Introduction
Pulmonary hypertension (PH) is a serious disease
that may result in mobility problems and mortality (1–3). PH
is predominantly caused by hypoxic pulmonary vasoconstriction (HPV)
and pulmonary vascular remodeling as a result of exposure to
chronic hypoxia (CH) in patients with respiratory disease (4). Elevation of intracellular calcium
concentration ([Ca2+]i) is essential for the
initiation of contraction and proliferation of vascular smooth
muscle cells (SMCs) (5,6). It has previously been reported that
in pulmonary artery (PA)SMCs, hypoxia leads to increases in
intracellular Ca2+ release and extracellular
Ca2+ influx (7).
Calcium influx is predominantly regulated by voltage-dependent
Ca2+ channels (VDCCs), and voltage-independent
nonselective cation channels, including store-operated
Ca2+ channels (SOCCs) and receptor-operated
Ca2+ channels (ROCCs). VDCC blockers cannot decrease
[Ca2+] during CH, indicating that Ca2+ influx
occurs via signaling pathways other than VDCCs (8–10).
In addition to VDCCs, HPV requires an influx of Ca2+ via
voltage-independent nonselective cation channels (6,11–15).
These channels contribute to membrane depolarization during
hypoxia.
Transient receptor potential (TRP) proteins form
nonselective cation channels in vascular SMCs (5,16,17).
TRPC1 and TRPC6 have been detected in endothelium-denuded
intrapulmonary arteries (12,18,19),
PASMCs (6,12,18,20),
and pulmonary venous SMCs (PVSMCs) (15,21).
TRPC1 and TRPC6 form predominantly SOCCs and ROCCs, respectively
(22–27). During CH, the levels of TRPC1 and
TRPC6 are increased in PASMCs (12) and the [Ca2+]i
is increased, indicating that TRPC1 and TRPC6 are important in
CH-induced HPV.
Intrapulmonary arteries and veins contribute to the
increase in pulmonary vascular resistance during hypoxia (28–31).
Therefore, the mechanism underlying the development of hypoxic PH
from the intrapulmonary veins has attracted increasing attention in
recent years. TRPC6 expression is increased in rat intrapulmonary
veins, as well as in PVSMCs with increased basal
[Ca2+]i and store-operated Ca2+
entry (15). However, the role of
TRPC6 in hypoxia-induced cation entry, and its signaling pathway in
PVSMCs, remains to be elucidated.
A previous study demonstrated that TRPC6 is key in
mediating hypoxia-induced increases in
[Ca2+]i in human PASMCs and that this is
linked to cellular energy status via activation of adenosine
monophosphate-activated protein kinase (AMPK) (6). The aim of the present study was to
determine the effect of hypoxia on Ca2+ entry pathways,
particularly on TRPC6 as an ROCC, as well as the relevant
activation and regulation signaling pathways in pulmonary veins
(PVs) by measuring [Ca2+]i during CH. The
proliferation and migration of PVSMCs, which are essential for PV
remodeling, were also examined. Data from the present study
suggested a key role for Ca2+ entry via TRPC6 in the
mediation of [Ca2+]i elevation, and the
proliferation and migration of rat distal PVSMCs, all of which may
be linked to the activation of AMPK.
Materials and methods
Reagents
Sodium pentobarbital, Hanks' Balanced Salt Solution
(HBSS), Earl's Balanced Salt Solution (EBSS), bovine serum albumin
(BSA) and dithiothreitol were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Collagenase I and papain were purchased from
Worthington Biochemical Corporation (Lakewood, NJ, USA). Basal
Smooth Muscle Cell Growth Medium-2 was purchased from PromoCell
(Heidelberg, Germany).
Ethics statement
Animal experiments conformed to the Guide for the
Care and Use of Laboratory Animals published by the US National
Institutes of Health (32). The
present study was approved by the Ethics Review Board for Animal
Studies of Institute of Southeast University (Nanjing, China).
Exposure of rats to CH
CH-induced PH (CHPH) was produced using an
established method (33). Briefly,
Sprague Dawley (SD) rats (weight, 250–300 g; Animal Center of
Southeast University, Nanjing, China) were randomized into CH
(n=12) and normoxic control groups (n=12). All animals were fed a
standard diet of rat chow and were settled in the laboratory animal
room at 21–26°C, 40–70% humidity and 15–20 Lux lighting. Rats in
the CH group were exposed to hypoxia for 3 weeks in a chamber that
was continuously flushed with a mixture of room air and
N2 to maintain 10% O2. The development of the
CHPH model was evaluated by measuring the mean PA pressure (MPAP),
right ventricle systolic pressure (RVSP), hematocrit, and weight
ratio of the RV/[left ventricle (LV) + septum (S)].
Rat distal pulmonary vein dissection and
PVSMC isolation
Male SD rats (weight, 250–300 g) were used for PV
and PVSMC isolation. PVs were isolated from rats maintained in
hypoxic and normoxic conditions for western blotting. Thereafter,
PVSMCs were enzymatically isolated from rats maintained in normoxic
conditions and cultured as described previously (15,34).
Briefly, rats were anesthetized with sodium pentobarbital (55
mg/kg; i.p.) and sacrificed by cervical dislocation. Rat lungs were
removed to a bath containing oxygenated modified Krebs solution
(118 mM NaCl, 4.7 mM KCl, 0.57 mM MgSO4, 1.18 mM
KH2PO4, 25 mM NaHCO3, 10 mM
dextrose, and 1.25 mM CaCl2) for dissection. Distal
pulmonary veins (PVs) (300–350 µm) were isolated from the
surrounding tissue, and the endothelium was denuded by gently
rubbing the luminal surface. The veins were digested at 37°C for 20
min in Ca2+-reduced HBSS supplemented with collagenase
type I (1,750 U/ml), papain (9.5 U/ml), BSA (2 mg/ml) and
dithiothreitol (1 mM). The solution was centrifuged at 100 × g and
25°C for 5 min, and the cells were resuspended in Basal Smooth
Muscle Cell Growth Medium-2 containing 10% supplement mix
(Promocell GmbH, Heidelberg, Germany) in an incubator containing 5%
CO2 at 37°C for the normoxic group, and 5%
CO2, 4% O2 and 91% N2 for the
hypoxic group. Cells were arrested in Basal Smooth Muscle Growth
Medium-2 with 0.1% supplement mix 24 h prior to treatment.
Primers and small interfering RNA (siRNA)
design
TRPC1, TRPC6 and β-actin gene sequences were
retrieved from the miRBase database (http://www.mirbase.org/) and used as a reference for
designing the polymerase chain reaction (PCR) primers. The primer
sequences are presented in Table
I. The siRNA were designed with a complementary sequence to rat
TRPC1 and TRPC6 mRNA as described previously (35). The designed primers, siRNA and
non-target siRNA (siNT) were synthesized by Invitrogen (Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
 | Table IPrimer and siRNA sequences. |
Table I
Primer and siRNA sequences.
| Primer/siRNA | Sequences,
5′→3′ |
|---|
| TRPC1 | F:
ATCTTCATGTGCGGTCACAGT
R: TACATCTCAAGCCGCAAGCA |
| TRPC6 | F:
TGGCAAGTCCAGCATACCTG
R: CTCCGTGTTTCTGCAGAGGT |
| β-actin | F:
CCCATCTATGAGGGTTACGC
R: TTTAATGTCACGCACGATTTC |
| siTRPC1 |
GAAUUUAAGUCGUCUGAAA |
| siTRPC6 |
GUCCAAAGUCCCUGCUUUA |
RNA interference
Cells were passaged onto coverslips in 500 µl
Opti-MEM (Invitrogen; Thermo Fisher Scientific, Inc.) 1 day prior
to transfection, and were allowed to reach 40–50% confluence by the
time of transfection. siRNA targeting TRPC1 or TRPC6 were
transfected using Lipofectamine™ 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at a final concentration of 1,000 ng/ml, as
described previously and according to the manufacturer's protocols
(6). The knockdown effects were
examined at 48 h and the results were compared with the
control.
Reverse transcription-quantitative PCR
(RT-qPCR)
PAs and PVs were de-endothelialized, frozen in
liquid nitrogen, and mechanically homogenized. Total RNA was
isolated from the tissues and cells using RNAiso Blood (Takara Bio,
Inc., Otsu, Japan), according to the manufacturer's protocols.
RT-qPCR experiments followed standard protocols. RNA (1 µg)
was first reverse transcribed to cDNA with random primers using the
RT Master Mix kit (Takara Biotechnology, Dalian, China). RT-PCR was
performed by using the PrimeScript RT reagent kit (Takara
Biotechnology) and the T100 Thermal Cycler (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) according to the manufacturer's
instructions. The RT-PCR cycling conditions were as follows: 37°C
for 15 min, 85°C for 5 sec and 4°C for holding. Real-time PCR
experiments followed standard protocols using the SYBR Green I qPCR
kit (Takara Biotechnology). The PCR cycling conditions were as
follows: 95°C for 30 sec, followed by 40 cycles at 95°C for 5 sec
and 60°C for 31 sec. The relative mRNA quantities of target genes
were normalized to the values of β-actin. The results were
expressed as fold changes of threshold cycle value relative to the
controls using the 2−ΔΔCq method (36).
Western blotting
Following serum starvation (0.1% supplement mix) for
24 h, cells were cultured in normoxic (5% CO2) or
hypoxic (5% CO2, 4% O2 and 91% N2)
conditions. Total proteins were prepared at the indicated
time-points using the protein extraction kit (TDY Biotech Co.,
Ltd., Beijing, China), containing complete protease inhibitor
(Roche Diagnostics, Indianapolis, IN, USA). Western blotting was
conducted as described previously (6). Cell lysates with equal quantities of
protein (50 µg) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred
electronically to polyvinylidene difluoride membranes. The
polyvinylidene fluoride membranes (Millipore, Darmstadt, Germany)
were blocked with 5% non-fat milk powder in 1X TBS containing 0.1%
Tween 20 for 1 h at room temperature, and were then incubated with
anti-TRPC1 (1:500; cat. no. sc15055; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), anti-TRPC6 (1:500; cat. no. sc19196; Santa
Cruz Biotechnology, Inc.) and anti-β-actin (1:5,000 cat. no. A5441;
Sigma-Aldrich) at 4°C overnight. The membranes were subsequently
incubated with horseradish peroxidase (HRP)-conjugated anti rabbit
secondary antibodies (1:5,000; cat. no. s004; TDY Biotech Co.,
Ltd.), HRP-conjugated anti-goat secondary antibodies (1:5,000; cat.
no. s008; TDY Biotech Co., Ltd.), or HRP-conjugated anti-mouse
secondary antibodies (1:5,000; cat. no. s001; TDY Biotech Co.,
Ltd), for 1 h at room temperature. The blots were visualized using
a Chemiluminescent Substrate kit (Pierce; Thermo Fisher Scientific,
Inc.). The membranes were scanned and the sum optical density was
quantitatively analyzed by Quantity One software v4.62 (Bio-Rad
Laboratories, Inc.).
Immunofluorescence staining
After washing three times in phosphate-buffered
saline (PBS), cells were fixed with 4% paraformaldehyde at room
temperature for 20 min. Cells were washed in PBS and treated with
0.3% Triton X-100 for 5 min on ice. Then cells were blocked with 5%
FBS for 30 min at room temperature and incubated with anti-smooth
muscle α-actin (1:100, Sigma-Aldrich) at 4°C overnight. Cells were
washed three times in PBS and incubated with Alexa Fluor
488-conjugated second antibody (Cell Signaling Technology, Beverly,
MA, USA) for 1 h. Finally, cells were washed with PBS, stained with
DAPI (Sigma-Aldrich) and observed under microscope (Olympus IX71,
Tokyo, Japan). Calcium imaging. Cells were washed with EBSS, and a
stock concentration of fura-2 AM [0.01 g in 50 µl dimethyl
sulfoxide (DMSO); Molecular Probes; Thermo Fisher Scientific,
Inc.]was prepared. A mixture of 1 µl stock dye in 200
µl EBSS was applied to the cells and incubated for at least
30 min prior to observation. Prior to placing the coverslips into
the recording chamber, the cells were rinsed in normal Tyrode's
medium (containing 137 mM NaCl, 5.4 mM KCl, 1.2 mM
MgCl2, 1.8 mM CaCl2, 1.2 mM
NaH2PO4, 10 mM D-glucose, 20 mM HEPES and 10
mM taurine), in order to remove residual dye. Data acquisition was
performed using an IX71 microscope (Olympus Corporation, Tokyo,
Japan). Fluorescent changes in fura-2 were measured with double
wavelength excitation at 340 and 380 nm, and emission at a
wavelength of 510 nm. Absolute Ca2+ was calibrated using
Fura-2 Calcium Imaging Calibration kit (Invitrogen; Thermo Fisher
Scientific, Inc.). Changes in Ca2+ concentration in the
region of interest were calculated according to a ratio of 340/380.
Time lapse recording initially captured the images at 1 sec
intervals; however, in order to minimize cell image bleaching in
the long experimental protocol, 2 or 3 sec intervals were applied
in the experiments. The majority of the data presented in the
figures were acquired at 3 sec intervals. The following chemicals
were used for calcium imaging: 5 µM nifedipine, 5 µM
SKF96365, 100 µM OAG and 10 µM U73122
(Sigma-Aldrich), and compound C (40 µM, Calbiochem; Merck
KGaA, Darmstadt, Germany). The chemicals were dissolved in ethanol
or DMSO and made as required on the day of experiments. Ethanol and
DMSO were tested alone in controls at the same vehicle
concentration and had no effect. Hypoxia medium was applied using
gas bubbled media (O2 level, 20 mmHg) as described
previously (6). Briefly, Tyrode's
solution was bubbled into para-film-sealed bottles. Then the medium
was applied through a 0.8 mm ID tube driven by a peristaltic pump
(minipuls 3, Gilson, Inc., Middleton, WI, USA) in which fluid flow
to the recording chamber (RG 26; Warner Instruments, LLC., Hamden,
CT, USA) was controlled at a rate of 5 ml/min.
Proliferation assays
An MTT assay was used to determine the proliferation
rate. Briefly, cells were seeded at a density of 5,000 per well in
96-well cell culture plates and starved with 0.1% FBS in SMC Growth
Medium-2 for 24 h. Cells were incubated under normoxic or hypoxic
conditions. After 24 h, 20 µl of MTT (5 mg/ml;
Sigma-Aldrich) was added to each well for 3 h incubation.
Subsequently, cells were dissolved in 150 µl DMSO, and the
absorbance was measured by a Multiscan FC (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 490 nm.
Migration assay
Migration was determined using the Transwell assay
(6.5-mm polycarbonate membrane with 8.0-µm pores; Corning
Inc., Corning, NY, USA). Briefly, suspensions containing
5×104 cells and fresh serum free media, were seeded on
the upper chamber. The cells were allowed to migrate through the
membrane to the lower surface for 6 h. Cells on the upper surface
of the membrane were scraped off with cotton swabs three times.
Cells that had migrated to the lower surface were fixed in 4%
paraformaldehyde, stained with 0.1% crystal violet and counted.
Migrated cell numbers were calculated as the number of migrated
cells per five different random fields. Cells were dissolved in 30%
acetic acid and measured at 570 nm using a microplate reader
(Thermo Fisher Scientific, Inc.).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Unpaired t-tests were performed for pairwise comparisons
of means. One-way analysis of variance, followed by the Tukey
post-hoc test, was used for multiple comparisons. The data were
analyzed using Prism 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA), and P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of CH on PA and right-side heart
pressures
Rats were exposed to CH (10% O2) for 21
days. The MPAP increased from 11.10±1.14 mmHg in normoxic rats to
18.42±0.77 mmHg in hypoxic rats (Fig.
1A), and the RVSP increased from 18.23±1.57 mmHg in normoxic
rats to 29.53±3.57 mmHg in hypoxic rats (Fig. 1B). In addition, RV/(LV + S)
increased from 0.23±0.04 in normoxic rats to 0.37±0.04 in hypoxic
rats (Fig. 1C), and the hematocrit
increased from 42.55±1.06% in normoxic rats to 51.87±1.90% in
hypoxic rats (Fig. 1D).
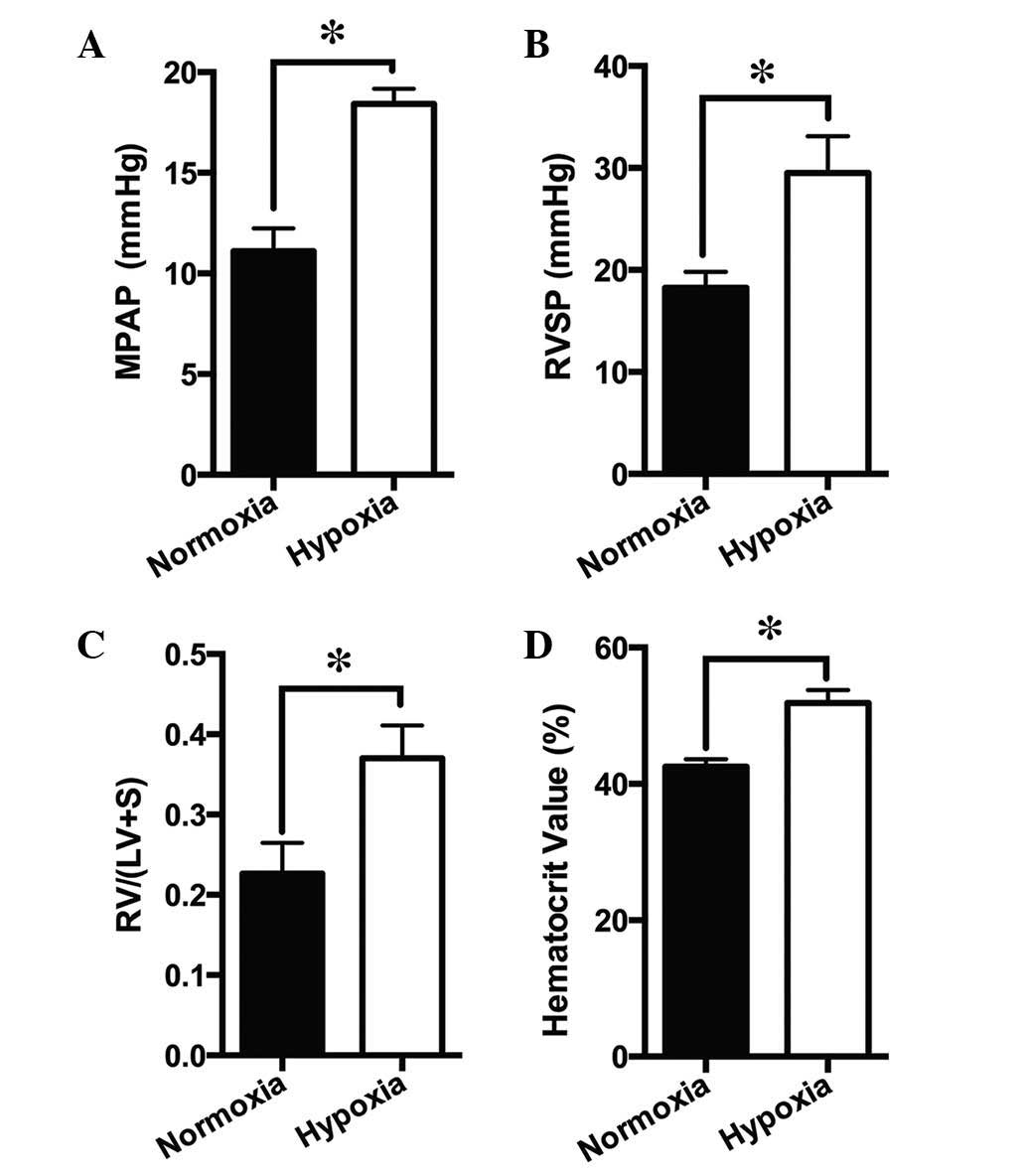 | Figure 1Effects of chronic hypoxia on PA and
right-side heart pressures. Animals were exposed to normoxia or
hypoxia (10% O2) for 21 days. The (A) MPAP, (B) RVSP,
(C) RV/(LV + S), and (D) hematocrit are presented as the mean ±
standard error of the mean (n=12, *P<0.05). PA,
pulmonary artery; MPAP, mean PA pressure; RV, right ventricle;
RVSP, right ventricle systolic pressure; LV, left ventricle; RV/(LV
+ S), RV/LV + septum mass ratio. |
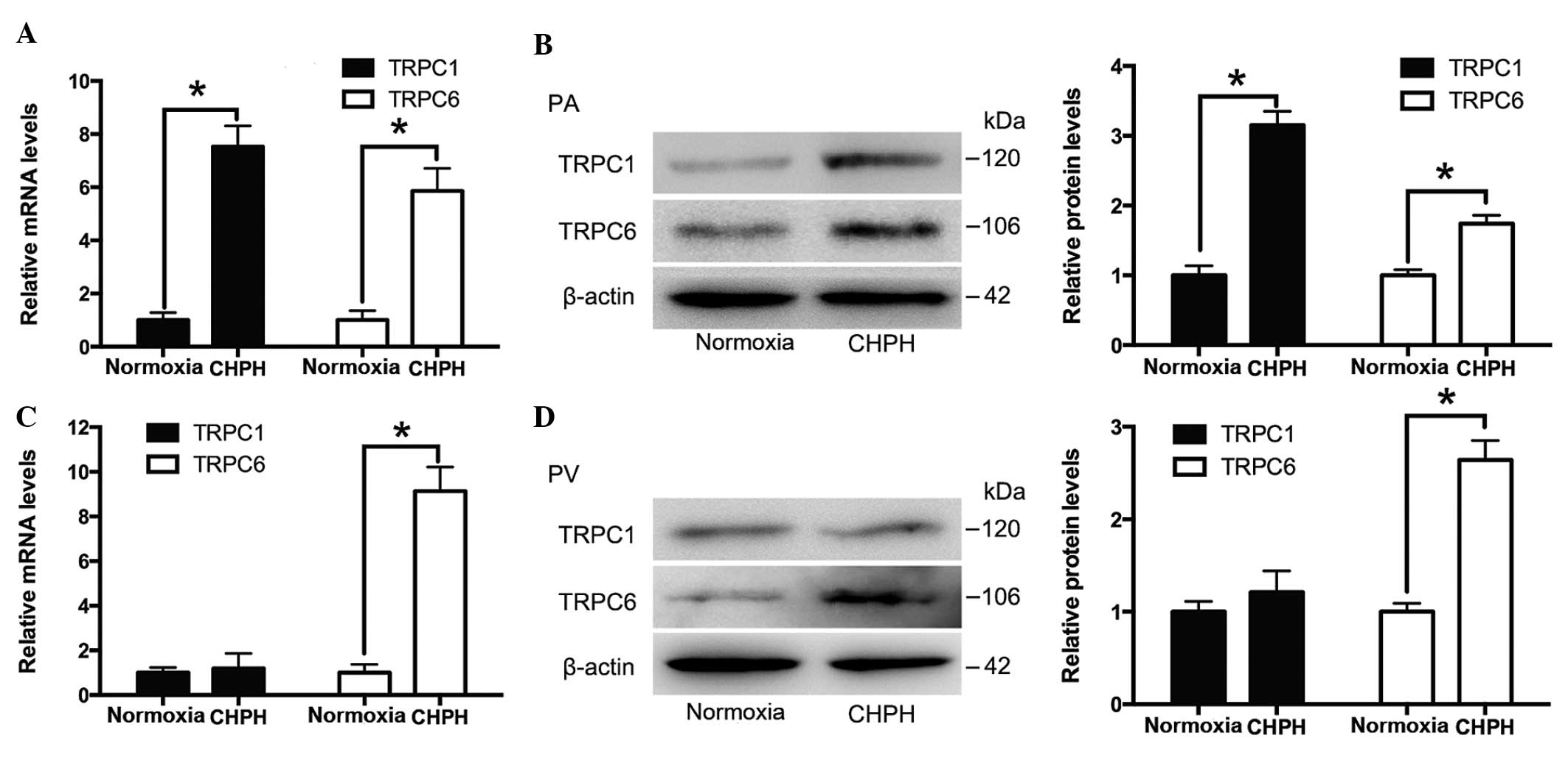
TRPC1 and TRPC6 expression in the distal
PA and PV tissue of normal and CHPH rats
Distal PAs and PVs were isolated from normal and
CHPH rats. The mRNA and protein expression levels of TRPC1 and
TRPC6 were detected by RT-qPCR and western blotting, respectively.
As presented in Fig. 2A and B, the
expression levels of TRPC1 and TRPC6 were increased in the distal
PA of the CHPH rats, whereas only TRPC6 expression levels were
increased in the distal PV of the CHPH rats, as compared with the
controls (Fig. 2C and D).
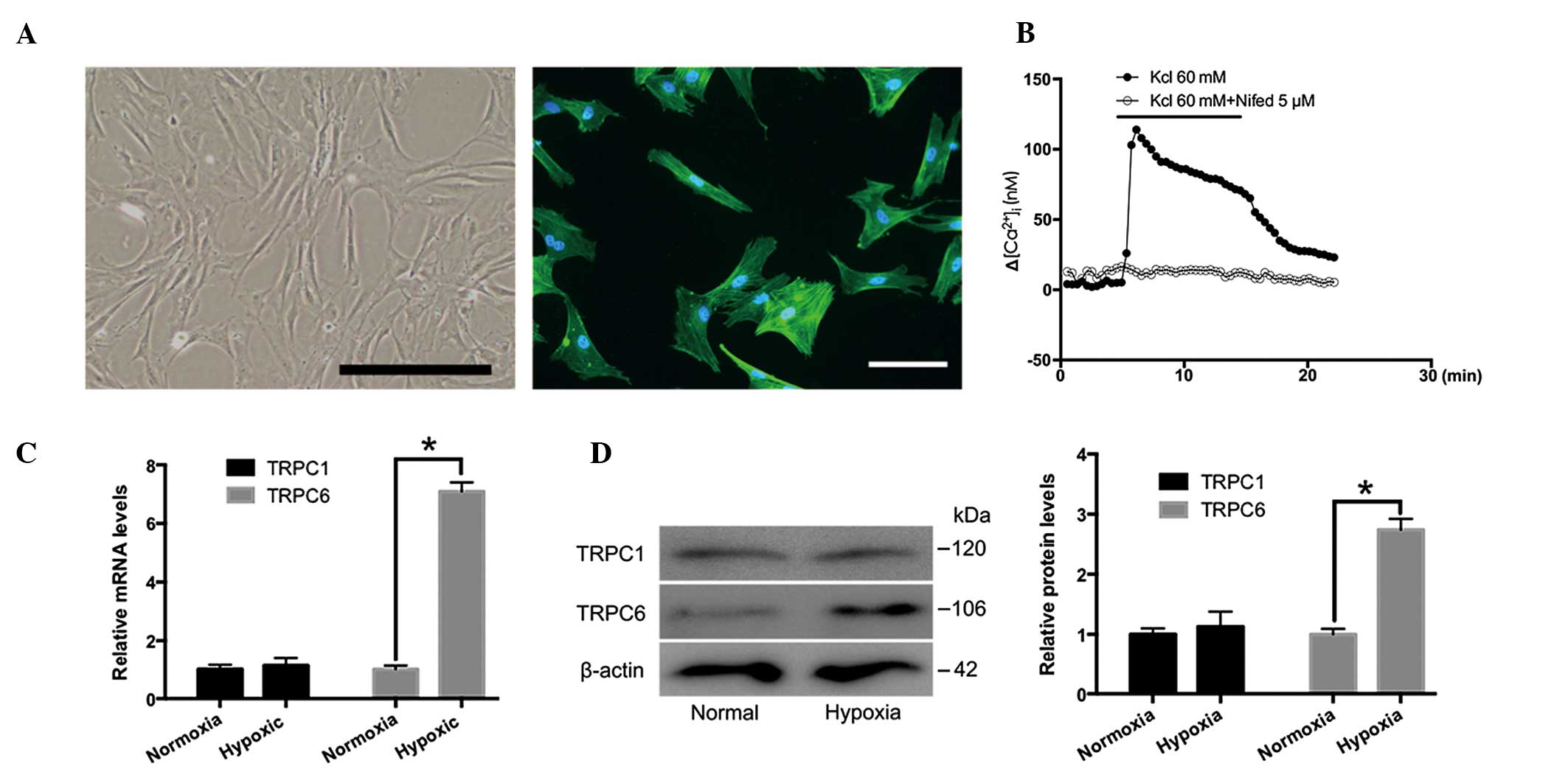
TRPC1 and TRPC6 expression in cultured
rat distal PVSMCs under normoxic and hypoxic conditions
Distal PVSMCs were isolated from normal rats and
analyzed. As presented in Fig. 3A,
PVSMCs were spindle-shaped and smooth muscle was α-actin-positive.
A high concentration K+ solution (60 µM) induced
[Ca2+]i elevation in PVSMCs, due to the opening of
L-type VGCCs (37). As shown in
Fig. 3B, nifedipine (5 µM)
inhibited KCl-induced [Ca2+]i elevation. Therefore,
nifedipine was used for further experiments. The expression levels
of TRPC1 and TRPC6 in rat distal PVSMCs cultured under normoxic and
hypoxic conditions (4% O2 for 72 h) were examined by
RT-qPCR and western blotting. The results demonstrated that TRPC6,
but not TRPC1, was increased in response to hypoxia, as compared
with the normoxic group (Fig. 3C and
D). These results indicate that TRPC6, but not TRPC1, may be
important in distal PVs in response to CH.
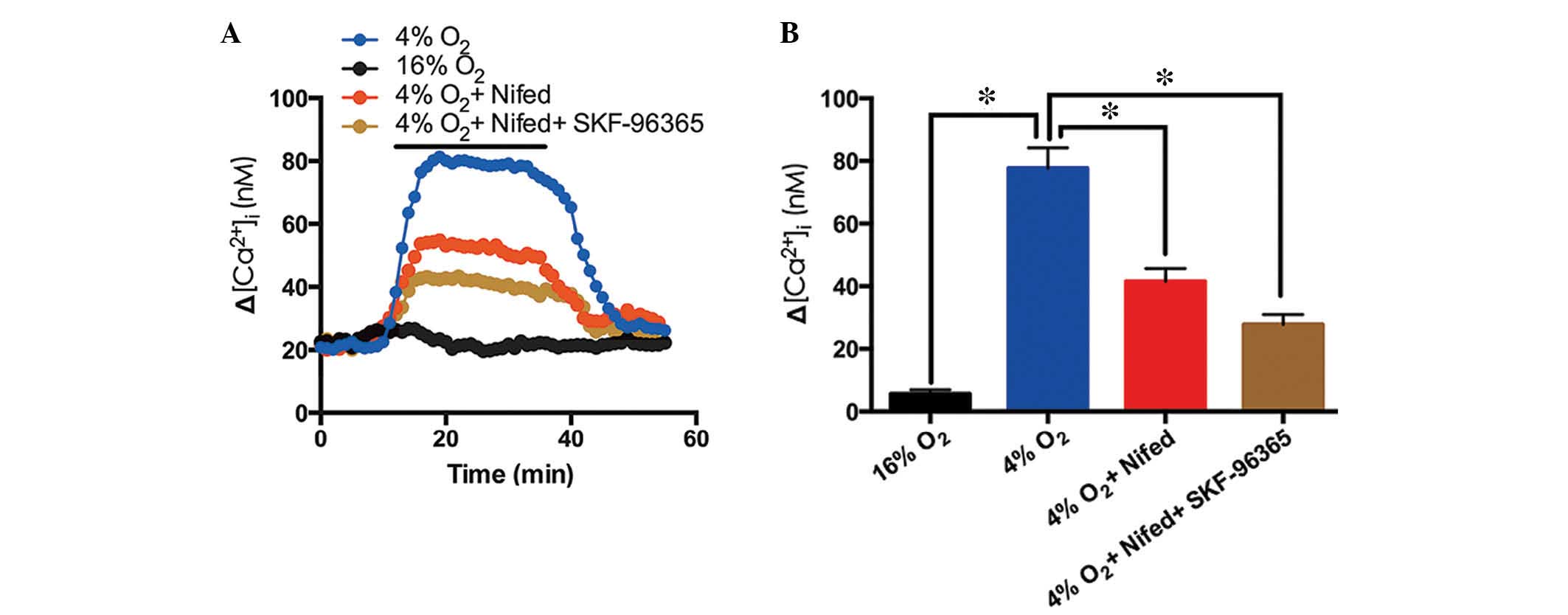
Role of voltage-gated calcium channels
(VGCCs) and TRPC6 blocker in hypoxia-induced Ca2+ entry
in PVSMCs
Following 5 min exposure to hypoxia (4%
O2) a significant Ca2+ elevation was induced
in cultured rat distal PVSMCs (Fig.
4A). Nifedipine (20 µM) inhibited the hypoxia-induced
[Ca2+]i elevation by 46.5%, whereas
nifedipine (20 µM) and SKF-96365 administered together
inhibited the hypoxia-induced [Ca2+]i
elevation by 64.2% (Fig. 4). These
results indicate that VGCCs and TRPC6 may contribute to
hypoxia-induced Ca2+ entry into PVSMCs.
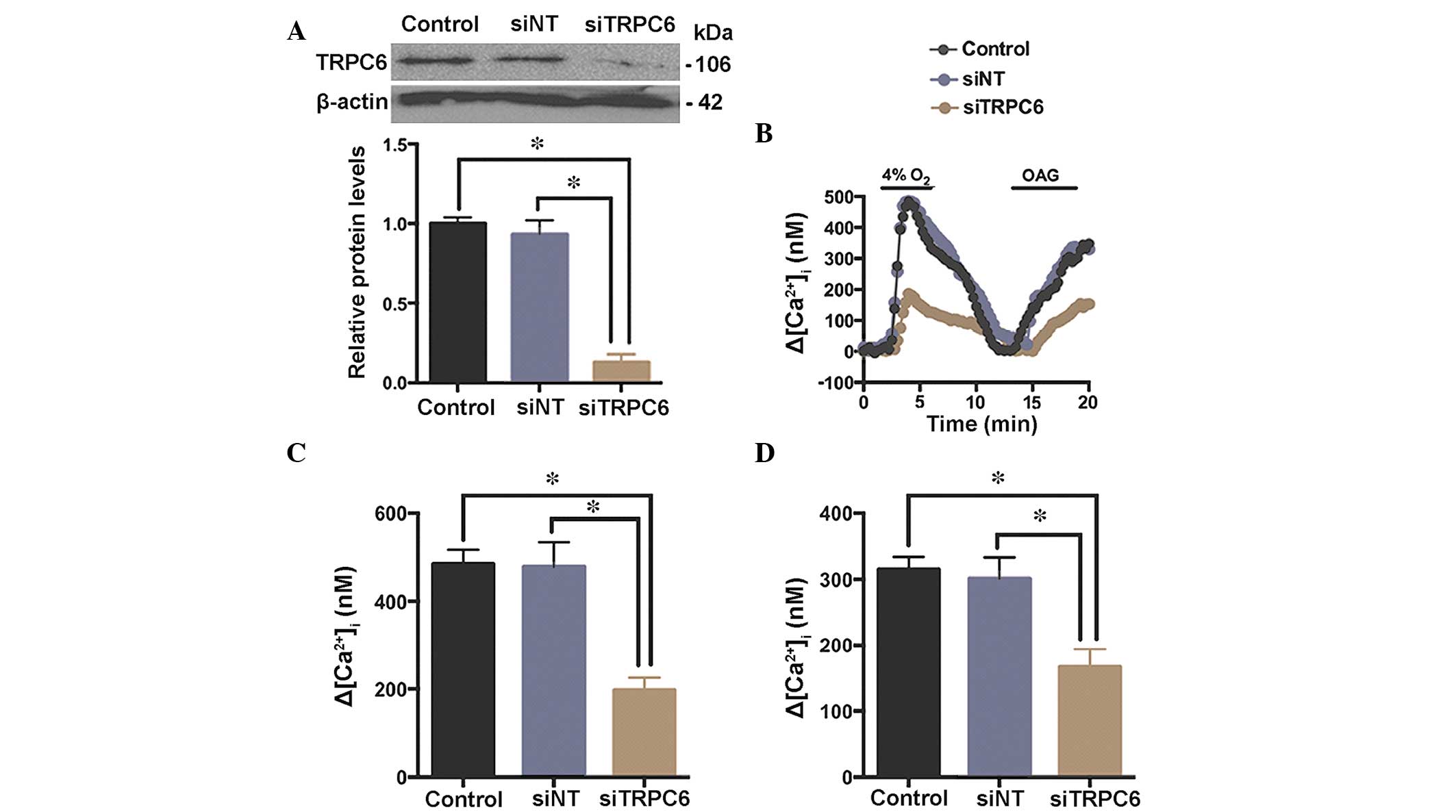
Effects of TRPC6 knockdown on
hypoxia-induced elevation of [Ca2+]i in rat distal
PVSMCs
A specific siRNA against TRPC6 (siTRPC6) was
synthesized, and a siNT served as a control. Rat distal PVSMCs were
isolated from normal rats and transfected with siRNA. TRPC6 protein
expression levels were determined by western blotting 48 h
post-transfection with siTRPC6 (Fig.
5A). As presented in Fig. 5B and
C, the hypoxia-induced [Ca2+]i elevation
(4% O2 for 5 min) was decreased by siTRPC6 (48 h),
indicating a role for TRPC6 in the hypoxia-induced elevation of
[Ca2+]i in PVSMCs. To determine the presence
of functional TRPC6 in rat distal PVSMCs,
1-oleoyl-2-acetyl-sn-glycerol (OAG), an analogue of diacylglycerol
(38), was used as a TRPC6 channel
activator (6). OAG (100 µM)
induced [Ca2+]i elevation, which was
significantly attenuated by siTRPC6 (Fig. 5D), implicating TRPC6 as a
functional ROCC in rat distal PVSMCs.
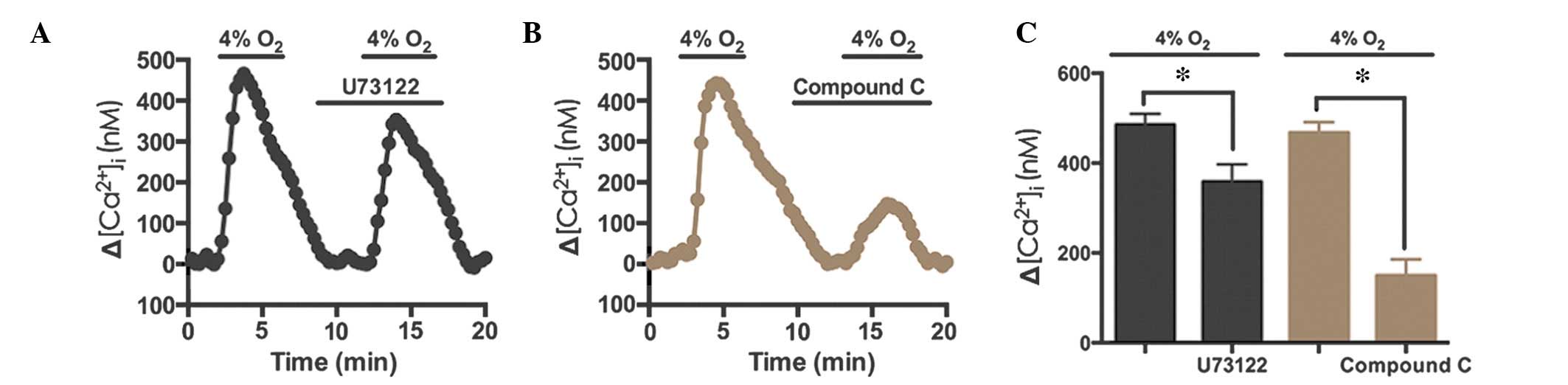
Hypoxia-induced Ca2+ elevation
occurs via phospholipase C (PLC) and AMPK
To determine the signaling pathway underlying TRPC6
activation, U73122 (10 µM), an antagonist of PLC, and
compound C (40 µM), an antagonist of AMPK, were used during
hypoxia-induced Ca2+ entry in rat distal PVSMCs. The
results demonstrated that U73122 inhibited the hypoxia-induced
Ca2+ response by 26.2% (n=72, N=3, P<0.05; Fig. 6), whereas compound C inhibited the
hypoxia-induced Ca2+ response by 68.1% (n=76, N=3,
P<0.05; Fig. 6B and C). These
results indicate that the AMPK signaling pathway may contribute to
hypoxia-induced Ca2+ elevation in PVSMCs.
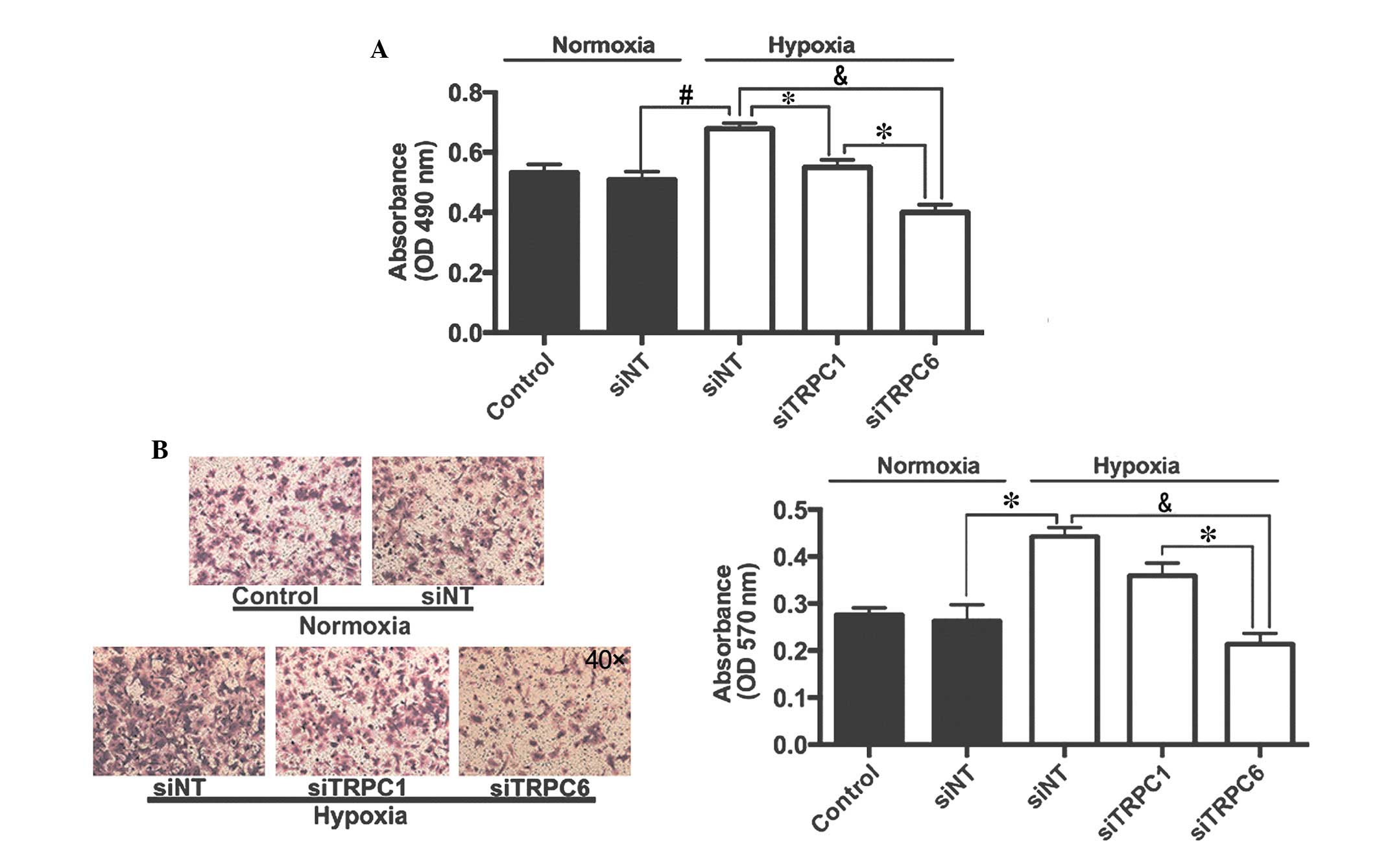
Effects of TRPC1 and TRPC6 on
hypoxia-induced proliferation and migration of rat distal
PVSMCs
Cells were exposed to normoxic or hypoxic (4%
O2, 72 h) conditions and were transfected with siNT or
siTRPC6. Proliferation and migration of PVSMCs was determined by
methyl thiazolyl tetrazolium (MTT) and Transwell assays,
respectively. As presented in Fig.
7A, hypoxia increased proliferation of PVSMCs from 0.51±0.03 in
the normoxic group to 0.68±0.02 in the hypoxic group (P<0.05).
Transfection with siTRPC1 only inhibited hypoxia-induced PVSMC
proliferation by 19.1%, whereas transfection with siTRPC6 inhibited
hypoxia-induced PVSMC proliferation by 41.2%, as compared with
siNT-transfected cells exposed to hypoxia. Similarly, hypoxia
increased migration of PVSMCs from 0.26±0.03 in the normoxic group
to 0.44±0.02 in the hypoxic group (P<0.05). Transfection with
siTRPC1 only inhibited hypoxia-induced PVSMC migration by 18.2%,
whereas transfection with siTRPC6 inhibited hypoxia-induced PVSMC
migration by 52.3%, as compared with siNT-transfected cells exposed
to hypoxia (Fig. 7B).
Discussion
In the present study, the expression and function of
TRPC1 and TRPC6 were examined in rat distal PVSMCs during CH in
vivo and in vitro. The major results of the present
study were as follows: i) TRPC6, not TRPC1, was functionally
upregulated in rat PVs and PVSMCs in response to CH; ii)
upregulated TRPC6 was accompanied by OAG-induced Ca2+
entry; iii) SKF96365 and siTRPC6 attenuated hypoxia-induced cation
entry; iv) hypoxia increased the proliferation and migration of
PVSMCs, and this effect was attenuated by siTRPC6; and v) AMPK was
suggested as the underlying pathway that links cellular energy
status and TRPC6 activation.
Hypoxia induces inflammation as well as
[Ca2+]i elevation, initiating PA contraction
and remodeling (39,40). However, PVs are affected prior to
PAs when oxygen partial pressure in the pulmonary circulation
decreases. PV contraction reportedly contributes to ~50% of the
total pulmonary resistance (41–43);
therefore, it is necessary to determine the effects of hypoxia on
PVs and to investigate the underlying molecular mechanisms. An
elevated [Ca2+]i is achieved predominantly by
cation entry via VDCCs, ROCCs, SOCCs, or Ca2+ release
from the sarcoplasmic reticulum. Previous studies have demonstrated
that hypoxia induces enhanced store-operated Ca2+ entry
and increases TRPC6 expression in rat PVSMCs (15,21).
TRPC6 protein forms voltage-independent
Ca2+-permeable cation channels, and is hypothesized to
be a major component of ROCCs (27,44).
In a previous study, the majority of the hypoxia-induced
[Ca2+]i elevation in human PASMCs was due to
cation entry via VDCCs and ROCCs (6), indicating that TRPC6 contributes to
membrane depolarization and [Ca2+]i
elevation. In addition, another previous study (15) detected TRPC1 and TRPC6 expression
in rat PVs and distal PVSMCs. In the present study, expression of
TRPC6, but not TRPC1, was increased in the PVs of CHPH rats, as
well as PVSMCs exposed to hypoxia, implicating an important role
for TRPC6 in hypoxia-induced cation entry.
Consistent with the results of previous studies
regarding PVs (15) and PAs
(6), the present study
demonstrated that blockade of VGCCs with nifedipine in PVSMCs
prevented hypoxia-induced [Ca2+]i elevation
by 46.5%, whereas coad-ministration of nifedipine and SKF96365 (a
TRPC blocker) prevented the hypoxia-induced
[Ca2+]i elevation by 64.2%. To overcome the
nonselectivity of the TRPC blocker, experiments using siTRPC6 to
knockdown TRPC6 expression in VSMCs were conducted. The expression
levels of TRPC6 and TRPC1 were examined by western blotting, in
order to investigate the specific effect of TRPC6 gene knockdown.
OAG, which is an analogue of diacylglycerol (38) and a TRPC6 activator, was used to
analyze the functional knockdown of TRPC6. The results demonstrated
that siTRPC6 significantly attenuated hypoxia-induced
[Ca2+]i elevation. These results suggested
that TRPC6 may have a key role in the response of PVSMCs to
hypoxia.
SMC proliferation and migration contribute to
pulmonary vessel remodeling, in which [Ca2+]i
is pivotal (5,6). In the present study, hypoxia
increased the proliferation and migration of rat distal PVSMCs, and
this effect was reduced by the knockdown of TRPC6. These results
further indicated that TRPC6 may participate in PV remodeling, and
may be considered a target for the treatment of PH.
AMPK is important in cellular energy homeostasis. In
a previous study, an AMPK antagonist attenuated hypoxia-induced
[Ca2+]i elevations (45). Furthermore, it has previously been
demonstrated that TRPC6 is activated via the AMPK signaling pathway
and not via the PLC signaling pathway in human PASMCs (46). Consistent with human PASMCs,
treatment with an AMPK antagonist in the present study attenuated
hypoxia-induced [Ca2+]i elevation in rat
PVSMCs, thus suggesting that AMPK may be a signal for TRPC6 in rat
PVSMCs.
In conclusion, the present study demonstrated a
functional role for hypoxia-induced TRPC6 upregulation in mediating
[Ca2+]i elevation, and the proliferation and
migration of rat distal PVSMCs. In addition, it was suggested that
TRPC6 may be activated via the AMPK signaling pathway.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81170105
and 81370225).
References
|
1
|
Hyduk A, Croft JB, Ayala C, Zheng K, Zheng
ZJ and Mensah GA: Pulmonary hypertension surveillance - United
States, 1980–2002. MMWR Surveill Summ. 54:1–28. 2005.PubMed/NCBI
|
|
2
|
George MG, Schieb LJ, Ayala C, Talwalkar A
and Levant S: Pulmonary hypertension surveillance: United States,
2001 to 2010. Chest. 146:476–495. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mehari A, Valle O and Gillum RF: Trends in
pulmonary hypertension mortality and morbidity. Pulm Med.
2014:1058642014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Naeije R: Pulmonary hypertension and right
heart failure in chronic obstructive pulmonary disease. Proc Am
Thorac Soc. 2:20–22. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sylvester JT, Shimoda LA, Aaronson PI and
Ward JP: Hypoxic pulmonary vasoconstriction. Physiol Rev.
92:367–520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tang C, To WK, Meng F, Wang Y and Gu Y: A
role for receptor-operated Ca2+ entry in human pulmonary
artery smooth muscle cells in response to hypoxia. Physiol Res.
59:909–918. 2010.
|
|
7
|
Salvaterra CG and Goldman WF: Acute
hypoxia increases cytosolic calcium in cultured pulmonary arterial
myocytes. Am J Physiol. 264:L323–L328. 1993.PubMed/NCBI
|
|
8
|
Shimoda LA, Sham JS, Shimoda TH and
Sylvester JT: L-type Ca(2+) channels, resting
[Ca(2+)](i), and ET-1-induced responses in chronically
hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol.
279:L884–L894. 2000.PubMed/NCBI
|
|
9
|
Berridge MJ: Capacitative calcium entry.
Biochem J. 312:1–11. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lewis RS: The molecular choreography of a
store-operated calcium channel. Nature. 446:284–287. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin MJ, Leung GP, Zhang WM, Yang XR, Yip
KP, Tse CM and Sham JS: Chronic hypoxia-induced upregulation of
store-operated and receptor-operated Ca2+ channels in
pulmonary arterial smooth muscle cells: A novel mechanism of
hypoxic pulmonary hypertension. Circ Res. 95:496–505. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang J, Weigand L, Lu W, Sylvester JT,
Semenza GL and Shimoda LA: Hypoxia inducible factor 1 mediates
hypoxia-induced TRPC expression and elevated intracellular
Ca2+ in pulmonary arterial smooth muscle cells. Circ
Res. 98:1528–1537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weissmann N, Dietrich A, Fuchs B, Kalwa H,
Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos y Schnitzler M,
Ghofrani HA, et al: Classical transient receptor potential channel
6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and
alveolar gas exchange. Proc Natl Acad Sci USA. 103:19093–19098.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoo HY, Park SJ, Seo EY, Park KS, Han JA,
Kim KS, Shin DH, Earm YE, Zhang YH and Kim SJ: Role of thromboxane
A2-activated nonselective cation channels in hypoxic
pulmonary vasoconstriction of rat. Am J Physiol Cell Physiol.
302:C307–C317. 2012. View Article : Google Scholar
|
|
15
|
Xu L, Chen Y, Yang K, Wang Y, Tian L,
Zhang J, Wang EW, Sun D, Lu W and Wang J: Chronic hypoxia increases
TRPC6 expression and basal intracellular Ca2+
concentration in rat distal pulmonary venous smooth muscle. PloS
One. 9:e1120072014. View Article : Google Scholar
|
|
16
|
Inoue R, Jensen LJ, Shi J, Morita H,
Nishida M, Honda A and Ito Y: Transient receptor potential channels
in cardiovascular function and disease. Circ Res. 99:119–131. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Groschner K, Rosker C and Lukas M: Role of
TRP channels in oxidative stress. Novartis Found Symp. 258:222–230;
discussion 231–225, 263–266. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu W, Wang J, Shimoda LA and Sylvester JT:
Differences in STIM1 and TRPC expression in proximal and distal
pulmonary arterial smooth muscle are associated with differences in
Ca2+ responses to hypoxia. Am J Physiol Lung Cell Mol
Physiol. 295:L104–L113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Shimoda LA and Sylvester JT:
Capacitative calcium entry and TRPC channel proteins are expressed
in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung
Cell Mol Physiol. 286:L848–L858. 2004. View Article : Google Scholar
|
|
20
|
Resnik ER, Keck M, Sukovich DJ, Herron JM
and Cornfield DN: Chronic intrauterine pulmonary hypertension
increases capacitative calcium entry in fetal pulmonary artery
smooth muscle cells. Am J Physiol Lung Cell Mol Physiol.
292:L953–L959. 2007. View Article : Google Scholar
|
|
21
|
Peng G, Ran P, Lu W, Zhong N and Wang J:
Acute hypoxia activates store-operated Ca(2+) entry and
increases intracellular Ca(2+) concentration in rat
distal pulmonary venous smooth muscle cells. J Thorac Dis.
5:605–612. 2013.PubMed/NCBI
|
|
22
|
Dietrich A, Kalwa H, Fuchs B, Grimminger
F, Weissmann N and Gudermann T: In vivo TRPC functions in the
cardiopulmonary vasculature. Cell Calcium. 42:233–244. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Inoue R, Okada T, Onoue H, Hara Y, Shimizu
S, Naitoh S, Ito Y and Mori Y: The transient receptor potential
protein homologue TRP6 is the essential component of vascular
alpha(1)-adrenoceptor-activated Ca(2+)-permeable cation
channel. Circ Res. 88:325–332. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parekh AB and Putney JW Jr: Store-operated
calcium channels. Physiol Rev. 85:757–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Villereal ML: Mechanism and functional
significance of TRPC channel multimerization. Semin Cell Dev Biol.
17:618–629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boulay G: Ca(2+)-calmodulin
regulates receptor-operated Ca(2+) entry activity of
TRPC6 in HEK-293 cells. Cell Calcium. 32:201–207. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang J, Yang K, Xu L, Zhang Y, Lai N,
Jiang H, Zhang Y, Zhong N, Ran P and Lu W: Sildenafil inhibits
hypoxia-induced transient receptor potential canonical protein
expression in pulmonary arterial smooth muscle via cGMP-PKG-PPARγ
axis. Am J Respir Cell Mol Biol. 49:231–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hasebe N, Onodera S, Yamashita H, Kawamura
Y, Haneda T and Tobise K: Site of hypoxic pulmonary
vasoconstriction in pulsatile perfused canine lung lobes. Jpn Circ
J. 56:837–846. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao Y and Raj JU: Role of veins in
regulation of pulmonary circulation. Am J Physiol Lung Cell Mol
Physiol. 288:L213–L226. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Raj JU, Hillyard R, Kaapa P, Gropper M and
Anderson J: Pulmonary arterial and venous constriction during
hypoxia in 3- to 5-wk-old and adult ferrets. J Appl Physiol (1985).
69:2183–2189. 1990.
|
|
31
|
Zhao Y, Packer CS and Rhoades RA:
Pulmonary vein contracts in response to hypoxia. Am J Physiol.
265:L87–L92. 1993.PubMed/NCBI
|
|
32
|
National Research Council (US) Institute
for Laboratory Animal Research: Guide for the Care and Use of
Laboratory Animals. Washington (DC): 1996
|
|
33
|
Yan J, Chen R, Liu P and Gu Y:
Docosahexaenoic acid inhibits development of hypoxic pulmonary
hypertension: in vitro and in vivo studies. Int J Cardiol.
168:4111–4116. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peng G, Wang J, Lu W and Ran P: Isolation
and primary culture of rat distal pulmonary venous smooth muscle
cells. Hypertens Res. 33:308–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ohba T, Watanabe H, Murakami M, Takahashi
Y, Iino K, Kuromitsu S, Mori Y, Ono J, Iijima T and Ito H:
Upregulation of TRPC1 in the development of cardiac hypertrophy. J
Mol Cell Cardiol. 42:498–507. 2007. View Article : Google Scholar
|
|
36
|
Thebault S, Zholos A, Enfissi A, Slomianny
C, Dewailly E, Roudbaraki M, Parys J and Prevarskaya N:
Receptor-operated Ca2+ entry mediated by TRPC3/TRPC6
proteins in rat prostate smooth muscle (PS1) cell line. J Cell
Physiol. 204:320–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Green KN, Boyle JP and Peers C: Hypoxia
potentiates exocytosis and Ca2+ channels in PC12 cells
via increased amyloid beta peptide formation and reactive oxygen
species generation. J Physiol. 541:1013–1023. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
39
|
Stenmark KR, Fagan KA and Frid MG:
Hypoxia-induced pulmonary vascular remodeling: Cellular and
molecular mechanisms. Circ Res. 99:675–691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Biddlestone J, Bandarra D and Rocha S: The
role of hypoxia in inflammatory disease (Review). Int J Mol Med.
35:859–869. 2015.PubMed/NCBI
|
|
41
|
Bonnet S and Archer SL: Potassium channel
diversity in the pulmonary arteries and pulmonary veins:
Implications for regulation of the pulmonary vasculature in health
and during pulmonary hypertension. Pharmacol Ther. 115:56–69. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schindler MB, Hislop AA and Haworth SG:
Postnatal changes in pulmonary vein responses to endothelin-1 in
the normal and chronically hypoxic lung. Am J Physiol Lung Cell Mol
Physiol. 292:L1273–1279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang TM, Luk HN, Sheu JR, Wu HP and Chiang
CE: Inducibility of abnormal automaticity and triggered activity in
myocardial sleeves of canine pulmonary veins. Int J Cardiol.
104:59–66. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Beech DJ: Emerging functions of 10 types
of TRP cationic channel in vascular smooth muscle. Clinical Exp
Pharmacol Physiol. 32:597–603. 2005. View Article : Google Scholar
|
|
45
|
Wyatt CN, Mustard KJ, Pearson SA, Dallas
ML, Atkinson L, Kumar P, Peers C, Hardie DG and Evans AM:
AMP-activated protein kinase mediates carotid body excitation by
hypoxia. J Biol Chem. 282:8092–8098. 2007. View Article : Google Scholar :
|
|
46
|
Tang C, To WK, Meng F, Wang Y and Gu Y: A
role for receptor operated Ca2+ entry in human pulmonary artery
smooth muscle cells in response to hypoxia. Physiol Res.
59:909–918. 2010.
|