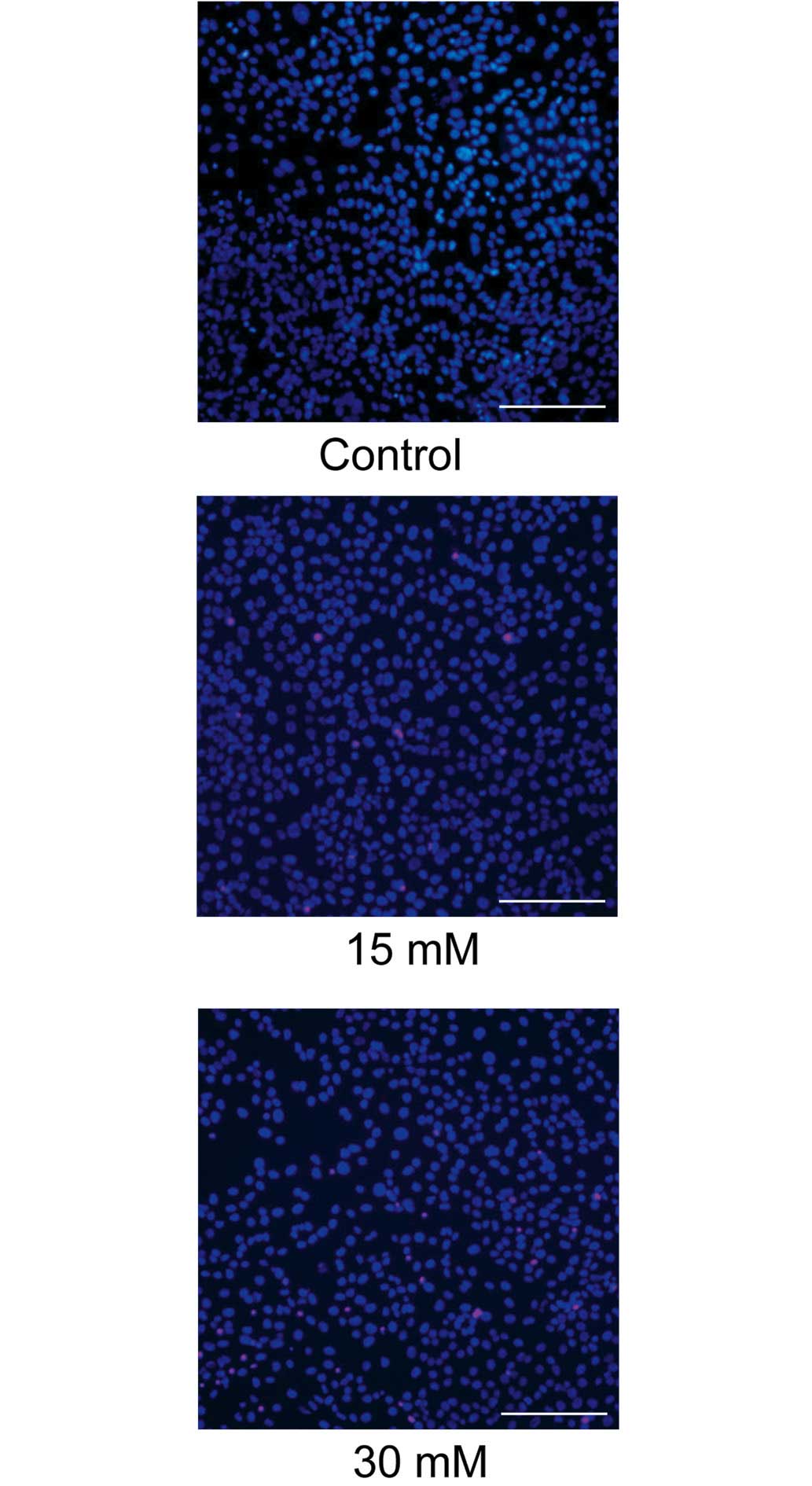
The cytotoxicity of sodium formate towards the 661W
cells was further assessed using Hoechst 33342 and PI double
staining. As shown in Fig. 2,
exposure to 30 mM sodium formate for 24 h resulted in a marked loss
of Hoechst-positive cells and numerous PI-positive cells,
indicating that apoptosis of the 661W cells was induced upon
treatment with 15 or 30 mM sodium formate.
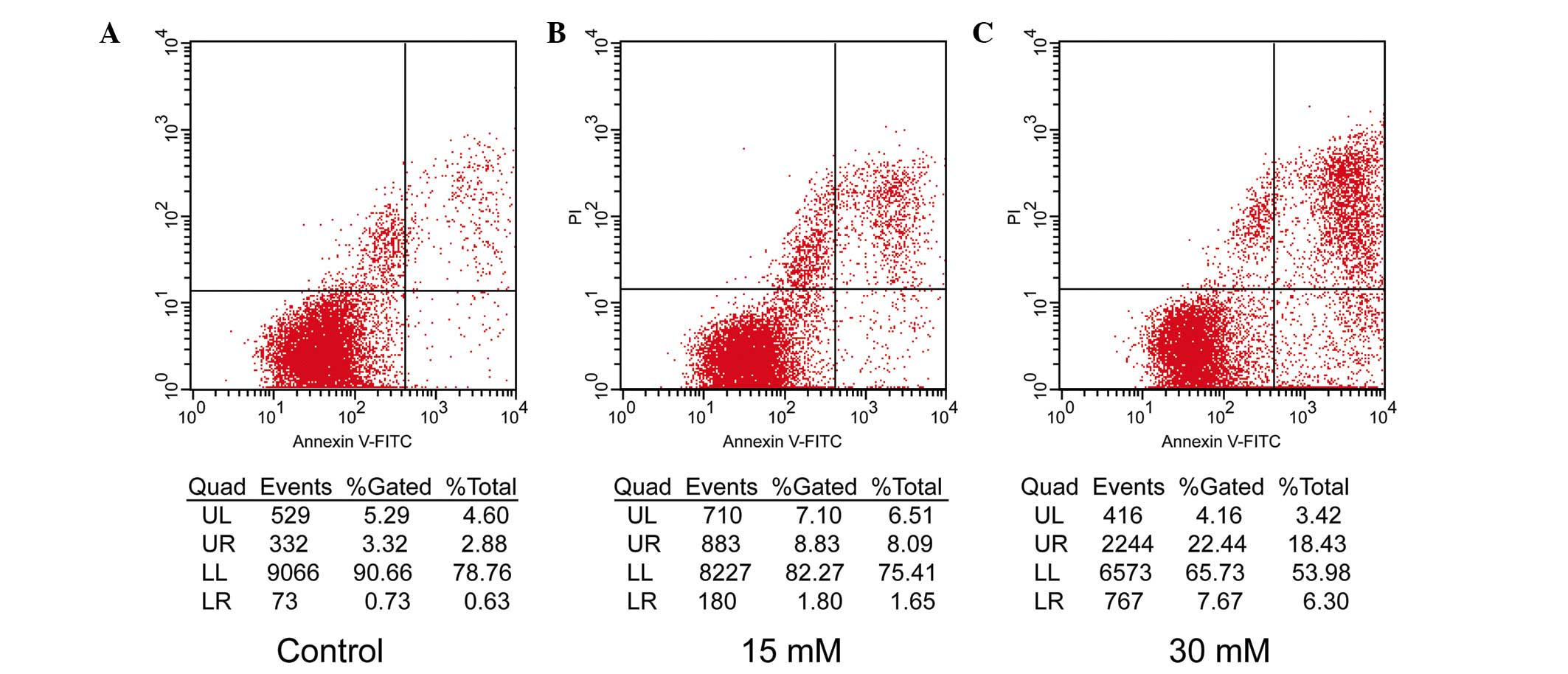
The apoptotic rates of the 661W cells were further
assessed by flow cytometric analysis using the annexin V-FITC
Apoptosis Detection kit, which readily distinguishes early
apoptotic events from late apoptosis/necrosis. In the premature
apoptotic cells, the membrane phospholipid, phosphatidylserine
(PS), is translocated from the inner to the outer leaflet of the
plasma membrane, and annexin V is a protein that has a high
affinity for PS. PI is used as a flow cytometric viability probe
for DNA content and can be used to differentiate viable from
non-viable cells. Following exposure to the two different doses (15
and 30 mM) of sodium formate for 24 h, markedly different
percentages of early apoptotic and late apoptotic/necrotic 661W
cells compared with control cells (0 µM sodium formate) were
identified, which increased from 2.88 to 18.43% and from 0.63 to
6.3%, respectively (Fig. 3). The
results indicated that the level of apoptosis of the 661W cells
treated with 15 or 30 mM sodium formate was markedly increased.
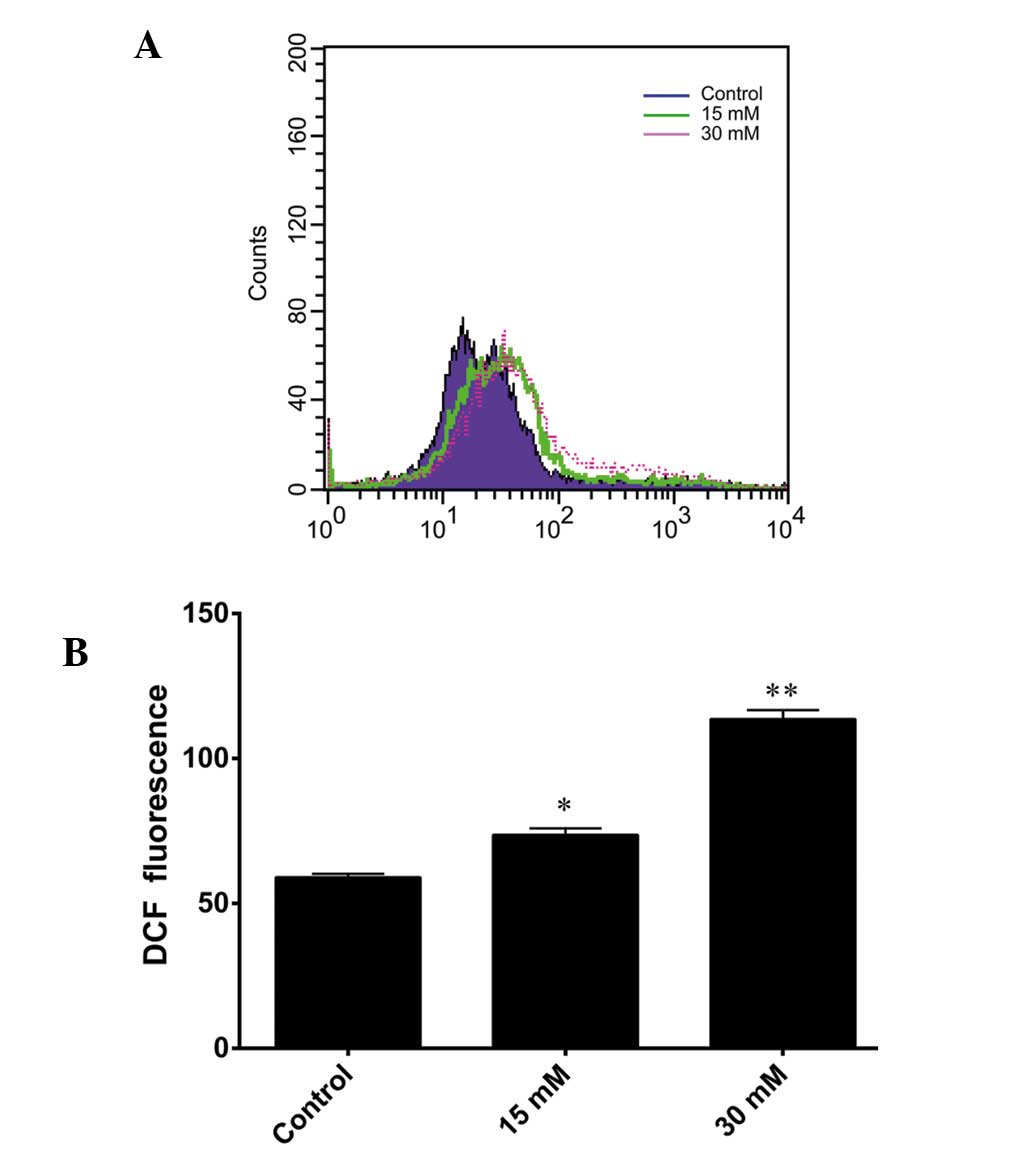
Sodium formate causes an increase in the
intracellular ROS levels
DCFH-DA staining was used to detect intracellular
ROS generation. DCFH-DA readily diffuses through the cell membrane
and is deacetylated by esterases to non-fluorescent DCFH.
Furthermore, DCFH is rapidly oxidized to its highly fluorescent
product, DCF, in the presence of ROS. Therefore, the DCF
fluorescence intensity is proportional to the quantity of
intracellular ROS present. As shown in Fig. 4A, a shift to the right was observed
in the fluorescence intensity signal for the 661W cells treated
with 15 and 30 mM sodium formate. The level of ROS increased
markedly following exposure of the cells to 15 or 30 mM sodium
formate for 24 h (Fig. 4B). These
results suggested that increased levels of ROS may be involved in
sodium formate-induced cytotoxicity in 661W cells.
JNK is activated in 661W cells following
exposure to sodium formate
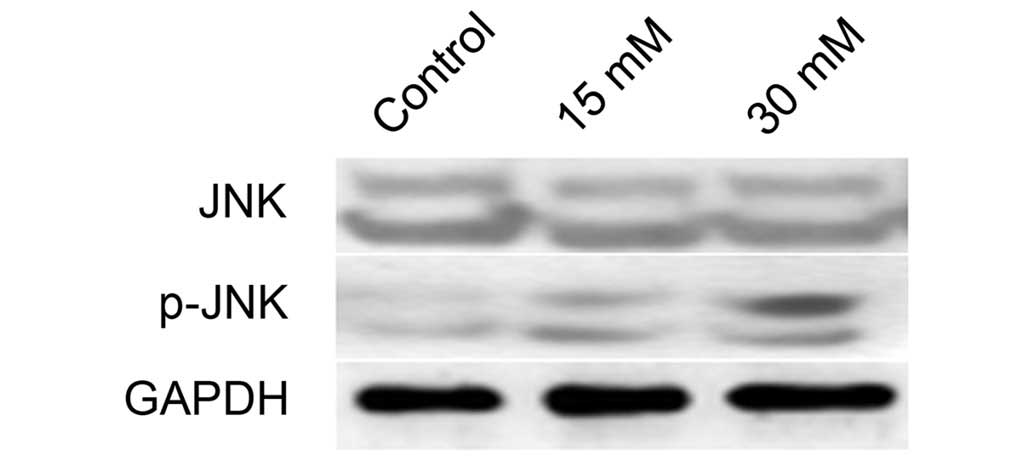
The protein expression of JNK and p-JNK in 661W
cells treated with sodium formate was subsequently examined. As
shown in Fig. 5, the protein
expression level of p-JNK increased in the 661W cells upon sodium
formate treatment, whereas no significant changes in the protein
expression level of JNK were identified following treatment of the
cells with either concentration of sodium formate (15 or 30 mM).
These results suggested that the JNK signaling pathway was
activated when 661W cells were treated with sodium formate.
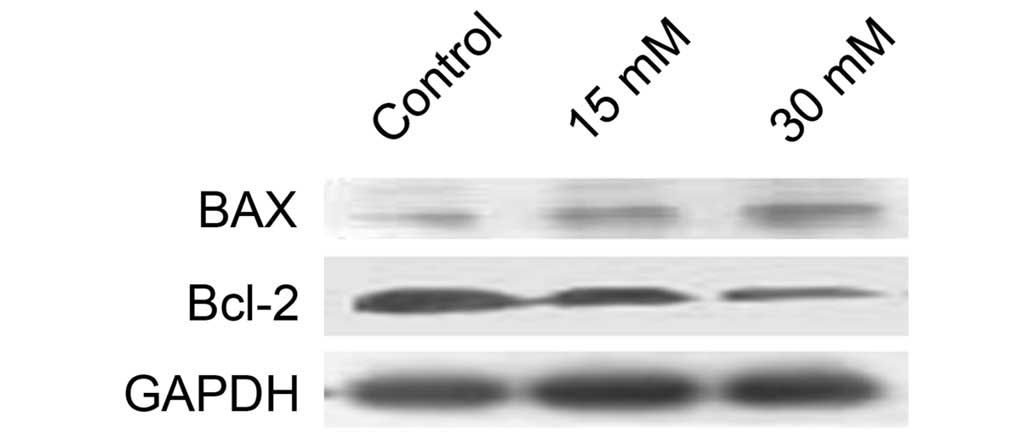
Sodium formate affects the protein
expression levels of Bax and Bcl-2 in 661W cells
Considering the role of the Bcl-2 family proteins in
mitochondria-dependent apoptosis, the effects of sodium formate on
the expression of the antiapoptotic protein, Bcl-2, and the
pro-apoptotic protein, Bax, were subsequently examined. As shown in
Fig. 6, Bcl-2 levels were markedly
decreased, and Bax levels were increased, in 661W cells treated
with 15 or 30 mM sodium formate, suggesting that the increase in
the ratio of Bax to Bcl-2 protein may be involved in
mitochondria-dependent apoptosis following exposure to sodium
formate.
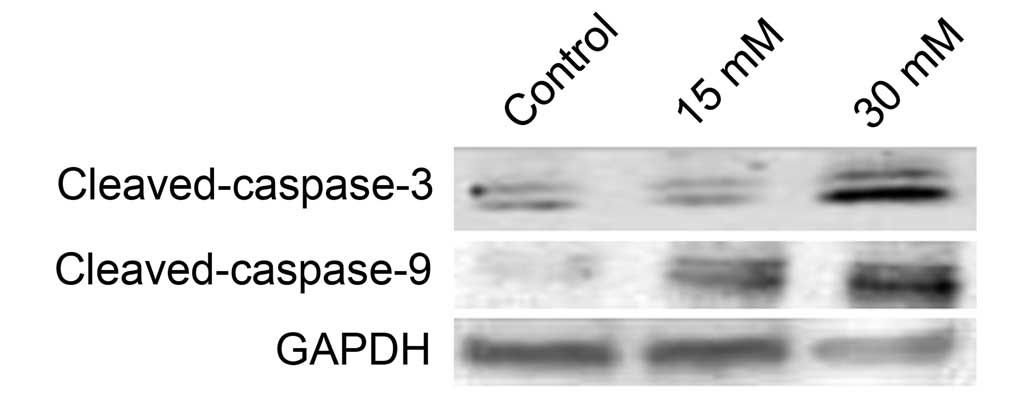
Sodium formate triggers caspase-dependent
apoptosis in 661W cells
Apoptosis is triggered by two types of apoptotic
caspases, namely initiator caspases (e.g. caspases-2, -8, -9 and
-10) and effector caspases (e.g. caspases-3, -6 and -7). Initiator
caspases cleave and activate inactive proforms of effector
caspases, and activated effector caspases cleave other protein
substrates to trigger the apoptotic process (12). In the present study, western
blotting indicated that the level of cleaved caspase-3 increased
when cells were treated with sodium formate (Fig. 7). Subsequently, the activity of
initiator caspases was investigated. As shown in Fig. 7, treatment with sodium formate
clearly increased the protein expression of cleaved caspase-9.
These data suggested that sodium formate may induce apoptosis in
661W cells, at least partly, via a caspase-dependent pathway.
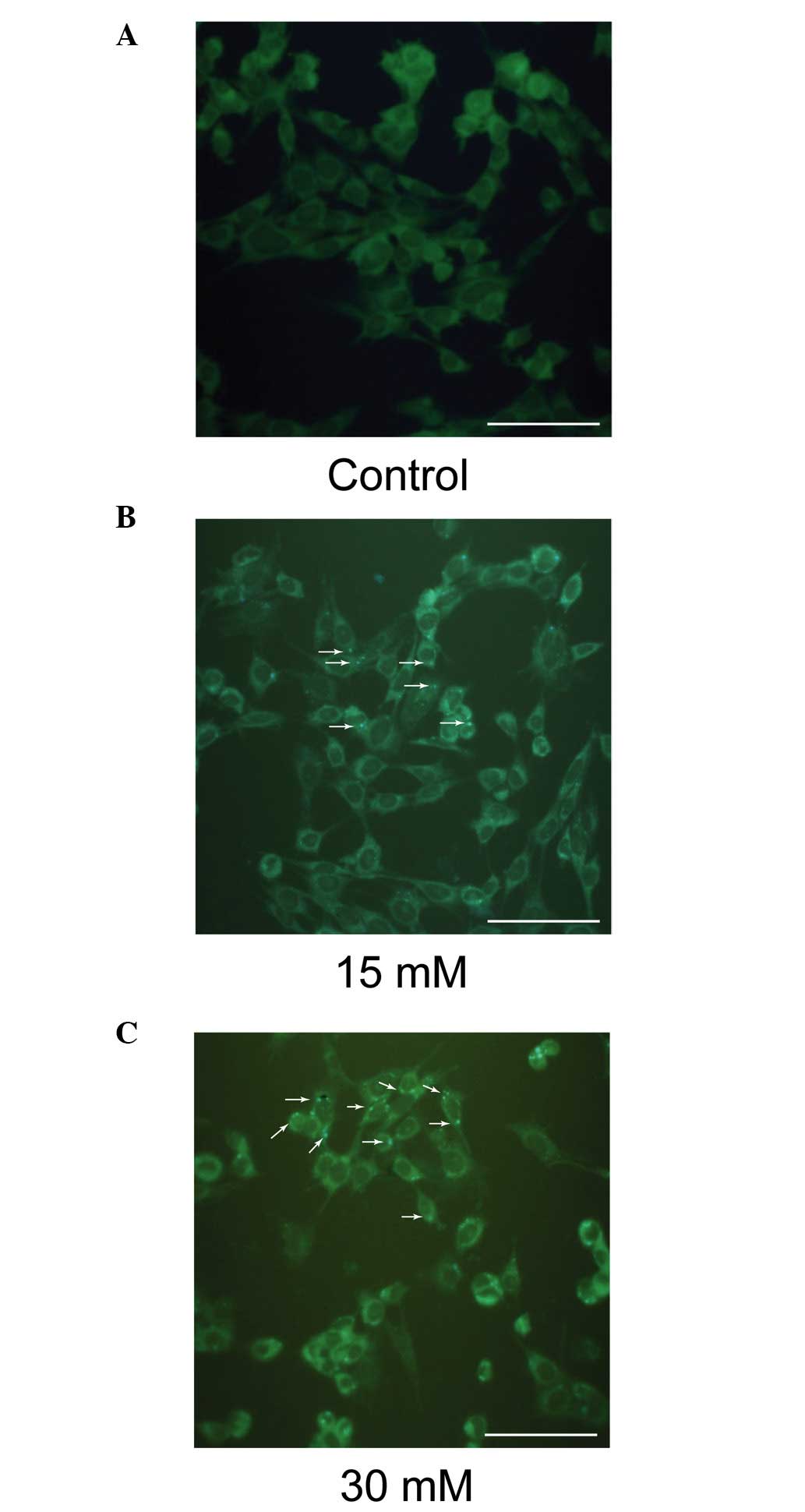
Sodium formate exposure induces autophagy
in 661W cells
MDC is a specific marker for autophagic vacuoles. As
shown in Fig. 8, the sodium
formate-treated cells exhibited a greater fluorescence intensity
and a greater number of MDC-labeled particles in the 661W cells
compared with the control, indicating that sodium formate increased
the recruitment of MDC to autophagosomes in the cell cytoplasm.
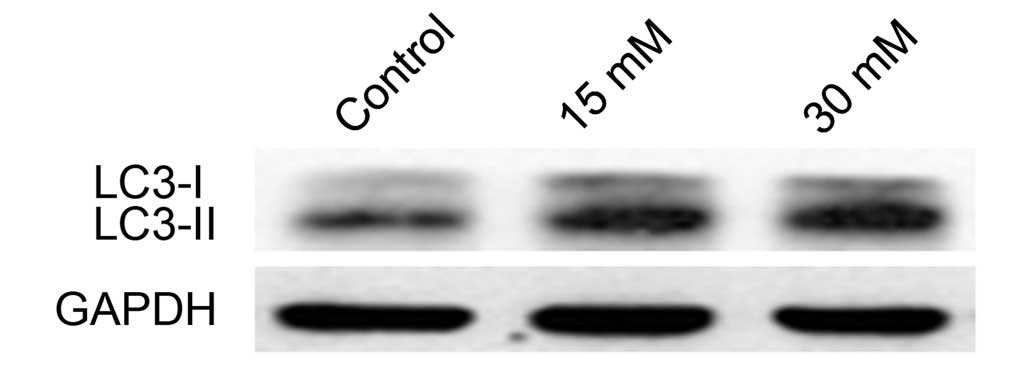
LC3II levels increase in 661W cells
treated with sodium formate
LC3-I is a cytosolic protein whose lipidated form,
LC3II (a phosphatidylethanolamine-modified form of LC3), is stably
associated with the autophagosomal membrane, and is therefore
considered an autophagy-specific marker. To determine whether
sodium formate induces autophagy in 661W cells, the expression
level of LC3 was investigated by western blotting. Compared with
the control, the levels of LC3-II were markedly increased in the
661W cells treated with 15 or 30 mM sodium formate (Fig. 9). No significant changes in the
level of LC3-I was observed during the course of the experiment.
These results suggested that autophagy may be involved in sodium
formate-induced cytotoxicity in 661W cells.
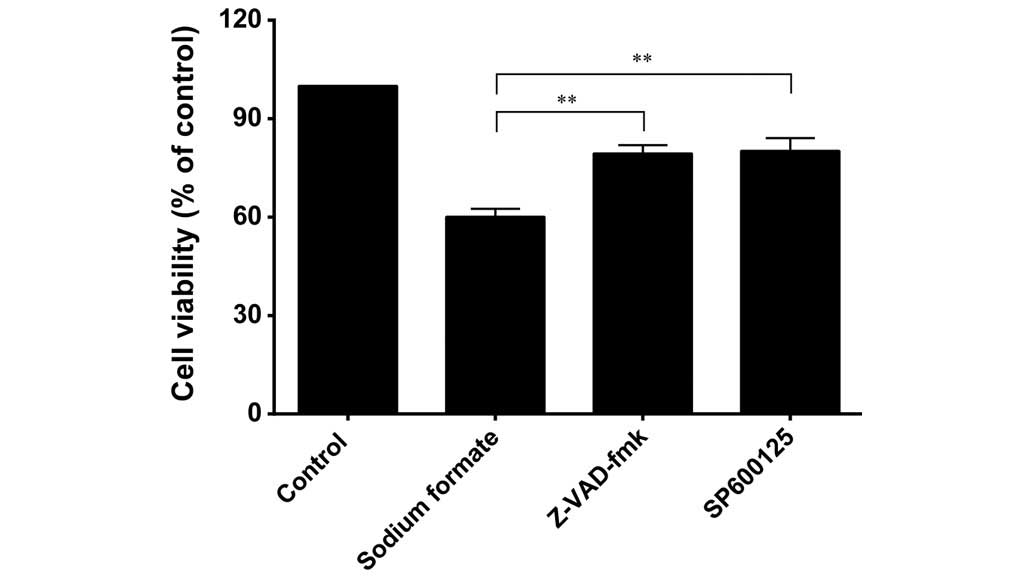
Effects of Z-VAD-fmk and SP600125 on cell
viability in sodium formate-induced 661W cell death
To assess the role of caspases in the cell death
process induced by sodium formate, 661W cells were pretreated with
a pan-caspase inhibitor, Z-VAD-fmk, for 0.5 h prior to treatment
with 30 mM sodium formate for 24 h. As shown in Fig. 10, Z-VAD-fmk at a concentration of
20 µM effectively circumvented the effects of sodium formate
on cell viability, resulting in only 19% growth inhibition, a value
significantly lower compared with that observed in the untreated
cells (40% inhibition; P<0.01). SP600125, at a concentration of
10 µM, clearly circumvented the effects of sodium formate on
cell viability, resulting in only 20% growth inhibition,
significantly lower compared with that observed in the untreated
cells (40% inhibition; P<0.01).
Discussion
Exposure to methanol is a major cause of acute
alcohol intoxication. Patients with acute methanol intoxication
experience acute neurological, visual and gastrointestinal
symptoms. Visual disturbances generally range from mild photophobia
and misty or blurred vision to markedly reduced visual acuity and
complete blindness. Despite the availability of efficient
treatments, methanol poisoning results in high rates of morbidity
and mortality (1,13).
It was reported that retinal ATP synthase decreases
following methanol intoxication. For example, proteomic analysis of
the rat retina revealed that sodium formate induces a marked
decrease in the levels of ATP in photoreceptor cells (7,14). A
marked decline in the viability of 661W cells following exposure to
sodium formate was also observed in the present study. It is well
known that the dysregulation of electron transport through the
mitochondrial respiratory chain, or impairments in the function of
antioxidant enzymes, may result in the accumulation of ROS
(15). Furthermore, ROS are
associated with high levels of biological activity and function as
secondary messengers in the JNK signaling pathway. As a member of
the mitogen-activated protein kinase family, JNK is involved in the
signal transduction of a variety of cellular pathways, including
those for apoptosis, inflammation and carcinogenesis (16,17).
In the present study, the JNK signaling pathway was activated and
the level of ROS increased when the 661W cells were treated with
sodium formate. ROS are potent activators of JNK via the process of
oxidative inactivation of endogenous JNK inhibitors, including JNK
phosphatases and glutathione S-transferase (18). Activation of the JNK pathway
induces ROS accumulation, which may in turn activate JNK in a
positive feedback manner. This activation may have contributed
towards 661W cell apoptosis following sodium formate treatment in
the present study. Activation of the JNK signaling pathway triggers
the apoptotic signaling pathway, including the intrinsic pathway
(also termed the mitochondrial pathway) and the extrinsic pathway
(also termed the death receptor pathway) (19). SP600125 is a specific JNK inhibitor
in common usage. It was demonstrated to reverse neuronal cell death
in rat hippocampal Cornu Ammonis 1 caused by transient brain
ischemia/reperfusion (20).
SP600125 also markedly improved the survival rate of retinal
ganglion cells against acute moderate ocular hypertension (21). In the present study, the
administration of SP600125 enhanced the cell viability, and negated
some of the effects elicited by sodium formate. Inhibiting JNK
activity by SP600125, or similar compounds, may provide a novel and
effective strategy to treat methanol intoxication.
The Bax/Bcl-2 ratio in cells regulates the
susceptibility of cells to apoptosis (22). The present study confirmed that the
expression of Bcl-2 was markedly decreased and the expression of
Bax was increased in the 661W cells treated with 15 or 30 mM sodium
formate. Increased levels of cleaved caspase-3 and cleaved
caspase-9 in the 661W cells treated with sodium formate were also
observed. These results suggested that the increases in the
Bax/Bcl-2 ratio and the level of cleaved caspases may be involved
in sodium formate-induced, mitochondrion-dependent apoptosis. The
pan-caspase inhibitor, Z-VAD-fmk, markedly enhanced the cell
viability and circumvented the effect of sodium formate treatment,
indicating that apoptosis of the 661W cells, induced by sodium
formate, occurs via a caspase-dependent pathway.
Autophagy is a cellular pathway responsible for the
clearance of proteins and organelles, and therefore a certain
degree of autophagy is necessary to maintain normal cellular
homeostasis (23). In the present
study, the autophagy of lysosomes and an increased expression level
of LC3II in the 661W cells was observed, on increasing the exposure
of the cells to sodium formate. Ramírez et al (24) demonstrated that hydroquinone
damages human retinal Müller cells via the oxidative, mitochondrial
and autophagic signaling pathways.
It is possible that cells, which have suffered a
mild insult can be rescued through the selective elimination of
damaged and proapoptotic mitochondria (those undergoing mitosis),
whereas a more profound insult resulting in damage to numerous
mitochondria would surpass the capacity for rescue. This would be
due to the fact that removal of an excessive number of the
mitochondria would leave behind a cell with an insufficient
capacity to produce ATP and release cytochrome c, and
ultimately the apoptosis of the photoreceptor cells would result.
Macroautophagy induced by formate may provide an explanation for
the decrease in ATP levels. Therefore, formate-induced autophagy
may be important in promoting mitochondrial dysfunction, and even
apoptosis, in 661W cells. Kunchithapautham and Rohrer (25) demonstrated that apoptotic and
autophagic genes may be coexpressed in photoreceptors undergoing
degeneration. It was suggested that autophagy is involved in
photoreceptor cell death, possibly by initiating apoptosis.
In conclusion, the present study provided evidence
that sodium formate induces the apoptosis of 661W cells, partly
through triggering the JNK pathway. Increased levels of autophagy
were observed during the process of cellular damage caused to the
661W cells by treatment with sodium formate. Autophagy has been
postulated to be involved in the pathogenesis of numerous retinal
diseases, including age-associated macular degeneration and
diabetic retinopathy. Therefore, studies of formate-induced 661W
cell dysfunction may provide valuable insights into the
pathogenesis of these retinal diseases.
Acknowledgments
The present study was supported by the Bethune Youth
Fund of Jilin University (no. 2013206043).
References
|
1
|
Massoumi G, Saberi K, Eizadi-Mood N,
Shamsi M, Alavi M and Morteza A: Methanol poisoning in Iran, from
2000 to 2009. Drug Chem Toxicol. 35:330–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harris C, Dixon M and Hansen JM:
Glutathione depletion modulates methanol, formaldehyde and formate
toxicity in cultured rat conceptuses. Cell Biol Toxicol.
20:133–145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harris C, Wang SW, Lauchu JJ and Hansen
JM: Methanol metabolism and embryotoxicity in rat and mouse
conceptuses: Comparisons of alcohol dehydrogenase (ADH1),
formaldehyde dehydrogenase (ADH3), and catalase. Reprod Toxicol.
17:349–357. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seme MT, Summerfelt P, Neitz J, Eells JT
and Henry MM: Differential recovery of retinal function after
mitochondrial inhibition by methanol intoxication. Invest
Ophthalmol Vis Sci. 42:834–841. 2001.PubMed/NCBI
|
|
5
|
Seme MT, Summerfelt P, Henry MM, Neitz J
and Eells JT: Formate-induced inhibition of photoreceptor function
in methanol intoxication. J Pharmacol Exp Ther. 289:361–370.
1999.PubMed/NCBI
|
|
6
|
Eells JT, Salzman MM, Lewandowski MF and
Murray TG: Formate-induced alterations in retinal function in
methanol-intoxicated rats. Toxicol Appl Pharmacol. 140:58–69. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Treichel JL, Henry MM, Skumatz CM, Eells
JT and Burke JM: Formate, the toxic metabolite of methanol, in
cultured ocular cells. Neurotoxicology. 24:825–834. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eells JT, Henry MM, Summerfelt P,
Wong-Riley MT, Buchmann EV, Kane M, Whelan NT and Whelan HT:
Therapeutic photobiomodulation for methanol-induced retinal
toxicity. Proc Natl Acad Sci USA. 100:3439–3444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kubáň P, Foret F and Bocek R: Capillary
electrophoresis with contactless conductometric detection for rapid
screening of formate in blood serum after methanol intoxication. J
Chromatogr A. 1281:142–147. 2013. View Article : Google Scholar
|
|
10
|
Sundaresan M, Yu ZX, Ferrans VJ, Irani K
and Finkel T: Requirement for generation of H2O2 for
platelet-derived growth factor signal transduction. Science.
270:296–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Zhou L, Bao YL, Wu Y, Yu CL,
Huang YX, Sun Y, Zheng LH and Li YX: Butyrate induces cell
apoptosis through activation of JNK MAP kinase pathway in human
colon cancer RKO cells. Chem Biol Interact. 185:174–181. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Los M, Wesselborg S and Schulze-Osthoff K:
The role of caspases in development, immunity, and apoptotic signal
transduction: Lessons from knockout mice. Immunity. 10:629–639.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barceloux DG, Bond GR, Krenzelok EP,
Cooper H and Vale JA; American Academy of Clinical Toxicology Ad
Hoc Committee on the Treatment Guidelines for Methanol Poisoning:
American Academy of Clinical Toxicology practice guidelines on the
treatment of methanol poisoning. J Toxicol Clin Toxicol.
40:415–446. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen JM, Zhu GY, Xia WT and Zhao ZQ:
Proteomic analysis of rat retina after methanol intoxication.
Toxicology. 293:89–96. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saoudi M, Ben Hsouna A, Trigui M, Jamoussi
K, Jaoua S and El Feki A: Differential oxidative stress responses
to methanol in intraperitoneally exposed rats: Ameliorative effects
of Opuntia vulgaris fruit extract. Toxicol Ind Health. 28:549–559.
2012. View Article : Google Scholar
|
|
16
|
Nakano H, Nakajima A, Sakon-Komazawa S,
Piao JH, Xue X and Okumura K: Reactive oxygen species mediate
crosstalk between NF-kappaB and JNK. Cell Death Differ. 13:730–737.
2006. View Article : Google Scholar
|
|
17
|
Lorin S, Pier ron G, Ryan KM, Codogno P
and Djavaheri-Mergny M: Evidence for the interplay between JNK and
p53-DRAM signalling pathways in the regulation of autophagy.
Autophagy. 6:153–154. 2010. View Article : Google Scholar
|
|
18
|
Zhang Y and Chen F: Reactive oxygen
species (ROS), troublemakers between nuclear factor-kappaB
(NF-kappaB) and c-Jun NH(2)-terminal kinase (JNK). Cancer Res.
64:1902–1905. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Niu YL, Li C and Zhang GY: Blocking Daxx
trafficking attenuates neuronal cell death following
ischemia/reperfusion in rat hippocampus CA1 region. Arch Biochem
Biophys. 515:89–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun H, Wang Y, Pang IH, Shen J, Tang X, Li
Y, Liu C and Li B: Protective effect of a JNK inhibitor against
retinal ganglion cell loss induced by acute moderate ocular
hypertension. Mol Vis. 17:864–875. 2011.PubMed/NCBI
|
|
22
|
Vander Heiden MG and Thompson CB: Bcl-2
proteins: Regulators of apoptosis or of mitochondrial homeostasis?
Nat Cell Biol. 1:E209–216. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
García-Escudero V and Gargini R: Autophagy
induction as an efficient strategy to eradicate tumors. Autophagy.
4:923–925. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramírez C, Pham K, Franco MF, Chwa M, Limb
A, Kuppermann BD and Kenney MC: Hydroquinone induces oxidative and
mitochondrial damage to human retinal Muller cells (MIO-M1).
Neurotoxicology. 39:102–108. 2013. View Article : Google Scholar
|
|
25
|
Kunchithapautham K and Rohrer B: Apoptosis
and autophagy in photoreceptors exposed to oxidative stress.
Autophagy. 3:433–441. 2007. View Article : Google Scholar : PubMed/NCBI
|