Introduction
High oxygen mechanical ventilation is widely used in
clinical settings to treat hypoxemia, which occurs in various
diseases (1). High oxygen
mechanical ventilation is important for treating severe respiratory
conditions, such as respiratory failure and acute respiratory
distress syndrome (ARDS) (2).
However, a study demonstrated that exposure to high levels of
oxygen for prolonged periods of time may actually result in acute
inflammatory reactions and lung injury (3). Therefore, there is a strong
requirement for a therapeutic strategy that will alleviate
hyperoxia-induced lung injury.
Interest in complement molecules has increased
globally and research has been conducted on the role of complement
anaphylatoxins in a number of diseases (4). Notably, complement anaphylatoxin C4a,
released from the N-terminal region of the parental protein
α-chain, has been investigated (5). It has been demonstrated that C4a is a
potent soluble anaphylotoxic and anti-chemotactic inflammatory
regulator, it inhibits the recruitment and activation of
monocytes/macrophages that are induced by C5a (6). Since C4a is able to inhibit
chemoattractants and secretagogues in inflammatory reactions, the
aim of the present study was to determine whether C4a inhibits
hyperoxic-induced lung injury. Excessive high oxygen mechanical
ventilation leads to lung injury, and provokes the secretion of
cytokines and chemokines, such as interleukin (IL)-1, IL-6 and
tumor necrosis factor-α (TNF-α) (7), which results in inflammatory cell
infiltration (8). C4a is released
from the fourth component of complement C4 during activation of the
complement classical pathway and lectin pathway (6). As it is a natural potential factor in
the human body, it is hypothesized that it may have potential in
clinical application. To the best of our knowledge, the present
study provides the first evidence of C4a-mediated effects during
hyperoxic lung injury, and the results indicated that C4a may prove
to be a novel therapeutic strategy for alleviating hyperoxic lung
injury.
Materials and methods
Animals
The experiments were conducted in the Animal
Experimental Center of the Mudanjiang Medical University
(Mudanjiang, China). All animal care and experimental protocols
were in accordance with the Animal Experiment Guidelines of the
Mudanjiang Medical University, and ethical approval was obtained
from the Mudanjiang Medical University. Male BALB/c mice (Kyudo
Co., Ltd., Saga, Japan), aged 6–8-weeks, were fed normal chow and
water ad libitum. The mice were divided into three groups (6
mice per group) and ventilated with 100% oxygen for 36 h, as
described previously (9).
Recombinant C4a (1 µg/25 g of body weight; BioMart, Nanjing,
China) was administered via an ALZET mini-osmotic pump (American
Health & Medical Supply International Corp. Co., Ltd.,
Scarsdale, NY, USA) immediately following exposure to 100% oxygen,
as previously described by Takao et al (10). Following 100% oxygen exposure for
36 h, the mice were sacrificed using chloral hydrate (0.03ml/10g
body weight; Sigma-Aldrich, St. Louis, MO, USA) and lung tissue
samples were harvested for experimental use. The body and relative
lung weights of the mice were determined.
Morphometric analysis
Paraffin-embedded lung tissue samples were observed
by conventional light microscopic examination. Lung tissue sections
(5 mm) were stained with hematoxylin and eosin (Sigma-Aldrich), and
assessed in a double-blinded manner. An automatic Provis AX-70
microscope with a camera (Olympus Corporation, Tokyo, Japan) was
used to capture the microscopy images of the lung samples.
Morphometric analyses of the lung tissue samples were performed
using NIH 1200 image analysis software (Image J version 1.45;
National Institutes of Health, Bethesda, MA, USA), as previously
described (11).
Western blot analysis
According to a method described by Laemmli (12), the proteins from the lung tissue
samples were isolated according to the method described in
(2) and separated using a vertical
slab gel containing 12% poly-acrylamide (Sigma-Aldrich). Proteins
were transferred to a nitrocellulose membrane (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) electrophoretically, as
previously described by Kyhse-Andersen (13), with minor modifications, using a
Semi-Dry Electroblotter (Sartorius AG, Göttingen, Germany) for 90
min at 15 V. The membrane was then treated with Block Ace™ (4%;
Sigma-Aldrich) for 30 min at 22°C. Polyclonal rabbit anti-human
immunoglobulin (IgG) anti-IL-1 (cat. no. SAB4503272), anti-IL-6
(cat. no. I2143) and goat IgG anti-TNF-α (cat. no. T1816) primary
antibodies (all Sigma-Aldrich) in phosphate-buffered saline
supplemented with 0.03% Tween® 20 (PBST; Sigma-Aldrich)
were added to the membranes at a dilution of 1:500 and incubated
for 1 h at 22°C. Following washing with PBST, membranes were
incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit
IgG, and anti-goat IgG (20 ng/ml) for 30 min at 22°C. Following
further washing, an enhanced chemiluminescence (ECL) reaction was
performed using an ECL Plus Western Blotting Detection system™ (GE
Healthcare Life Sciences, Shanghai, China). β-actin served as the
internal control (Sigma-Aldrich). The images of the gels on film
were acquired and analyzed automatically with Gel-Pro Analyzer 4.0
software (Media Cybernetics, Inc., Rockville, MD, USA).
Preparation of the bronchoalveolar lavage
fluid (BALF)
Following sacrifice, surgery was performed on the
mouse chests to expose the lung. Whole lung lavage was performed
four times with injections of 0.5 ml sterile saline through a 21 G
flat syringe needle (BioMart, Shanghai, China) cannulated 0.7 cm
into the trachea. BALF was recovered from each mouse for
quantitative cell counting. The total number of cells was counted
using a hemocytometer (Countess® II Automated Cell
Counter, AMQAX1000; Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), and cell differentiation was determined for
>500 cells placed on cytocentrifuge slides treated with
Wright-Giemsa staining (Sigma-Aldrich), as previously described
(14). A 100 ml aliquot of BALF
was used for the total cell count, and the remainder of the BALF
was immediately centrifuged at 1,000 × g for 10 min. Macrophages
(contained in the precipitate) were isolated from the BALF. The
BALF supernatants were stored at −80°C for further cytokine and
chemokine analysis.
Measurement of histamine release
The histamine release reaction was assessed as
previously described (15).
Briefly, total cells in BALF (2×106 cells/ml) were kept
in cell medium (containing 150 mM NaCl, 3.7 mM KCl, 3 mM
Na2HPO4, 3.5 mM KH2PO4,
5.6 mM glucose, 0.1% gelatin, 0.1% bovine serum albumin, and 1 mM
CaCl2, pH 6.8). Following centrifugation at 10,000 × g
for 1 min at 22°C, the supernatant was collected, and the
precipitated cells were lysed in mast cell medium (Guidechem,
Shanghai, China) supplemented with 2% Triton X-100 (1 ml;
Sigma-Aldrich) for 30 min at 37°C. This analysis was performed
using the F-2500 quantification system with FL Solutions (Hitachi
Ltd., Tokyo, Japan) by calculating the ratio of fluorescent signal
to histamine concentration. Histamine concentrations in the
supernatant and in the cell lysate were measured as the fluorescent
signal observed at 22°C with excitation and emission wavelengths of
360 nm and at 450 nm, respectively. The results were presented as
the percent ratio of released histamine in the supernatant to the
total cellular histamine content, using the following equation:
Amount of histamine in the supernatant/total amount of histamine in
the supernatant and in the cell lysate × 100.
Enzyme-linked immunosorbent assay
(ELISA)
BALF was collected for the IL-1, IL-6 and TNF-α
assays, which were conducted using the respective mouse ELISA kits
(Sigma-Aldrich). The ELISA plates were coated with 100 µl
capture antibody per well at 4°C overnight. Following washing, with
washing buffer contained in the ELISA kit, 200 µl assay
dilution buffer (from the ELISA kit) was added per well for
blocking at room temperature for 1 h. The serially diluted samples
were added to the plate and incubated at 4°C overnight.
HRP-conjugated avidin was added after coating with detection
antibody, and the samples were incubated at room temperature for 30
min. The substrate 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich)
was added and the solution was incubated for 15 min. Subsequently,
2NH2SO4 was added to stop the reaction and
absorbance was measured at 450 nm using an ELISA MTP-800 microplate
reader (Corona Electric, Tokyo, Japan).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the lung tissue samples
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) and
relative mRNA expression was normalized to glyceraldehyde
3-phosphate dehydrogenase (GAPDH). The following primers were used:
CD68 forward, 5′-CATCAGAGCCCGAGTACAGTCTACC-3′, and reverse,
5′-AATTCTGCGCCATGAATGTCC-3′; F4/80 forward,
5′-GAGATTGTGGAAGCATCCGAGAC-3′, and reverse,
5′-GATGACTGTACCCACATGGCTGA-3′; CD64 forward,
5′-CTTCTCCTTCTATGTGGGCAGT-3′, and reverse,
5′-GCTACCTCGCACCAGTATGAT-3′; CD19 forward,
5′-CCACAAAGTCCCAGCTGAAT-3′, and reverse,
5′-GGGGTCCCAGATTTCAAAGT-3′; CD3 forward, 5′-CAGCCTCTTTCTGAGGGAAA-3′
and reverse, 5′-AGGATTCCATCCAGCAGGTA-3′; and GAPDH forward,
5′-GCACCGTCAAGGCTGAGAAC-3′, and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′.
An RNase-Free kit (Roche Diagnostics (Shanghai) Ltd., Shanghai,
China) was used with the following reaction conditions: 37°C for 20
min, 75°C for 10 min and maintained at 4°C, and a Transcriptor
First Strand cDNA Synthesis kit (Roche Diagnostics (Shanghai) Ltd.)
was used with the following reaction conditions: 55°C for 30 min,
85°C for 5 min and maintained at 4°C. RT-qPCR was performed using
the ABI 7300 Fast Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.).
Statistical analysis
The paired Student's t-test was used to analyze the
data. Analyses were conducted using SPSS 17.0 (SPSS, Inc., Chicago,
IL, USA) and the data are expressed as the mean ± standard
deviation, and each experiment was repeated in triplicate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
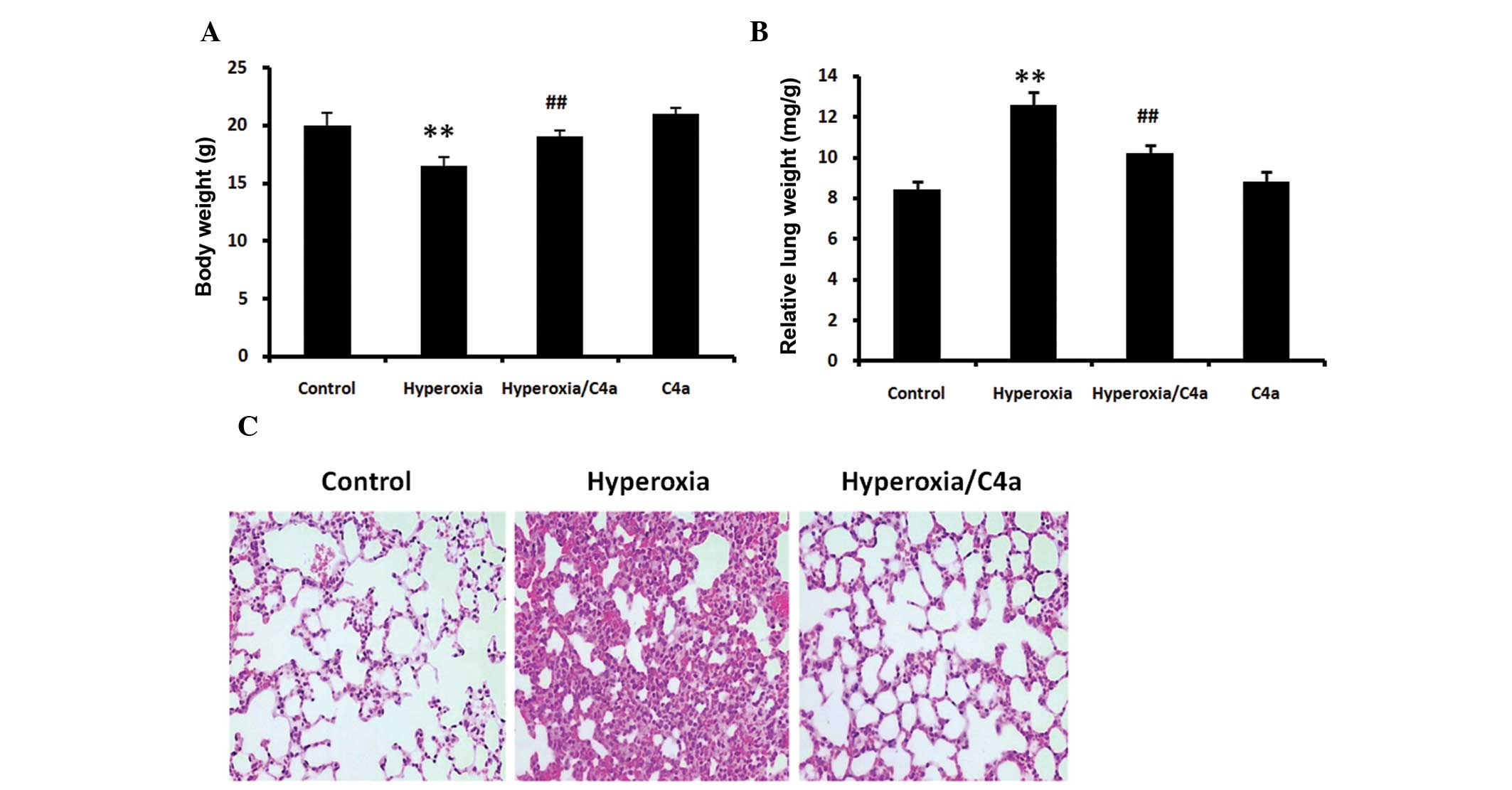
Hyperoxia-induced body and lung weight
alterations are reduced by C4a
The BALB/c mice were ventilated with 100% oxygen for
36 h with or without C4a treatment in conjunction. Following
hyperoxic treatment alone, the body weights of the mice were
significantly reduced (Fig. 1A,
P<0.01) and the relative lung weight was significantly increased
(Fig. 1B, P<0.01). C4a
significantly reduced these changes in body weight and relative
lung weigh compared with the hyperoxia alone-treated mice (Fig. 1A and B).
Hyperoxia-induced morphological changes
in lung tissue are attenuated by C4a
The BALB/c mice were ventilated with 100% oxygen for
36 h with or without C4a treatment and lung morphological changes
were analyzed. Hyperoxia induced increases in thickness of the
alveolar walls and inflammatory cell infiltration in the lung
tissue. However, treatment with C4a attenuated the
hyperoxia-induced morphological changes, as shown in Fig. 1C.
Hyperoxia-induced inflammatory reaction
in lung tissue and BALF samples is attenuated by C4a
The BALB/c mice were ventilated with 100% oxygen for
36 h with or without C4a treatment, prior to lung tissue and BALF
sample harvesting. The expression levels of IL-1, IL-6 and TNF-α in
the lung tissue samples (Fig. 2A and
B) and BALF (Fig. 2C), and the
histamine release levels in the total cells in the BALF (Fig. 2D), were significantly increased
following treatment with 100% oxygen. Hyperoxia-induced expression
of IL-1, IL-6 and TNF-α in lung tissue and BALF and histamine
release of total cells in BALF were significantly reduced by
treatment with C4a (Fig. 2;
P<0.01).
 | Figure 2C4a attenuates the hyperoxia-induced
inflammatory reaction in lung tissue and BALF samples. The BALB/c
mice were ventilated with 100% oxygen for 36 h with or without C4a
treatment, prior to lung tissue and BALF harvesting. (A and B) The
protein expression levels of IL-1, IL-6 and TNF-α in lung tissue,
determined by western blot analysis. and (C) in BALF. (D) The
histamine release levels of the total cells in the BALF and the
expression levels of IL-1, IL-6 and TNF-α were significantly
increased following treatment with 100% oxygen. Treatment with C4a
significantly reduced the expression levels of IL-1, IL-6 and TNF-α
in the lung tissue and BALF samples, as well as the histamine
release levels of total cells in the BALF, as compared with those
of the hyperoxia-only group. (C) The levels of histamine release
were normalized with cell numbers. The data are expressed as the
mean ± standard deviation (n=3). *P<0.05,
**P<0.01 vs. the control mice; #P<0.05,
##P<0.01 vs. the hyperoxia-treated mice. IL,
interleukin; TNF-α, tumor necrosis factor-α; BALF, bronchoalveolar
lavage fluid. |
C4a attenuated the hyperoxia-induced
macrophage infiltration, but not neutrophil or lymphocyte
infiltration in BALF
BALF samples were collected following ventilation
with 100% oxygen for 36 h with or without C4a treatment (Fig. 3). Hyperoxia significantly induced
inflammatory cell infiltration in BALF (P<0.01). The
hyperoxia-induced macrophage infiltration was significantly reduced
by treatment with C4a (Fig. 3B;
P<0.01). However, C4a treatment did not reduce neutrophil or
lymphocyte infiltration (Fig. 3C and
D).
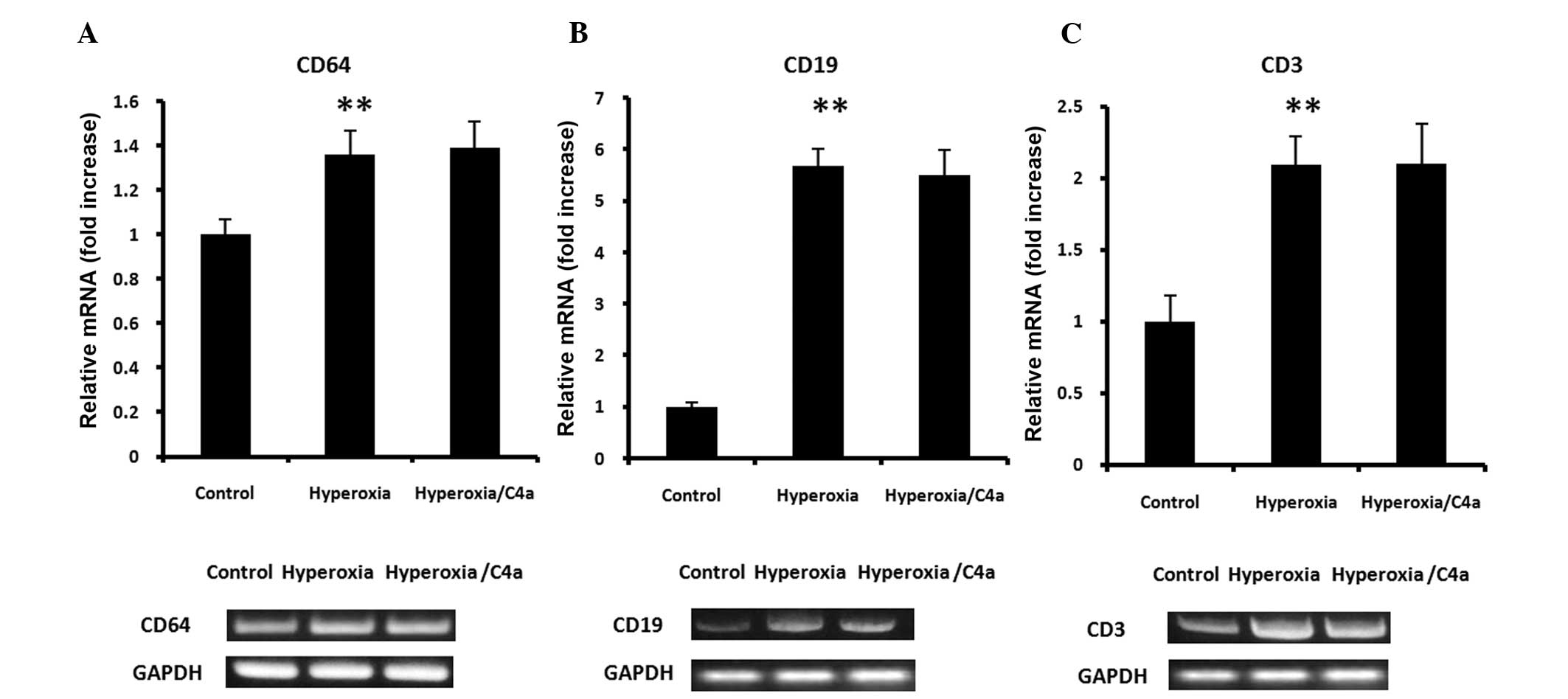
C4a attenuated the hyperoxia-induced
macrophage infiltration, but not neutrophil or lymphocyte
infiltration in lung tissue samples
As markers of macrophages, CD68 and F4/80 mRNA
expression levels were detected in the lung tissue samples. The
expression of CD68 and F4/80 mRNA in the lung tissue was
significantly induced by hyperoxia, however, treatment with C4a
significantly suppressed CD68 and F4/80 mRNA expression levels
(Fig. 4; P<0.01). Furthermore,
mRNA expression of CD64 (neutrophil marker), CD19 (B lymphocyte
marker) and CD3 (T lymphocyte marker) were detected in the lung
tissue samples. Hyperoxia significantly increased the mRNA
expression levels of CD64, CD19 and CD3 in the lung tissue samples.
However, the hyperoxia-induced CD64, CD19 and CD3 mRNA expression
levels were not suppressed by C4a treatment (Fig. 5).
Discussion
To the best of our knowledge, the present study
reported the first evidence of complement anaphylatoxin
C4a-mediated action during hyperoxic lung injury in mice. In recent
years, complement anaphylatoxin research has intensified,
particularly in a number of diseases, such as hepatitis C,
cardiovascular disease, immunological diseases and tumors (4,6,14,16).
Complement anaphylatoxin C4a, an important member of the complement
anaphylatoxin family, has also been investigated (5). C4a, which is liberated from the
N-terminal region of the parental protein α-chain, was previously
isolated from the inflammatory joint fluid of patients with
rheumatoid arthritis (16,17). C4a is able to inhibit the effects
of chemoattractants and secretagogues in inflammatory reactions
(5,14). However, the role of C4a in
hyperoxic lung injury has yet to be elucidated.
Respiratory failure and acute respiratory distress
syndrome (ARDS) are common diseases in the clinic (1). As a clinical therapy, mechanical
ventilation with high oxygen is widely used in the treatment of the
hypoxemia associated with various diseases (2,18).
However, as shown in a previous study, prolonged exposure to
hyperoxia can lead to inflammatory reactions and lung injury
(3). As a consequence, identifying
novel therapeutic strategies to alleviate hyperoxic lung
inflammatory and injury is required. As C4a is able to inhibit
chemoattractants and secretagogues in inflammatory reactions
(5,6,14),
it was hypothesized that C4a could inhibit hyperoxic lung
injury.
The present study provided, to the best of our
knowledge, the first analysis of the effects of C4a against
hyperoxic lung injury. Excessive high oxygen mechanical ventilation
leads to lung injury and provokes the secretion of cytokines and
chemokines, such as histamine, IL-1, IL-6 and TNF-α (6,19,20),
which results in inflammatory cell infiltration (7). IL-1 and IL-6 are involved in pro- and
anti-inflammatory responses via the regulation of leukocyte
function and apoptosis (19,21).
IL-1 and IL-6 have been reported to beneficially regulate
neutrophil adhesion and migration (22). Elevated IL-1 and IL-6 expression
levels have been demonstrated in the majority of lung injury
models, suggesting they may be biological markers of lung injury
(23,24). TNF-α is also a canonical
inflammatory cytokine that promotes the inflammatory response, such
as inflammatory cell migration and proliferation (25). Treatment with 100% oxygen led to
lung injury and marked morphological changes. To improve the
current understanding of hyperoxia-induced lung injury, the
expression levels of IL-1, IL-6 and TNF-α in lung tissue samples
and BALF, and the histamine release from the cells in BALF were
examined. Hyperoxia significantly increased the expression levels
of IL-1, IL-6 and TNF-α in lung tissue samples and BALF, and also
increased the histamine release levels in total cells.
Hyperoxia-induced lung injury, lung morphological changes and
inflammatory reaction in the lung tissue samples and BALF were all
significantly attenuated following treatment with C4a.
This study showed that hyperoxia significantly
increased the total cell count, as well as the numbers of
macrophages, neutrophils and lymphocytes in the BALF. C4a
significantly attenuated the hyperoxia-induced increase in the
number of macrophages. These results are consistent with previous
results, which demonstrated that hyperoxia could recruit
inflammatory cells, such as macrophages, lymphocytes and
neutrophils (26). However, the
increased numbers of neutrophils and lymphocytes were not affected
by treatment with C4a. Furthermore, the mRNA expression levels of
CD68 and F4/80, indicators of macrophage accumulation; CD64, a
neutrophil marker; CD19, a B lymphocyte marker; and CD3, a lung
tissue T lymphocyte marker, were significantly increased by
hyperoxia. The mRNA expression levels of CD68 and F4/80 in lung
tissue samples significantly reduced following treatment with C4a,
as compared with the hyperoxia-only group. Conversely, treatment
with C4a did not affect the mRNA expression levels of CD64, CD19 or
CD3 in the murine lung tissue samples. These results were
concordant with those of a previous study which demonstrated that
C4a is a monocyte and macrophage migration inhibitory factor, and
the C4a receptor is restricted to monocytes and macrophages
(27). As shown in Fig. 6, hyperoxia induced lung injury,
lung morphological changes, and inflammatory reactions, which were
attenuated by treatment with C4a. C4a acted via a
macrophage-dependent but not a neutrophil/lymphocyte-dependent
signaling pathway. However, as a novel therapeutic strategy for
hyperoxic lung injury, treatment with C4a requires further
investigation.
In conclusion, to the best of our knowledge, the
present study is the first to demonstrate the crucial role of C4a
in the attenuation of hyperoxia-induced lung injury, and provides a
rationale for the potential use of C4a in clinical settings to
treat acute exacerbation of hyperoxic lung injury.
References
|
1
|
Vitacca M, Bianchi L, Bazza A and Clini
EM: Advanced COPD patients under home mechanical ventilation and/or
long term oxygen therapy: Italian healthcare costs. Monaldi Arch
Chest Dis. 75:207–214. 2011.
|
|
2
|
Xu Y, Tian Z and Xie P: Targeting
complement anaphylatoxin C5a receptor in hyperoxic lung injury in
mice. Mol Med Rep. 10:1786–1792. 2014.PubMed/NCBI
|
|
3
|
Vosdoganes P, Lim R, Koulaeva E, Chan ST,
Acharya R, Moss TJ and Wallace EM: Human amnion epithelial cells
modulate hyperoxia-induced neonatal lung injury in mice.
Cytotherapy. 15:1021–1029. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Imakiire K, Uto H, Sato Y, Sasaki F,
Mawatari S, Ido A, Shimoda K, Hayashi K, Stuver SO, Ito Y, et al:
Difference in serum complement component C4a levels between
hepatitis C virus carriers with persistently normal alanine
aminotransferase levels or chronic hepatitis C. Mol Med Rep.
6:259–264. 2012.PubMed/NCBI
|
|
5
|
Hugli TE: Biochemistry and biology of
anaphylatoxins. Complement. 3:111–127. 1986.PubMed/NCBI
|
|
6
|
Zhao Y, Xu H, Yu W and Xie BD: Complement
anaphylatoxin C4a inhibits C5a-induced neointima formation
following arterial injury. Mol Med Rep. 10:45–52. 2014.PubMed/NCBI
|
|
7
|
Pushparaj PN, Tay HK, Wang CC, Hong W and
Melendez AJ: VAMP8 is essential in anaphylatoxin-induced
degranulation, TNF-alpha secretion, peritonitis and systemic
inflammation. J Immunol. 183:1413–1418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davies J, Karmouty-Quintana H, Le TT, Chen
NY, Weng T, Luo F, Molina J, Moorthy B and Blackburn MR: Adenosine
promotes vascular barrier function in hyperoxic lung injury.
Physiol Rep. 2:e121552014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mikawa K, Nishina K, Maekawa N and Obara
H: Attenuation of hyperoxic lung injury in rabbits with superoxide
dismutase: Effects on inflammatory mediators. Acta Anaesthesiol
Scand. 39:317–322. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takao Y, Mikawa K, Nishina K, Maekawa N
and Obara H: Lidocaine attenuates hyperoxic lung injury in rabbits.
Acta Anaesthesiol Scand. 40:318–325. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Runzi M, Raptopoulos V, Saluja AK, Kaiser
AM, Nishino H, Gerdes D and Steer ML: Evaluation of necrotizing
pancreatitis in the opossum by dynamic contrast-enhanced computed
tomography: Correlation between radiographic and morphologic
changes. J Am Coll Surg. 180:673–682. 1995.PubMed/NCBI
|
|
12
|
Laemmli UK: Cleavage of structural
proteins during the assemblyof the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kyhse-Andersen J: Electroblotting of
multiple gels: A simple apparatus without buffer tank for rapid
transfer of proteins from polyacrylamide to nitrocellulose. J
Biochem Biophys Methods. 10:203–209. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsuruta T, Yamamoto T, Matsubara S,
Nagasawa S, Tanase S, Tanaka J, Takagi K and Kambara T: Novel
function of C4a anaphylatoxin. Release from monocytes of protein
which inhibits monocyte chemotaxis. Am J Pathol. 142:1848–1857.
1993.PubMed/NCBI
|
|
15
|
Nishiura H, Tokita K, Li Y, Harada K,
Woodruff TM, Taylor SM, Nsiama TK, Nishino N and Yamamoto T: The
role of the ribosomal protein S19 C-terminus in Gi
protein-dependent alternative activation of p38 MAP kinase via the
C5a receptor in HMC-1 cells. Apoptosis. 15:966–981. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsubara S, Yamamoto T, Tsuruta T, Takagi
K and Kambara T: Complement C4-derived monocyte-directed chemotaxis
inhibitory factor. A molecular mechanism to cause polymorphonuclear
leukocyte-predominant infiltration in rheumatoid arthritis synovial
cavities. Am J Pathol. 138:1279–1291. 1991.PubMed/NCBI
|
|
17
|
Murakami Y, Yamamoto T, Imamichi T and
Nagasawa S: Cellular responses of guinea pig macrophages to C4a;
inhibition of C3a-induced O2-generation by C4a. Immunol Lett.
36:301–304. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Levitt JE, Calfee CS, Goldstein BA, Vojnik
R and Matthay MA: Early acute lung injury: Criteria for identifying
lung injury prior to the need for positive pressure
ventilation*. Crit Care Med. 41:1929–1937. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jones SA: Directing transition from innate
to acquired immunity: Defining a role for IL-6. J Immunol.
175:3463–3468. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bao JP, Jiang LF, Li J, Chen WP, Hu PF and
Wu LD: Visceral adipose tissue-derived serine protease inhibitor
inhibits interleukin-1β-induced catabolic and inflammatory
responses in murine chondrocytes. Mol Med Rep. 10:2191–2197.
2014.PubMed/NCBI
|
|
21
|
Kim HS, Park JW, Kwon OK, Kim JH, Oh SR,
Lee HK, Bach TT, Quang BH and Ahn KS: Anti-inflammatory activity of
a methanol extract from Ardisia tinctoria on mouse macrophages and
paw edema. Mol Med Rep. 9:1388–1394. 2014.PubMed/NCBI
|
|
22
|
Wolters PJ, Wray C, Sutherland RE, Kim SS,
Koff J, Mao Y and Frank JA: Neutrophil-derived IL-6 limits alveolar
barrier disruption in experimental ventilator-induced lung injury.
J Immunol. 182:8056–8062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Halbertsma FJ, Vaneker M, Scheffer GJ and
van der Hoeven JG: Cytokines and biotrauma in ventilator-induced
lung injury: A critical review of the literature. Neth J Med.
63:382–392. 2005.PubMed/NCBI
|
|
24
|
Frank JA, Parsons PE and Matthay MA:
Pathogenetic significance of biological markers of
ventilator-associated lung injury in experimental and clinical
studies. Chest. 130:1906–1914. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Piguet PF, Collart MA, Grau GE, Sappino AP
and Vassalli P: Requirement of tumour necrosis factor for
development of silica-induced pulmonary fibrosis. Nature.
344:245–247. 1990. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Janssen WJ, Barthel L, Muldrow A,
Oberley-Deegan RE, Kearns MT, Jakubzick C and Henson PM: Fas
determines differential fates of resident and recruited macrophages
during resolution of acute lung injury. Am J Respir Crit Care Med.
184:547–560. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dolinay T, Wu W, Kaminski N, Ifedigbo E,
Kaynar AM, Szilasi M, Watkins SC, Ryter SW, Hoetzel A and Choi AM:
Mitogen-activated protein kinases regulate susceptibility to
ventilator-induced lung injury. PLoS One. 3:e16012008. View Article : Google Scholar : PubMed/NCBI
|