Introduction
Atherosclerosis (AS) is a progressive disease
characterized by accumulation of lipids and fibrous elements in the
arterial tunica intima, which results in atherosclerotic plaque
formation and arterial narrowing (1). During the progression of AS,
macrophages are critical (2). The
development of AS is strongly associated with engulfment of
oxidized-low-density lipoprotein (Ox-LDL) by macrophages. It has
previously been reported that macrophages engulf Ox-LDL via
scavenger receptors, resulting in the deposition of a large
quantity of lipids, which further promote the progression of AS
(1). Recently, mitogen-activated
protein kinase (MAPK) signaling cascades have been investigated in
macrophages in vitro, in order to evaluate their importance
(3).
Extracellular signal-regulated kinases 1/2 (ERK1/2)
are members of the MAPK family, which are involved in mediating
various processes, including cell cycle progression, cell
migration, survival, differentiation and proliferation (4). However, the association between
ERK1/2 signaling and lipid metabolism remains to be elucidated.
ERK1/2 inhibitors have been reported to synergize
with liver X receptor (LXR) ligand to induce ATP-binding cassette
transporter (ABC)A1 expression and regulate cholesterol efflux
(5). In addition, ABCA1 and ABCG1
have been demonstrated to stimulate the efflux of lipids via the
reverse cholesterol transport (RCT) pathway (6). As a specific macrophage scavenger
receptor, cluster of differentiation (CD)36 recognizes and
internalizes Ox-LDL particles, resulting in cellular cholesterol
uptake, lipid accumulation in intracellular space and the
production of foam cells (7). It
has therefore been hypothesized that ABCA1/G1 and CD36 are crucial
to the maintenance of intracellular cholesterol stability or the
efficacy of the RCT process (8).
However, the regulation of ABCA1/G1 and CD36 expression by ERK1/2
in macrophages remains to be elucidated. The aim of the present
study was to investigate the association between ERK1/2 and lipid
metabolism, and evaluate the effect of ERK1/2 on the expression
levels of ABCA1/G1 and CD36 in macrophages.
Materials and methods
Animals
A total of 40 male Sprague Dawley rats (weight,
200–220 g; age, 8–9 weeks) were purchased from the Laboratory
Animal Center of Fujian Medical University (Fuzhou, China). The
rats were housed under a 12 h light/dark cycle at 37°C in an
atmosphere containing 5% CO2, and were given ad
libitum access to food and water. All animal experiments in the
present study were performed in accordance with the recommendations
of the Guidelines for the Care and Use of Laboratory Animals of the
Ministry of Science of People's Republic of China. The present
study was approved by the Animal Care and Use Committee and
Institutional Review Board of Fujian University of Traditional
Chinese Medicine (Fuzhou, China) (permission no. IRB201100117) and
the Ethics Review Committee of Fujian University of Traditional
Chinese Medicine (permission no. FJZYYDXERC2011010).
Isolation and treatment of rat peritoneal
macrophages (RPMs)
The rats were anesthetized with 10% chloral hydrate
(Peking Union-Biology Co., Ltd., Beijing, China) and were
sacrificed by cervical dislocation, and soaked in 75% ethanol for 5
min. RPMs were harvested by lavaging the peritoneal cavity with 10
ml Dulbecco's modified Eagle's medium/Ham's F12 (DMEM-F12; Hyclone;
GE Healthcare Life Sciences, Logan, UT, USA). Cells were
centrifuged at 4°C, 1,000 × g for 15 min, and cultured in DMEM
supplemented with 1% glutamine and penicillin/streptomycin (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), and 10% fetal
bovine serum (FBS; Hyclone; GE Healthcare Life Sciences) at 37°C in
an incubator containing 5% CO2. RPMs in the exponential
growth phase were seeded onto 6-well plates at a density of
5×106 cells/well for the indicated time. RPMs were
randomly assigned to three groups. Cells in the control group were
incubated in DMEM-F12 supplemented with 10% FBS for 12, 24 and 48
h; cells in the Ox-LDL group were treated with 50 mg/l Ox-LDL
(Peking Union-Biology Co., Ltd.) or 10 µg/ml DiI-Ox-LDL
(Peking Union-Biology Co., Ltd.) for 12, 24 and 48 h, respectively
(8–12); whereas cells in the Ox-LDL + U0126
group were incubated in DMEM-F12 containing 10% FBS supplemented
with 10 µM U0126 (Sigma-Aldrich, St. Louis, MO, USA) and
Ox-LDL or DiI-Ox-LDL for 12, 24 or 48 h.
Morphological observation
RPMs were incubated with DMEM-F12 containing 10% FBS
in 6-well plates. Once the cells had adhered to the culture plates,
RPMs were pretreated with Ox-LDL alone, or in combination with
U0126, for 12, 24 and 48 h, whereas the untreated cells served as
controls. Cellular lipid accumulation was detected using oil red O
staining and DiI fluorescence. Cells were washed three times with
phosphate-buffered saline (PBS), fixed in 5% formalin solution for
15 min at room temperature, washed once with PBS and stained with
oil red O (Wuhan Boster Biological Technology, Ltd., Wuhan, China)
for 10 min, followed by hematoxylin (Wuhan Boster Biological
Technology, Ltd.) staining for 5 min. Cells were then observed
under a Leica DM IL compact inverted stereo microscope (Leica
Microsystems GmbH, Wetzlar, Germany), and images were captured at
magnification ×400. In addition, RPMs in monolayer cultures were
exposed to DiI-Ox-LDL alone, or in combination with U0126, for 12,
24 and 48 h. The medium was removed and cells were washed with PBS,
mounted on cover slips and analyzed using fluorescent
microscopy.
Cytotoxicity assay
The viability of RPMs post-treatment with Ox-LDL
(10, 30, 50, 70 and 90 mg/l) was assessed using an MTS assay
(Promega Corporation, Madison, WI, USA). The RPMs were grown in
96-well plates and were incubated with Ox-LDL (10, 30, 50, 70 or 90
mg/l) for 12, 24 or 48 h. The MTS assay was used to measure the
viability of the cells. Cells were incubated at 37°C with MTS (1.90
mg/ml) for 4 h, and absorbance was measured using a microplate
reader (Multiskan GO; Thermo Fisher Scientific Inc.) at a
wavelength of 490 nm. Three wells were set for each concentration
of Ox-LDL. All experiments were repeated twice.
Reverse transcription-polymerase chain
reaction (RT-PCR) assay
Total RNA was isolated from the RPMs using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.),
and total RNA (500 ng) was reverse transcribed in a total volume of
20 µl containing oligo (dT) primers at 42°C for 30 min using
TransScript® One-Step gDNA Removal and cDNA Synthesis
SuperMix (Beijing Transgen Biotech Co., Ltd., Beijing, China).
Primers used for PCR (Thermo Fisher Scientific, Inc.) are presented
in Table I. PCR was performed on
an Applied Biosystems® 2720 Thermal Cycler (Applied
Biosystems; Thermo Fisher Scientific, Inc.) in a 20 µl
reaction system containing cDNA (500 ng/µl), each specific
primer, and 2X EasyTaq® PCR SuperMix (Beijing Transgen
Biotech Co., Ltd.) under the following conditions: Pre-denaturation
at 94°C for 5 min; 35 cycles at 94°C for 30 sec, 58°C for 30 sec
and 72°C for 1 min; followed by final extension at 72°C for 7 min.
The PCR products were separated by 1.5% agarose gel
electrophoresis, and the relative mRNA expression levels were
determined as the ratio of the grayscale value of the target gene
to GAPDH (Image-Lab version 5.0; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The experiment was repeated three times.
 | Table ISequences of primers used for reverse
transcription-polymerase chain reaction. |
Table I
Sequences of primers used for reverse
transcription-polymerase chain reaction.
| Gene | Primer sequence,
5′→3′ |
|---|
| CD36 | F:
ACTCCAGAACCCAGACAACCAC |
| R:
ACCAAGTAAGACCATCTCAACCAG |
| ABCA1 | F:
CCTAAGCATTATCAAGGAGGGAAG |
| R:
AGAGATGACAAGGAGGACGGAAG |
| ABCG1 | F:
GTCCTGGGCATCTTCTTCATCTC |
| R:
CCAACTCAGCCAACACTCCTCTC |
| GAPDH | F:
ACGGCAAGTTCAACGGCACAG |
| R:
GAAGACGCCAGTAGACTCCACGAC |
Western blot analysis
RPMs were digested with pancreatin (Gibco; Thermo
Fisher Scientific, Inc.), pippetted evenly, and centrifuged. The
supernatant was transferred to a 1.5 ml centrifuge tube, lysed on
ice in radioimmunoprecipitation assay lysis buffer (Beyotime
Institute of Biotechnology, Shanghai, China) for 30 min, and
fractionated three times with ultrasound. The mixture was then
centrifuged at 13,800 × g for 15 min, and the supernatant was
stored at −80°C for subsequent experiments. Protein isolated from
the RPMs was quantified using the Bicinchoninic Acid assay (Wuhan
Boster Biological Technology, Ltd). Approximately 30 µg
protein was separated by 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis at 110 V for 2.5 h at room temperature, and was
then transferred to polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA) for a further 1.5 h at 110 V and
4°C. The membrane was blocked with 5% non-fat milk in Tris-buffered
saline for 1 h at room temperature and probed with mouse monoclonal
anti-ABCA1 (1:1,000; cat. no. ab18180), rabbit monoclonal
anti-ABCG1 (1:1,000; cat. no. ab52617), rabbit polyclonal anti-CD36
(1:500; cat. no. ab78054) (Abcam, Cambridge, UK), rabbit polyclonal
phosphorylated (p)-p44/42 MAPK (ERK1/2) (1:1,000; cat. no. 9101),
rabbit polyclonal p44/42 MAPK (ERK1/2) (1:1,000; cat. no. 9102) and
anti-β-actin (1:1,000; cat. no. 4967) (Cell Signaling Technology,
Inc., Danvers, MA, USA) antibodies at 4°C overnight. The membrane
was then washed with PBS, and incubated with horseradish
peroxidase-conjugated goat anti-rabbit (1:5,000; cat. no. A0208)
and anti-mouse (1:5,000; cat. no. A0216) secondary antibodies
(Beyotime Institute of Biotechnology) for a further 2 h at room
temperature. Enhanced chemiluminescence reagents (Beyotime
Institute of Biotechnology) and chemiluminescent substrates were
used to visualize immunoreactive bands. The grayscale values of the
protein bands were estimated using the image processing software
Image-Lab version 5.0 (Bio-Rad Laboratories, Inc.). The ratio of
the grayscale value of the target protein band to β-actin protein
band was defined as the protein expression level. All experiments
were repeated three times.
Statistical analysis
All data are presented as the mean ± standard
deviation. Differences between means were determined using one-way
analysis of variance followed by Dunnett's test, or Student's
t-test. Statistical analyses were conducted using SPSS version 18.0
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Oil red O staining
RPMs were treated with 50 mg/l Ox-LDL for 12, 24 and
48 h, washed twice with PBS, and stained with oil red O and
hematoxylin. Intense oil red O staining (red) was detected in RPMs
treated with Ox-LDL, indicating intracellular lipid accumulation.
However, staining was markedly reduced in RPMs co-treated with
Ox-LDL and 10 µM U0126, suggesting reduced lipid deposition
(Fig. 1A).
 | Figure 1ERK1/2 inhibitor, U0126, markedly
reduces lipid deposition in RPMs treated with 50 mg/l Ox-LDL alone
or in combination with 10 µM U0126 for 12, 24 and 48 h. (A)
Cells were stained with oil red O and examined under an inverted
microscope (magnification, ×400). RPMs were observed to engulf a
large quantity of lipids (red). (B) No red fluorescence was
detected in untreated RPMs, whereas red fluorescence was detected
in DiI-Ox-LDL-treated RPMs, suggesting lipid deposition in
macrophages (magnification, ×400). ERK, extracellular
signal-regulated kinases; RPMs, rat peritoneal macrophages; Ox-LDL,
oxidized-low-density lipoprotein. |
Fluorescence
To examine the effects of the ERK1/2 inhibitor on
DiI-Ox-LDL uptake by the RPMs, cells were cultured with DiI-Ox-LDL
for 12, 24 and 48 h. Treatment with DiI-Ox-LDL markedly increased
cytoplasmic fluorescence of RPMs, as compared with untreated
controls (Fig. 1B), and engulfment
of DiI-Ox-LDL in RPMs was demonstrated by observation of
intracellular accumulation of DiI-labeled lipids. However, markedly
reduced DiI-Ox-LDL fluorescence was observed in RPMs following the
addition of U0126, indicating that treatment with U0126 may result
in reduced lipid deposition.
MTS assay
To evaluate the hypothesis that Ox-LDL may result in
necrosis and apoptosis of RPMs, the effect of Ox-LDL on cell
viability was evaluated by MTS assay. A greater viability of RPMs
was observed following treatment with Ox-LDL at a concentration of
50 mg/l, as compared with Ox-LDL treatment at 10, 30, 70 and 90
mg/l for 12 and 48 h. Treatment with Ox-LDL at a concentration of
50 mg/l resulted in greater viability of RPMs, as compared with
Ox-LDL treatment at 70 and 90 mg/l for 24 h. These results suggest
that cell viability was increased in response to 50 mg/l Ox-LDL
treatment (Fig. 2A).
Expression levels of ERK and p-ERK
Western blot analysis detected comparative ERK
expression in RPMs treated with Ox-LDL for 12, 24 and 48 h, whereas
p-ERK expression levels were markedly increased in Ox-LDL-treated
RPMs, as compared with untreated RPMs (Fig. 2B). The addition of 10 µM
U0126 led to a marked reduction in the expression levels of p-ERK,
as compared with treatment with Ox-LDL alone, indicating that the
ERK1/2 inhibitor attenuates ERK1/2 phosphorylation.
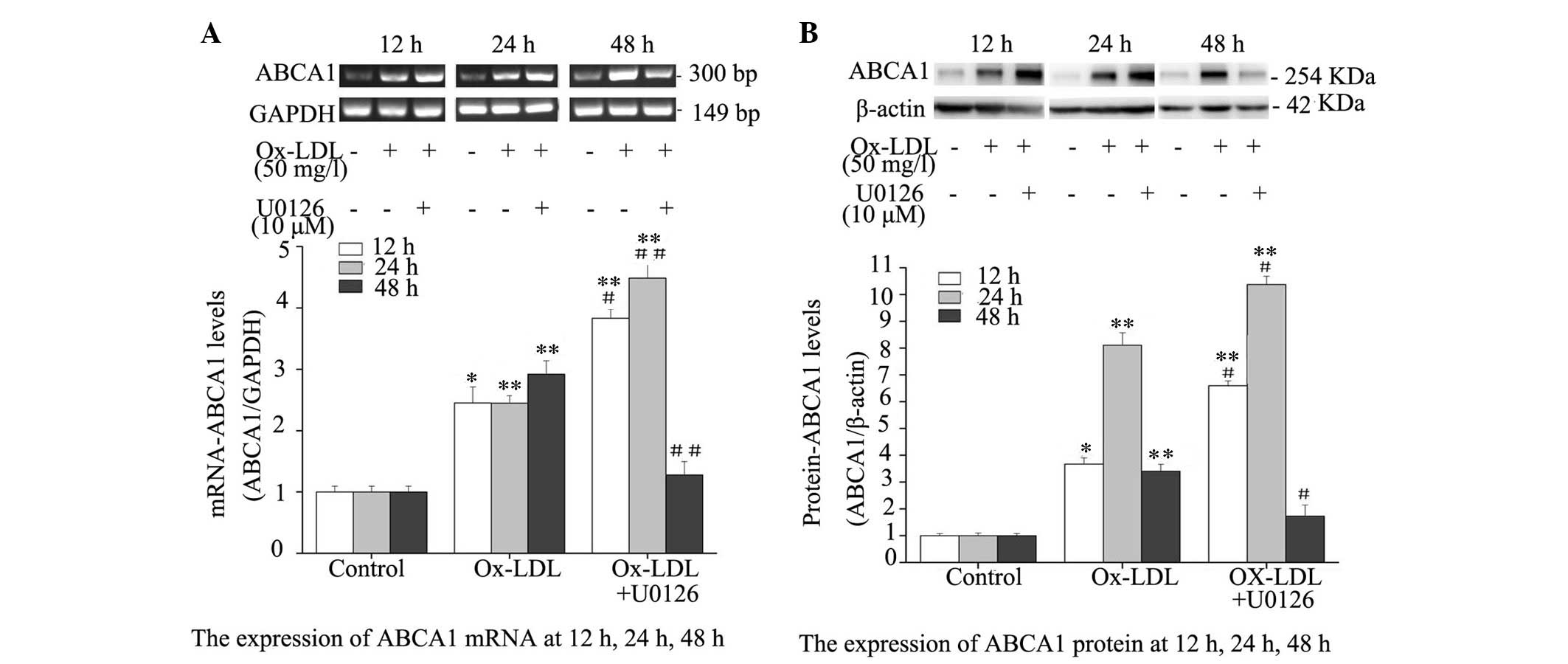
Inhibition of ERK1/2 upregulates
Ox-LDL-induced ABCA1 expression
To evaluate the effect of ERK1/2 inhibition on ABCA1
expression, mRNA and protein expression levels of ABCA1 were
determined in Ox-LDL-treated RPMs. ABCA1 expression levels were
markedly elevated in RPMs treated with Ox-LDL alone or Ox-LDL +
U0126 at the mRNA (Fig. 3A) and
protein (Fig. 3B) level. In
addition, the mRNA and protein expression levels of ABCA1 were
notably increased in RPMs following co-treatment with Ox-LDL +
U0126 for 12 and 24 h; however, the mRNA and protein expression
levels of ABCA1 were not significantly greater in RPMs treated with
Ox-LDL + U0126 for 48 h, as compared with RPMs treated with Ox-LDL
alone. These results indicate that ERK1/2 inhibition may upregulate
mRNA and protein expression levels of ABCA1 in Ox-LDL-treated
macrophages.
Inhibition of ERK1/2 augments
Ox-LDL-induced ABCG1 expression
Western blotting and RT-PCR indicated a significant
increase in ABCG1 expression at the transcriptional (Fig. 4A) and translational (Fig. 4B) level following Ox-LDL treatment.
In addition, mRNA and protein expression levels of ABCG1 were
significantly higher in RPMs co-treated with Ox-LDL and U0126 for
12 and 24 h, as compared with those treated with Ox-LDL alone.
Conversely, no significant difference was detected in ABCG1 mRNA
and protein expression levels at 48 h post-treatment with Ox-LDL
alone or in combination with U0126, and the mRNA and protein
expression levels of ABCG1 were not higher in RPMs treated with
Ox-LDL + U0126, as compared with RPMs treated with Ox-LDL alone.
These results indicate that ERK1/2 inhibition augments mRNA and
protein expression levels of ABCG1 in Ox-LDL-stimulated
macrophages.
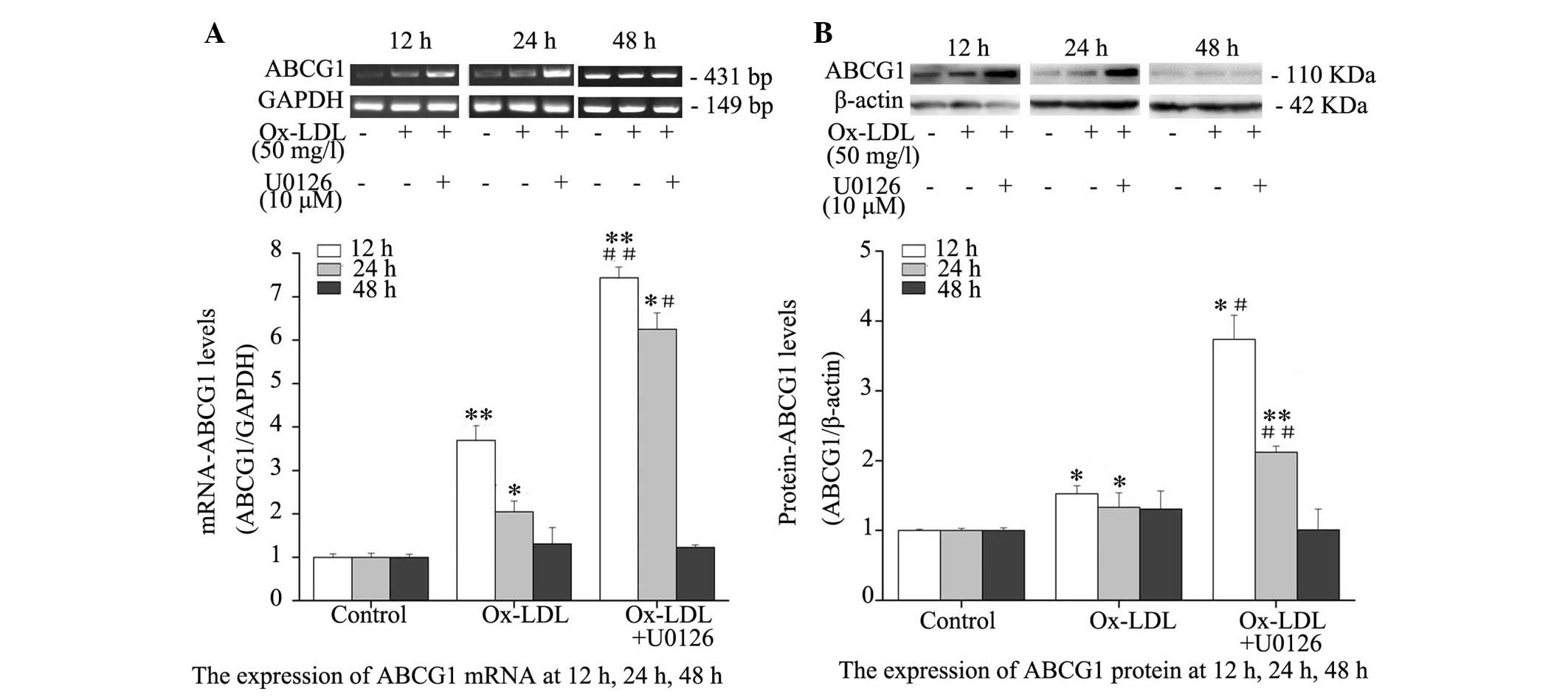 | Figure 4Effects of ERK1/2 inhibitor, U0126,
on ABCG1 mRNA and protein expression levels in RPMs. RPMs were
treated with 50 mg/l Ox-LDL alone or in combination with 10
µM U0126 for 12, 24 and 48 h. (A) Reverse
transcription-polymerase chain reaction was used to determine ABCG1
mRNA expression. GAPDH served as an internal control. (B) Whole
cell lysates were used for western blot analysis to determine
protein expression. Data are presented as the mean ± standard
deviation. *P<0.05, **P<0.01, the
Ox-LDL group vs. the control group; #P<0.05,
##P<0.01, the Ox-LDL + U0126 group vs. the Ox-LDL
group. RPMs, rat peritoneal macrophages; ERK, extracellular
signal-regulated kinases; Ox-LDL, oxidized-low-density lipoprotein;
ABCG1, ATP-binding cassette transporter G-1; GAPDH, glyceraldehyde
3-phosphate dehydrogenase. |
Inhibition of ERK1/2 reduces CD36
expression in Ox-LDL-treated macrophages
The effect of ERK1/2 inhibition on Ox-LDL-induced
CD36 expression was evaluated by RT-PCR and western blotting.
Treatment with Ox-LDL resulted in a significant elevation in the
expression levels of CD36, as compared with the untreated controls
(P<0.05), and co-treatment with Ox-LDL and U0126 for 12, 24 and
48 h resulted in significant reductions in mRNA (Fig. 5A) and protein (Fig. 5B) expression levels of CD36, as
compared with treatment with Ox-LDL alone. These results suggest
that Ox-LDL markedly induces CD36 expression and inhibition of
ERK1/2 reduces CD36 expression in Ox-LDL-treated macrophages.
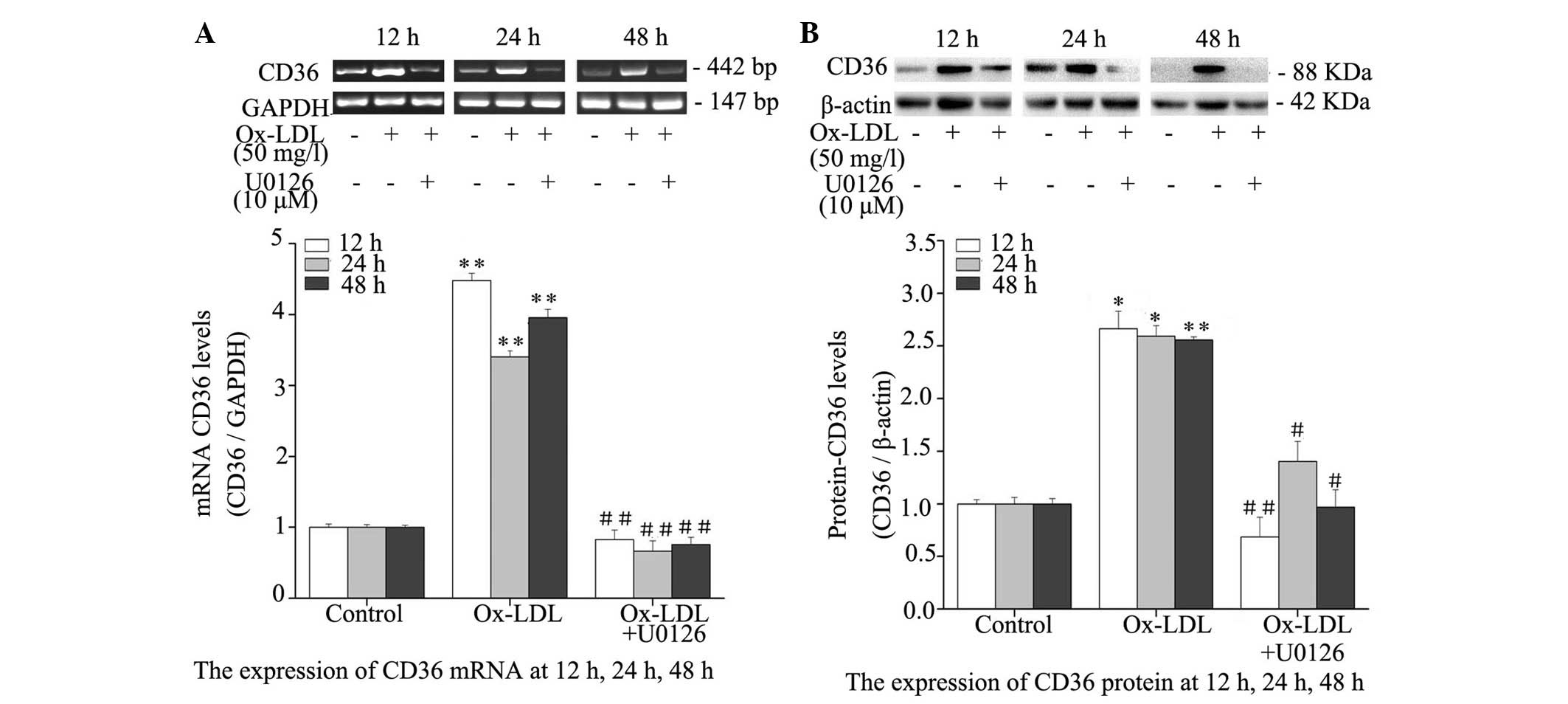 | Figure 5Effects of ERK1/2 inhibitor, U0126,
on CD36 mRNA and protein expression levels in RPMs. RPMs were
treated with 50 mg/l Ox-LDL alone or in combination with 10
µM U0126 for 12, 24 and 48 h. (A) Reverse
transcription-polymerase chain reaction was used to determine CD36
mRNA expression. GAPDH served as an internal control. (B) Whole
cell lysates were used for western blot analysis to determine CD36
protein expression. Data are presented as the mean ± standard
deviation. *P<0.05, **P<0.01, the
Ox-LDL group vs. the control group; #P<0.05,
##P<0.01, the Ox-LDL + U0126 group vs. the Ox-LDL
group. CD, cluster of differentiation; Ox-LDL, oxidized-low-density
lipoprotein; RPMs, rat peritoneal macrophages; ERK, extracellular
signal-regulated kinases; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
Discussion
Atherosclerotic lesions are characterized by
accumulation of cholesterol in the arterial tunica intima.
Macrophages are central to the development and progression of AS
(13,14); they have been observed to remove
cholesterol deposits in arteries, which is beneficial in the
initial stages of AS; however, continued cholesterol accumulation
may result in the formation of foam cells, which are involved in AS
progression (15,16). The response of RPMs to modified LDL
is considered a good model for AS research (17,18).
The results of the present study indicated that RPMs engulfed a
large quantity of Ox-LDL, as demonstrated by DiI fluorescence and
oil red O staining. In addition, the present study demonstrated
that inhibition of ERK1/2 markedly reduced Ox-LDL deposition in the
RPMs. It has previously been reported that ERK1/2 is associated
with cell migration, survival, differentiation and proliferation
(4). However, the association
between ERK1/2 and lipid metabolism remains to be elucidated.
Cholesterol uptake is a pathway by which
extracellularly modified LDLs are ingested by macrophages via
scavenger receptors. As a major scavenger receptor of Ox-LDL
(19,20), CD36 binds to >50% of Ox-LDL in
macrophages (21), which is
crucial to the progression of AS. Absence of CD36 on the cell
surface of macrophages has been demonstrated to be protective
against AS (22). In addition,
treatment with a CD36 competitive peptide ligand (EP80317) that
blocks the Ox-LDL-binding site of CD36 resulted in a marked
reduction (up to 51%) of atherosclerotic lesions in apolipoprotein
E-deficient mice (23). It has
therefore been hypothesized that inhibition of CD36 expression in
macrophages has potential anti-atherosclerotic effects. It has been
reported that the CD36 signaling pathway may be initiated by
internalization of Ox-LDL (3,7).
Consistent with these previous findings, the present study
indicated that Ox-LDL resulted in a notable increase in CD36
expression. Increasing evidence suggests that Ox-LDL increases ERK
phosphorylation (3,20,24).
The results of the present study indicated that inhibition of
ERK1/2 markedly reduced Ox-LDL-induced CD36 expression in RPMs.
Furthermore, a p38 MAPK inhibitor, SB203580, has previously been
demonstrated to suppress CD36 expression in THP-1 cell lines
(24) and RAW264.7 cells (20). The present study demonstrated that
treatment with an ERK1/2 inhibitor, U0126, attenuated
Ox-LDL-induced CD36 expression at the transcriptional and
translational level. It is therefore assumed that ERK1/2 may be
important in mediating CD36 expression in macrophages.
Macrophages have been reported to maintain cellular
lipid homeostasis via cholesterol efflux pathways. The principle
molecules associated with cholesterol efflux in macrophages are
ABCA1 and ABCG1 (25,26). In THP-1 cells, Ox-LDL has been
shown to upregulate ABCA1 expression (27). The present study demonstrated that
the mRNA and protein expression levels of ABCA1 were significantly
increased following Ox-LDL treatment, and that inhibition of ERK1/2
increased ABCA1 mRNA and protein expression levels in macrophages.
It has been reported that ERK1/2 inhibitors synergize with LXR
activation, in order to induce ABCA1 expression in a RAW macrophage
cell line (5), and U0126, an
ERK1/2 inhibitor, has been demonstrated to delay the degradation of
ABCA1 mRNA and protein in macrophages (28). However, a previous study indicated
that inhibition of ERK1/2 was able to promote ABCA1 and ABCG1
protein degradation in tumor cells, whereas inhibition of ERK1/2
upregulated ABCA1 and ABCG1 expression in human THP-1 macrophages
(29). It is therefore
hypothesized that the effect of ERK1/2 inhibition on ABCA1 and
ABCG1 expression may depend on cell specificity. The results of the
present study demonstrated that inhibition of ERK1/2 increased
ABCA1 expression in Ox-LDL-induced macrophages at 12 and 24 h, and
gradually decreased ABCA1 expression at 48 h. The half-life of
ABCA1 protein is 1–2 h, and ABCA1 is frequently present during
homeostasis between expression and degradation. Inhibition of
ERK1/2 has been demonstrated to suppress the degradation of ABCA1
mRNA and protein (5) or increase
its stability (5). However,
macrophages secrete various inflammatory cytokines, which affect
ABCA1 expression (30,31). Further studies are required to
investigate the mechanisms underlying the effects of ERK1/2
inhibition on the regulation of cytokines that affect ABCA1
expression. The present study demonstrated that alterations to
ABCG1 mRNA and protein expression levels were similar to those
observed in ABCA1 expression at 12 and 24 h post-treatment with
Ox-LDL. It has been reported that macrophages may overexpress
unsaturated fatty acids and products of 12/15-lipoxygenase, which
increase the degradation of ABCG1 protein (32,33).
In addition, ABCG1 expression is affected by cell activity and
cytokines secreted from macrophages (34).
The present study indicated that inhibition of
ERK1/2 markedly suppressed lipid deposition in macrophages by
promoting ABCA1 and ABCG1 expression, which are associated with
cholesterol efflux. ABCA1 and ABCG1 expression may be induced by
ERK1/2 inhibition or LXRα (35),
thus indicating that an association may exist between LXR and ERK.
Since ABCA1 and ABCG1 are nuclear receptor LXR target genes
involved in cholesterol efflux, activation of LXR may increase
ABCA1 and ABCG1 expression in macrophages (36,37).
However, it has been reported that ERK1/2 inhibitors have no effect
on LXRα and LXRβ expression (5).
It is conceivable that ERK1/2 inhibition may directly increase
ABCA1 and ABCG1 expression by methods other than upregulating LXR
expression. ERK1/2 controls transcriptional and
post-transcriptional regulation of LXR and peroxisome
proliferator-activated receptor (PPAR)α/γ (29,38–40),
and mediates expression of their target genes. Further studies are
required to elucidate the association between ERK and
PPARs/LXRs-ABCA1/G1, and to understand the underlying
mechanisms.
In conclusion, inhibition of ERK1/2 increases
macrophage lipid efflux by upregulating ABCA1 and ABCG1 expression,
and suppresses lipid engulfment by downregulating CD36 expression
at the transcriptional and translational level. These findings
suggest that inhibition of ERK1/2 may exert potential
anti-atherosclerotic effects, and involvement of the ERK1/2 pathway
in lipid metabolism may provide additional knowledge for the
development of novel treatment strategies for AS.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81001543
and 81473744) and the Personal Supporting Projects of Health
Department in Fujian Province of China (grant no. 2013-ZQN-ZD-28).
The funders had no role in study design, data collection and
analysis, decision to publish or preparation of the manuscript.
Abbreviations:
|
AS
|
atherosclerosis
|
|
RPMs
|
rat peritoneal macrophages
|
|
Ox-LDL
|
oxidized-low-density liprotein
|
|
DiI-ox-LDL
|
DiI-labeled oxidized-low-density
lipoprotein
|
|
ABCA1
|
ATP-binding cassette transporter
A-1
|
|
ABCG1
|
ATP-binding cassette transporter
G-1
|
|
MAPK
|
mitogen-activated protein kinase
|
|
ERK1/2
|
extracellular signal-regulated kinases
1/2
|
|
p-ERK
|
phosphorylated ERK
|
|
PBS
|
phosphate-buffered saline
|
References
|
1
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Febbraio M and Silverstein RL: CD36:
Implications in cardiovascular disease. Int J Biochem Cell Biol.
39:2012–2030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rahaman SO, Lennon DJ, Febbraio M, Podrez
EA, Hazen SL and Silverstein RL: A CD36-dependent signaling cascade
is necessary for macrophage foam cell formation. Cell Metab.
4:211–221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roskoski RJ Jr: ERK1/2 MAP kinases:
Structure, function and regulation. Pharmacol Res. 66:105–143.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou X, Yin Z, Guo X, Hajjar DP and Han J:
Inhibition of ERK1/2 and activation of liver X receptor
synergistically induce macrophage ABCA1 expression and cholesterol
efflux. J Biol Chem. 285:6316–6326. 2010. View Article : Google Scholar :
|
|
6
|
Ye D, Lammers B, Zhao Y, Meurs I, Van
Berkel TJ and Van Eck M: ATP-binding cassette transporters A1 and
G1, HDL metabolism, cholesterol efflux, and inflammation: Important
targets for the treatment of atherosclerosis. Curr Drug Targets.
12:647–660. 2011. View Article : Google Scholar
|
|
7
|
Silverstein RL and Febbraio M: CD36 and
atherosclerosis. Curr Opin Lipidol. 11:483–491. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu HY, Cui HB, Chen XM, Chen XY, Wang SH,
Du WP, Zhou HL, Zhao RC, Zhou Y, Liu YH, et al: Imbalanced response
of ATP-binding cassette transporter A1 and CD36 expression to
increased oxidized low-density lipoprotein loading contributes to
the development of THP-1 derived foam cells. J Biochem. 155:35–42.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu W, Jiang J, Yan D, Li D, Li W, Ma Y,
Yang L, Qu Z and Ruan Q: Pentraxin 3 promotes oxLDL uptake and
inhibits cholesterol efflux from macrophage-derived foam cells. Exp
Mol Pathol. 96:292–299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jing Q, Xin SM, Zhang WB, Wang P, Qin YW
and Pei G: Lysophosphatidylcholine activates p38 and p42/44
mitogen-activated protein kinases in monocytic THP-1 cells, but
only p38 activation is involved in its stimulated chemotaxis. Circ
Res. 87:52–59. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng X, Liu X, Song L, He Y, Li X and
Zhang H: Atorvastatin inhibits macrophage-derived foam cell
formation by suppressing the activation of PPARγ and NF-κB pathway.
Nan Fang Yi Ke Da Xue Xue Bao. 34:896–900. 2014.In Chinese.
PubMed/NCBI
|
|
12
|
Bhandary B, Lee GH, So BO, Kim SY, Kim MG,
Kwon JW, Song JY, Lee HK, Kim HR, Chae SW and Chae HJ: Rubus
coreanus inhibits oxidized-LDL uptake by macrophages through
regulation of JNK activation. Am J Chin Med. 40:967–978. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Glass CK and Witztum JL: Atherosclerosis.
The road ahead Cell. 104:503–516. 2001.
|
|
14
|
Tiwari RL, Singh V and Barthwal MK:
Macrophages: An elusive yet emerging therapeutic target of
atherosclerosis. Med Res Rev. 28:483–544. 2008. View Article : Google Scholar
|
|
15
|
Ouimet M: Autophagy in obesity and
atherosclerosis: Interrelationships between cholesterol
homeostasis, lipoprotein metabolism and autophagy in macrophages
and other systems. Biochim Biophys Acta. 1831:1124–1133. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Webb NR and Moore KJ: Macrophage-derived
foam cells in atherosclerosis: Lessons from murine models and
implications for therapy. Curr Drug Targets. 8:1249–1263. 2007.
View Article : Google Scholar
|
|
17
|
Morio H, Saito H, Hirai A, Tamura Y and
Yoshida S: Effect of modified LDL on the release of NO and PGI2
from rat peritoneal macrophages. J Atheroscler Thromb. 2:41–45.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hirai A, Kino T, Tokinaga K, Tahara K,
Tamura Y and Yoshida S: Regulation of sterol carrier protein 2
(SCP2) gene expression in rat peritoneal macrophages during foam
cell formation. A key role for free cholesterol content. J Clin
Invest. 94:2215–2223. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nicholson AC and Hajjar DP: CD36, oxidized
LDL and PPAR gamma: Pathological interactions in macrophages and
atherosclerosis. Vascul Pharmacol. 41:139–146. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Min KJ, Um HJ, Cho KH and Kwon TK:
Curcumin inhibits oxLDL-induced CD36 expression and foam cell
formation through the inhibition of p38 MAPK phosphorylation. Food
Chem Toxicol. 58:77–85. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Febbraio M, Podrez EA, Smith JD, Hajjar
DP, Hazen SL, Hoff HF, Sharma K and Silverstein RL: Targeted
disruption of the class B scavenger receptor CD36 protects against
atherosclerotic lesion development in mice. J Clin Invest.
105:1049–1056. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Febbraio M, Guy E and Silverstein RL: Stem
cell transplantation reveals that absence of macrophage CD36 is
protective against atherosclerosis. Arterioscler Thromb Vasc Biol.
24:2333–2338. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marleau S, Harb D, Bujold K, Avallone R,
Iken K, Wang Y, Demers A, Sirois MG, Febbraio M, Silverstein RL, et
al: EP 80317, a ligand of the CD36 scavenger receptor, protects
apolipoprotein E-deficient mice from developing atherosclerotic
lesions. FASEB J. 19:1869–1871. 2005.PubMed/NCBI
|
|
24
|
Zhao M, Liu Y, Wang X, New L, Han J and
Brunk UT: Activation of the p38 MAP kinase pathway is required for
foam cell formation from macrophages exposed to oxidized LDL.
APMIS. 110:458–468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang N, Lan D, Chen W, Matsuura F and Tall
AR: ATP-binding cassette transporters G1 and G4 mediate cellular
cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci
USA. 101:9774–9779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang N, Silver DL, Costet P and Tall AR:
Specific binding of ApoA-I, enhanced cholesterol efflux, and
altered plasma membrane morphology in cells expressing ABC1. J Biol
Chem. 275:33053–33058. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang CK, Yi GH, Yang JH, Liu LS, Wang Z,
Ruan CG and Yang YZ: Oxidized LDL upregulated ATP binding cassette
transporter-1 in THP-1 macrophages. Acta Pharmacol Sin. 25:581–586.
2004.PubMed/NCBI
|
|
28
|
Chang YC, Sheu WH, Chien YS, Tseng PC, Lee
WJ and Chiang AN: Hyperglycemia accelerates ATP-binding cassette
transporter A1 degradation via an ERK-dependent pathway in
macrophages. J Cell Biochem. 114:1364–1373. 2013. View Article : Google Scholar
|
|
29
|
Mulay V, Wood P, Manetsch M, Darabi M,
Cairns R, Hoque M, Chan KC, Reverter M, Alvarez-Guaita A, Rye KA,
et al: Inhibition of mitogen-activated protein kinase Erk1/2
promotes protein degradation of ATP binding cassette transporters
A1 and G1 in CHO and HuH7 cells. PLoS One. 8:e626672013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hao XR, Cao DL, Hu YW, Li XX, Liu XH, Xiao
J, Liao DF, Xiang J and Tang CK: IFN-gamma down-regulates ABCA1
expression by inhibiting LXRalpha in a JAK/STAT signaling
pathway-dependent manner. Atherosclerosis. 203:417–428. 2009.
View Article : Google Scholar
|
|
31
|
Chen M, Li W, Wang N, Zhu Y and Wang X:
ROS and NF-kappaB but not LXR mediate IL-1beta signaling for the
downregulation of ATP-binding cassette transporter A1. Am J Physiol
Cell Physiol. 292:C1493–C1501. 2007. View Article : Google Scholar
|
|
32
|
Uehara Y, Miura S, von Eckardstein A, Abe
S, Fujii A, Matsuo Y, Rust S, Lorkowski S, Assmann G, Yamada T and
Saku K: Unsaturated fatty acids suppress the expression of the
ATP-binding cassette transporter G1 (ABCG1) and ABCA1 genes via an
LXR/RXR responsive element. Atherosclerosis. 191:11–21. 2007.
View Article : Google Scholar
|
|
33
|
Nagelin MH, Srinivasan S, Lee J, Nadler JL
and Hedrick CC: 12/15-Lipoxygenase activity increases the
degradation of macrophage ATP-binding cassette transporter G1.
Arterioscler Thromb Vasc Biol. 28:1811–1819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang C, Cui K, Diao Y, Du M and Wang S:
Effect of selenium-enriched garlic pil against cytotoxicity induced
by OX-LDL in endothelial cells. Evid Based Complement Alternat Med.
2014:5376522014. View Article : Google Scholar
|
|
35
|
Xue X, Chen T, Wei W, Zhou X, Lin Z and
Chen L: Effects of Alisma Decoction on lipid metabolism and
inflammatory response are mediated through the activation of the
LXRα pathway in macrophage-derived foam cells. Int J Mol Med.
33:971–977. 2014.PubMed/NCBI
|
|
36
|
Tang SL, Chen WJ, Yin K, Zhao GJ, Mo ZC,
Lv YC, Ouyang XP, Yu XH, Kuang HJ, Jiang ZS, et al: PAPP-A
negatively regulates ABCA1, ABCG1 and SR-B1 expression by
inhibiting LXRα through the IGF-I-mediated signaling pathway.
Atherosclerosis. 222:344–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma AZ, Song ZY and Zhang Q: Cholesterol
efflux is LXRα isoform-dependent in human macrophages. BMC
Cardiovasc Disord. 14:802014. View Article : Google Scholar
|
|
38
|
Bhatt KH, Sodhi A and Chakraborty R:
Peptidoglycan induced expression of peroxisome
proliferator-activated receptor γ in mouse peritoneal macrophages:
Role of ERK and JNK MAP kinases. Cytokine. 60:778–786. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stachowska E, Kijowski J, Dziedziejko V,
Siennicka A and Chlubek D: Conjugated linoleic acid regulates
phosphorylation of PPARγ by modulation of ERK 1/2 and p38 signaling
in human macrophages/fatty acid-laden macrophages. J Agric Food
Chem. 59:11846–11852. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mogilenko DA, Shavva VS, Dizhe EB, Orlov
SV and Perevozchikov AP: PPARγ activates ABCA1 gene transcription
but reduces the level of ABCA1 protein in HepG2 cells. Biochem
Biophys Res Commun. 402:477–482. 2010. View Article : Google Scholar : PubMed/NCBI
|