Introduction
Although the incidence of gastric cancer has
decreased in the past 50 years in most developed countries, it
remains one of the most important health issues in developing
countries (1). According to the
International Gastric Cancer Society, gastric cancer affects
>800,000 people and accounts for 65,000 cancer-associated
mortalities annually (2),
therefore ranking as the fourth most common cancer type and the
second leading cause of cancer-associated mortality (3,4).
Accumulating evidence has shown that aberrant cellular metabolism,
involving multiple factors and steps, is a pivotal feature during
tumorigenesis and cancer progression (5). However, the precise regulatory
mechanisms underlying the development and progression of gastric
cancer remain to be elucidated.
Non-muscle myosin IIA (NMIIA) belongs to the myosin
II sub-family and is encoded by the MYH9 gene. It is an actin-based
molecular motor that includes skeletal, cardiac, smooth muscle and
non-muscular myosins (6,7). Although the functions of NMIIA may be
different in various cell types, interactions with actin
microfilaments, microtubules, S100A4 as well as cadherin- and
integrin complexes, have been identified, which may affect cellular
activities, including tumor invasion (8).
Previous studies have indicated that NMIIA has a
vital role in adhesion, invasion and migration of cancer cells,
including breast cancer (8,9),
esophageal squamous cancer (7),
anaplastic large cell lymphoma (10) and gastric cancer (6,11),
as well as patient prognosis. The association between the
overexpression of NMIIA and the progression as well as poor
prognosis of gastric cancer has been clarified (4,6);
however, the signaling pathways of the involvement of NMIIA in
gastric cancer have remained elusive.
The present study determined the levels of NMIIA
expression in clinical gastric cancer tissues and matched
non-tumorous gastric tissue specimens. Through in vitro
Transwell and wound-healing assays, the present study assessed the
invasive and migratory capacity of gastric cancer cells following
MYH9 gene silencing by RNA interference (RNAi). In addition, the
underlying molecular mechanisms of the roles of NMIIA in the
invasion and migration of gastric cancer cells were explored. The
present study provided insight into the role of NMIIA in gastric
cancer progression.
Materials and methods
Patients and tissue specimens
Frozen clinical gastric cancer tissue specimens and
matched non-tumorous gastric tissue specimens were collected from
63 gastric cancer patients from at the Department of
Gastrointestinal Surgery (Zhengzhou, China). None of the patients
received radiotherapy, chemotherapy or biotherapy prior to surgery.
The present study was approved by the Ethics Committee of the First
Affiliated Hospital of Zhengzhou University (Zhengzhou, China).
Written informed consent was provided by the patients prior to
commencement.
Cell lines and culture
The SGC-7901 and MGC-803 human gastric cancer lines
were purchased from the Shanghai Institute of Cell Biology
(Shanghai, China). The two cell lines were cultivated in RPMI 1640
(cat no. 21870-076; Gibco-BRL, Invitrogen Life Technologies, Inc.,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(HyClone, Logan, UT, USA), 4 mM glutamine (cat no. 25030-149;
Invitrogen Life Technologies), 100 U/ml penicillin (Sigma-Aldrich,
St. Louis, MO, USA) and 100 µg/ml streptomycin
(Sigma-Aldrich) in an incubator with a humidified atmosphere
containing 5% CO2 at 37°C (12).
Silencing of NMIIA
NMIIA isoform-specific small interfering (si)RNA and
control siRNA were smart pools from Qiagen (Hilden, Germany). siRNA
transfection was performed using HiPerfect transfection reagent
(Qiagen) according to the manufacturer's instructions. Cells were
plated in 12-well plates 72 h after transfection with 2 µM
siRNA. Following incubation for 24 h, cells were harvested and
subjected to subsequent experiments.
RNA extraction and reverse-transcription
polymerase chain reaction (PCR) analysis
Total RNA was extracted from minced tissues using
TRIzol reagent (Invitrogen Life Technologies, Inc.), followed by
cDNA synthesis using the TaqMan reverse transcription kit (cat. no.
4304134; Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The primers used for the amplification of the
cDNAs were as follows: NMIIA forward, 5′-AGA GCT CAC GTG CCT
CAACG-3′ and reverse, 5′-TGA CCA CAC AGA ACA GGC CTG-3′; β-actin
forward, 5′-ATT GCC GAC AGG ATG CAGA-3′ and reverse, 5′-GAG TAC TTG
CGC TCA GGA GGA-3′ (Sangon Biotech, Shanghai, China). The PCR
mixture (10 µl) was comprised of 5 µl TaqMan Fast
Advanced Master Mix (Thermo Fisher Scientific, Inc.), 1 µl
cDNA (1:50 dilution) and 2 µl of each forward and reverse
primer (1 mM). PCR was performed using the following cycling
parameters: Denaturation at 95°C for 5 min in the first cycle and
for 30 sec in the second cycle, annealing at 57°C for 30 sec and
elongation at 72°C for 30 sec, with a final extension at 72°C for 5
min. The β-actin gene was used as an internal control. The PCR
products were separated using agarose gel (Beyotime Institute of
Biotechnology, Haimen, China) electrophoresis. Data were analyzed
by 2−[∆Ct sample − ∆Ct control].
Western blot analysis
The expression of NMIIA in the frozen clinical
tissue specimens and cultured cells at 24 h after siRNA
transfection was examined using western blot analysis. The tissue
specimens and cultured cells were homogenized and lysed with
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology), containing 100 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1%
Triton X-100, 1mM EDTA, 10mM glycerophosphate, 2 mM sodium vandate
and protease inhibitor. Protein concentration was determined using
a Micro-BCA protein kit (Pierce Biotechnology, Inc., Rockford, IL,
USA), The protein (40 µg/lane) was then resolved by 12%
SDS-PAGE (Beyotime Institute of Biotechnology). Following
electrophoresis, the blots were transferred onto a polyvinylidene
fluoride membrane (EMD Millipore, Billerica, MA, USA). The
membranes were incubated with rabbit polyclonal NMIIA antibody
(1:500; cat. no. ab24762; Abcam, Cambridge, UK) at 4°C overnight or
anti-β-actin mouse monoclonal antibody (1:1,000; cat. no. A1978;
clone AC-15; Sigma-Aldrich) at 37°C for 2 h. After washing with
Tris-buffered saline containing Tween 20, the blots were visualized
using an enhanced chemiluminescence kit (cat. no. sc-2048; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA). In addition, the levels
of JNK and c-Jun in SGC-7901 cells were detected by western blot
analysis. In this assay, antibodies against phosphorylated (p)-JNK
(rabbit monoclonal; cat. no. 4668; clone 81E11; 1:1,500; Cell
Signaling Technology, Inc., Danvers, MA, USA) and p-c-Jun (mouse
monoclonal; cat. no. 2315; clone L70B11; 1:1,000; Cell Signaling
Technology, Inc.), JNK (mouse monoclonal; cat. no. 610627; clone
37/pan-JNK/SAPK1; 1:1,000; BD Transduction Laboratories, Franklin
Lakes, NJ, USA) and c-Jun (mouse monoclonal; cat. no. 610327; clone
3/Jun; 1:1,000; BD Transduction Laboratories) were used, which were
incubated at 4°C overnight. β-actin (Sigma-Aldrich) were used. All
procedures were repeated at least three times. Following antibody
incubation, the membranes were washed with Tris-buffered saline
containing Tween 20 (pH 7.4) three times. Following enhanced
chemiluminescence, the blots were exposed to Kodak X-OMAT BT film
(Kodak, Rochester, NY, USA). The bands were visualized using
densitometry with Image-Pro Plus version 6.0 (Media Cybernetics,
Inc., Rockville, MD, USA)
Transwell invasion assay
Briefly, 1×104 cells/well in serum-free
medium were seeded into the upper chamber of a Transwell plate (cat
no. 3422, Corning Inc., Corning, NY, USA) that was filter-coated
with Matrigel (cat no. E1207; Sigma-Aldrich). At the same time, the
bottom compartment of the chamber was filled with medium containing
10% FBS as a chemoattractant. After 24 h of incubation at 37°C with
5% CO2, the cells remaining in the upper chamber were
carefully removed using a cotton swab and the cells that had
transgressed through the Matrigel and were located at the bottom of
the membrane were fixed with 100% methanol and stained with
hematoxylin. Quantification was performed by counting the number of
cells transgressed through the matrigel using an inverted
microscope (GX41; Olympus Corporation, Tokyo, Japan) at 200×
magnification.
Wound-healing assay
Cells were seeded into six-well tissue culture
plates at 2×106 cells per well and grown in serum-free
RPMI 1640 medium for 24 h to form a confluent monolayer. A wound
across the cell monolayer was created using a 100-µl pipette
tip (Sinoland, Qingdao, China). Cell migration into the wound area
was then inspected under an IX70 inverted phase-contrast microscope
(Olympus Corporation) at 100× magnification. The distance of wound
closure was calculated for quantitative analysis.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Student's t-test was used for comparing significant
differences between the means of the two groups. Statistical
analysis was performed using SPSS 13.0 software (SPSS, Inc.,
Chicago, IL, USA). A P-value from a two-tailed test of <0.05 was
considered to indicate a statistically significant distance between
values.
Results
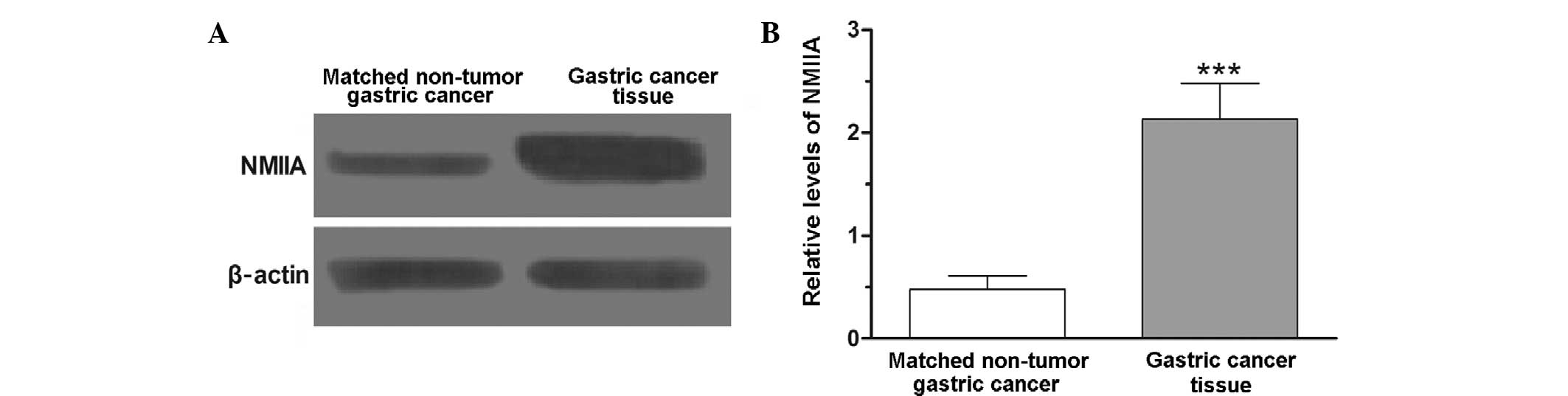
Overexpression of NMIIA in gastric
cancer
To investigate the levels of NMIIA expression in
gastric cancer, clinical gastric cancer tissue specimens and
matched non-tumorous gastric tissue specimens were subjected to
western blot analysis. The results showed that the levels of NMIIA
protein expression in 51 out of 63 gastric cancer tissue specimens
were increased compared to those in their matched non-tumorous
gastric tissue specimens. Statistical analysis showed that
differences in NMIIA expression between gastric cancer tissues and
matched non-tumorous gastric tissues were statistically significant
(P<0.001) (Fig. 1).
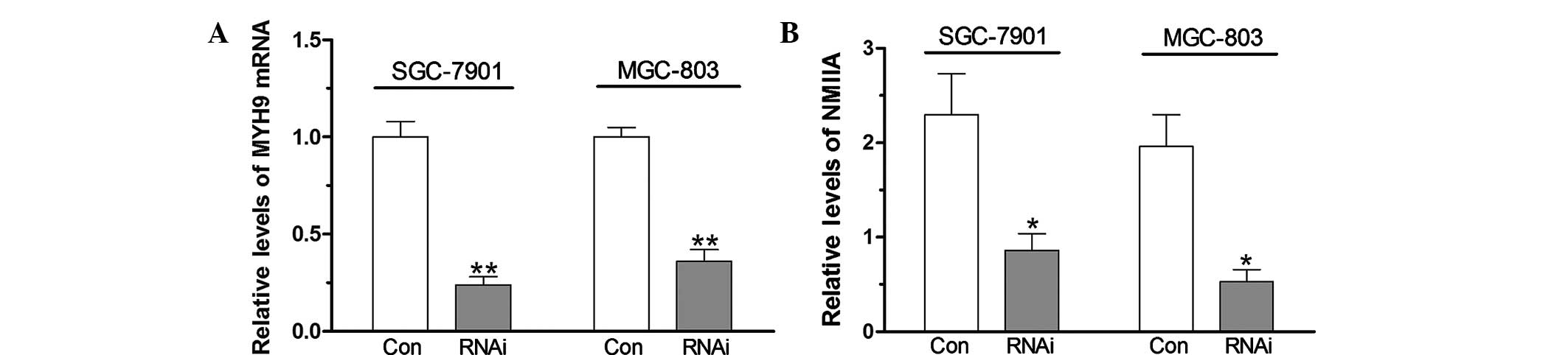
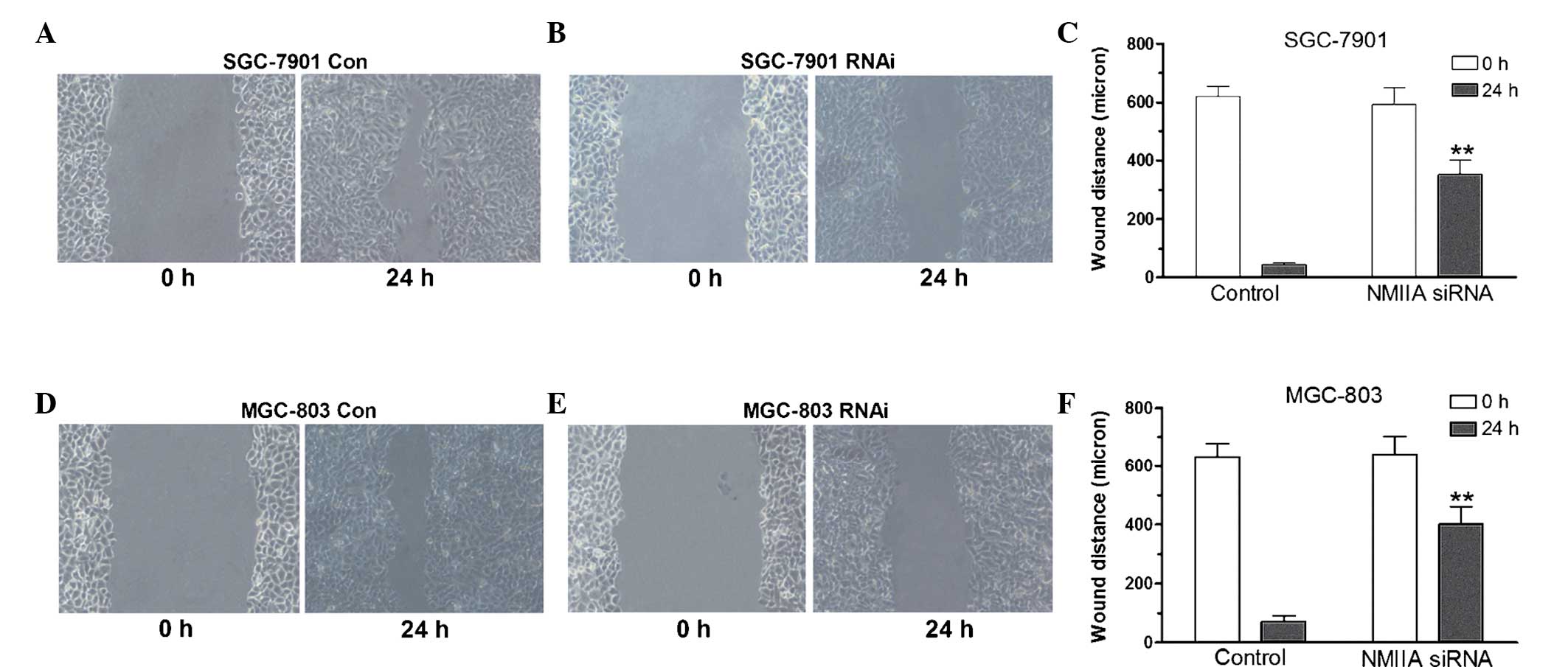
Suppression of NMIIA expression by RNAi
blocks gastric cancer cell migration
As shown in Fig. 2,
the MYH9 gene was silenced in SGC-7901 and MGC-803 with specific
siRNAs and the levels of NMIIA protein decreased compared with
those in the control group (P<0.05). A wound-healing assay was
performed to assess the migration capacity of gastric cancer cells.
As shown in Fig. 3, knockdown of
NMIIA inhibited the migratory capacity of SGC-7901 and MGC-803
cells, as indicated by a decreased amount of wound closure of
gastric cancer cells following knockdown of the MYH9 gene
(P<0.01), indicating that NMIIA is involved gastric cancer cell
migration.
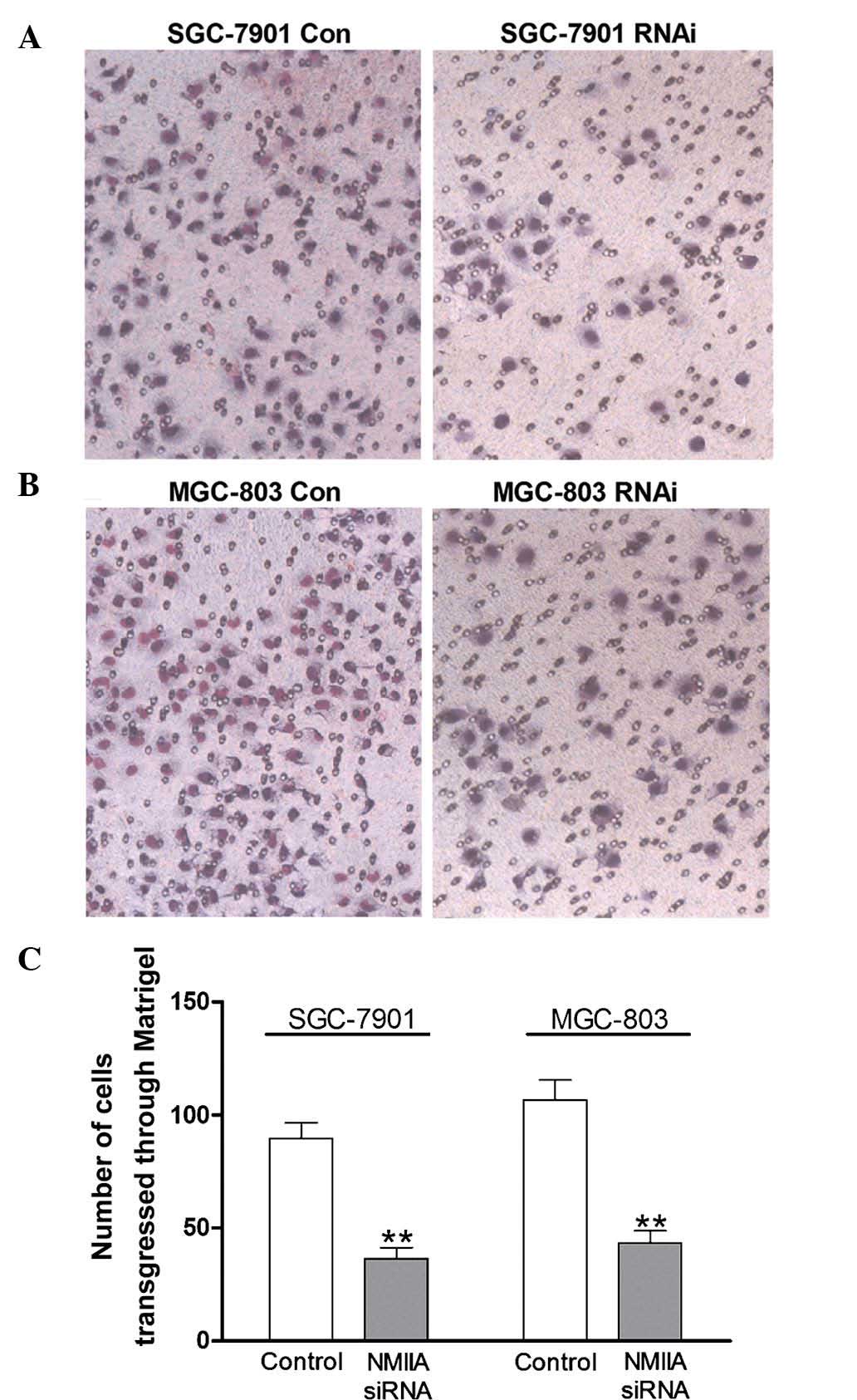
Downregulation of NMIIA expression
inhibits the invasion of gastric cancer cells
A Matrigel invasion assay was performed to observe
the effects of NMIIA knockdown on the invasive capacity of gastric
cancer cells. As shown in Fig. 4A and
B, knockdown of NMIIA inhibited the number of SGC-7901 and
MGC-803 cells transgressing though the Matrigel. Quantification of
the results showed that NMIIA knockdown significantly reduced the
invasive capacities of the two cell lines (P<0.01) (Fig. 4C).
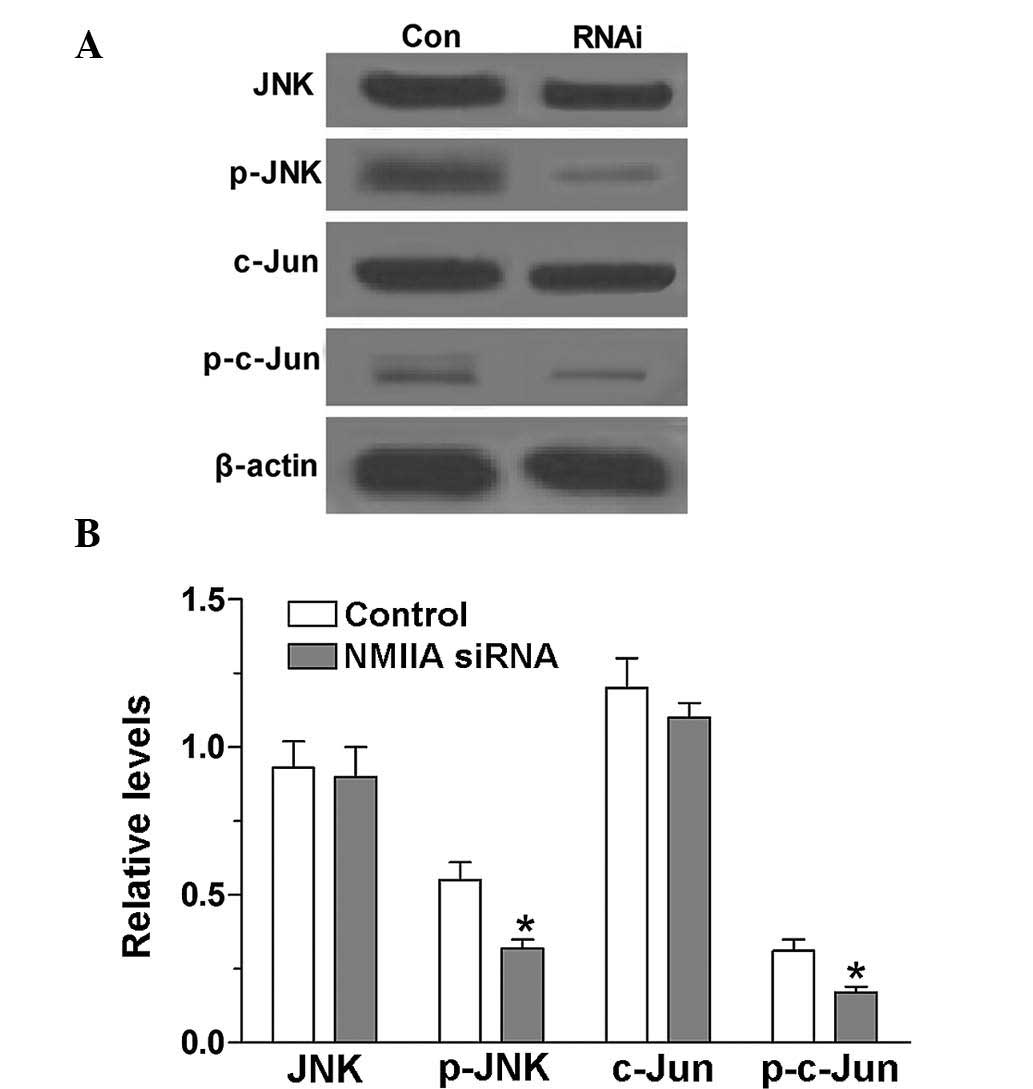
NMIIA is an activator of the JNK
signaling pathway
The levels of p-JNK and p-c-Jun in SGC-7901 cells
were detected using western blot analysis prior to and following
knockdown of NMIIA expression by siRNA. The results showed that the
levels of p-JNK and p-c-Jun in SGC-7901 cells were high. However,
after inhibition of NMIIA expression by siRNA, the levels of p-JNK
and p-c-Jun were significantly decreased (P<0.05), which
indicated that NMIIA is an activator of JNK and c-Jun (Fig. 5).
Discussion
Gastric cancer is one of the most frequent cancer
types in the world (13). The
mechanisms responsible for the occurrence, development and
prognosis of gastric cancer have been investigated from various
perspectives. For instance, Wang et al (3) investigated the underlying regulatory
signaling pathways in gastric cancer by integrating gene expression
profiles and transcriptional regulatory element databases, showing
that the five transcription factors hypoxia-inducible factor-1α,
nuclear factor-κB1, breast cancer 1, signal transducer and
activator of transcription (STAT)3 and STAT1 were able to regulate
82 differentially-expressed genes in gastric cancer. In addition,
these genes formed 95 regulation modes, among which MMP1, TIMP1,
TLR2, FCGR3A, IRF1, FAS and TFF3 were central genes that were
simultaneously regulated by at least two of these five
transcription factors, and were associated with hypoxia,
inflammation and immune disorders. In addition, a recent study
revealed that the expression of CXC motif receptor 1 and 2 proteins
promoted MMP-9 expression by activating JNK/c-Jun and extracellular
signal-regulated kinase (ERK)/Ets-1 pathways, resulting in a more
aggressive phenotype in gastric cancer cells (12). The present study examined the
effects of NMIIA on the invasion and migration of gastric cancer
cells, based on previous evidence showing that NMIIA was
overexpressed in certain types of cancer (7,9,10).
The results of the present study showed that NMIIA was
overexpressed in gastric cancer tissues, which was consistent with
the results of a previous study (6).
JNK, a member of the mitogen-activated protein
kinase family that regulates a range of pathological processes
involved in tumor and brain development and neurological disorders
(14), is encoded by three genes:
JNK1 and 2, which are ubiquitously expressed, and JNK3, which is
restricted to the testis, heart and brain (15,16).
The functions of JNK isoforms in diseases have been most thoroughly
demonstrated in cancer (14).
According to previous studies, JNK1 deficiency significantly
decreased hepatocellular carcinoma in a mouse model (17), while JNK2 was shown to act as a
tumor promoter in skin cancer formation (18). A number of studies have explored
the JNK signaling pathway in gastric cancer. When SGC-7901 cells
were treated with vitamin E succinate, transforming growth factor-β
was activated, which in turn increased the activity of JNK, which
then induced c-Jun phosphorylation; finally, p-c-Jun initiated
apoptosis of gastric cancer cells (19). Similarly, the antioxidant analogue
α-tocopheryl succinate induced apoptosis by activating ERK1/2 and
JNK via c-Jun in the gastric cancer cell line SGC-7901 (20). In addition, the fact that the
specific JNK inhibitor SP-600125 inhibited cell viability, induced
apoptosis and caused cell cycle arrest in gastric cancer cells was
most likely associated with its inhibition of JNK2 phosphorylation,
which attenuated the JNK signaling pathway (21). The present study found that
knockdown of NMIIA inhibited the migration and invasion of gastric
cancer cells, while simultaneously decreasing the protein levels of
JNK and c-Jun as well.
It is known that JNK is able to phosphorylate c-Jun
on serines 63 and 73 at the N-terminal activating sites, which
leads to increased stability of c-Jun and an increase in its
transactivation potential and DNA-binding affinity (22–24).
The results of the present study indicated that NMIIA inhibited the
activation of JNK, resulting in the inhibition of c-Jun
phosphorylation, which, in turn, attenuated the migratory and
invasive capacities of gastric cancer cells. In fact, the JNK
signaling pathway is involved in the migration and invasion of
cancer cells, which has been demonstrated by various studies on the
basis of a range of in vivo and in vitro experimental
models. For instance, isoliquiritigenin decreases the
phosphorylation of JNK and c-Jun and certain other regulatory
factors, which inhibits the migration, adhesion and invasion of
prostate cancer cells.
In conclusion, the present study demonstrated that
NMIIA was overexpressed in gastric cancer and knockdown of NMIIA by
RNAi inhibited the migration and invasion of gastric cancer cells
in vitro, which may proceed via the JNK signaling pathway.
The present study may be useful for the development of novel
strategies for the clinical control of gastric cancer
metastasis.
References
|
1
|
Herszényi L and Tulassay Z: Epidemiology
of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci.
14:249–258. 2010.PubMed/NCBI
|
|
2
|
Compare D, Rocco A and Nardone G: Risk
factors in gastric cancer. Eur Rev Med Pharmacol Sci. 14:302–308.
2010.PubMed/NCBI
|
|
3
|
Wang J, Ni Z, Duan Z, Wang G and Li F:
Altered expression of hypoxia-inducible factor-1α (HIF-1α) and its
regulatory genes in gastric cancer tissues. PLoS One. 9:e998352014.
View Article : Google Scholar
|
|
4
|
Liu X, Liu Q, Fan Y, Wang S, Liu X, Zhu L,
Liu M and Tang H: Downregulation of PPP2R5E expression by miR-23a
suppresses apoptosis to facilitate the growth of gastric cancer
cells. FEBS Lett. 588:3160–3169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu D, Zhang L, Shen Z, Tan F, Hu Y, Yu J
and Li G: Clinicopathological Significance of NMIIA overexpression
in human gastric cancer. Int J Mol Sci. 13:15291–15304. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xia ZK, Yuan YC, Yin N, Yin BL, Tan ZP and
Hu YR: Nonmuscle myosin IIA is associated with poor prognosis of
esophageal squamous cancer. Dis Esophagus. 25:427–436. 2012.
View Article : Google Scholar
|
|
8
|
Derycke L, Stove C, Vercoutter-Edouart AS,
De Wever O, Dollé L, Colpaert N, Depypere H, Michalski JC and
Bracke M: The role of non-muscle myosin IIA in aggregation and
invasion of human MCF-7 breast cancer cells. Int J Dev Biol.
55:835–840. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Betapudi V, Licate LS and Egelhoff TT:
Distinct roles of nonmuscle myosin II isoforms in the regulation of
MDA-MB-231 breast cancer cell spreading and migration. Cancer Res.
66:4725–4733. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lamant L, Gascoyne RD, Duplantier MM,
Armstrong F, Raghab A, Chhanabhai M, Rajcan-Separovic E, Raghab J,
Delsol G and Espinos E: Non-muscle myosin heavy chain (MYH9): A new
partner fused to ALK in anaplastic large cell lymphoma. Gene
Chromosomes Cancer. 37:427–432. 2003. View Article : Google Scholar
|
|
11
|
Liang S, He L, Zhao X, Miao Y, Gu Y, Guo
C, Xue Z, Dou W, Hu F, Wu K, et al: MicroRNA let-7f inhibits tumor
invasion and metastasis by targeting MYH9 in human gastric cancer.
PLoS One. 6:e184092011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Z, Wang Y, Dong S, Ge C, Xiao Y, Li R,
Ma X, Xue Y, Zhang Q, Lv J, et al: Association of CXCR1 and 2
expressions with gastric cancer metastasis in ex vivo and tumor
cell invasion in vitro. Cytokine. 69:6–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Matsuoka J, Yashiro M, Sakurai K, Kubo N,
Tanaka H, Muguruma K, Sawada T, Ohira M and Hirakawa K: Role of the
stemness factors sox2, oct3/4 and nanog in gastric carcinoma. J
Surg Res. 174:130–135. 2012. View Article : Google Scholar
|
|
14
|
Davies C and Tournier C: Exploring the
function of the JNK (c-Jun N-terminal kinase) signalling pathway in
physiological and pathological processes to design novel
therapeutic strategies. Biochem Soc Trans. 40:85–89. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Johnson GL and Nakamura K: The c-jun
kinase/stress-activated pathway: Regulation, function and role in
human disease. Biochim Biophys Actas. 1773:1341–1348. 2007.
View Article : Google Scholar
|
|
16
|
Ma J, Zhang L, Han W, Shen T, Ma C, Liu Y,
Nie X, Liu M, Ran Y and Zhu D: Activation of JNK/c-Jun is required
for the proliferation, survival and angiogenesis induced by EET in
pulmonary artery endothelial cells. J Lipid Res. 53:1093–1105.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hui L, Zatloukal K, Scheuch H, Stepniak E
and Wagner EF: Proliferation of human HCC cells and chemically
induced mouse liver cancers requires JNK1-dependent p21
downregulation. J Clin Invest. 118:3943–3953. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen N, Nomura M, She QB, Ma WY, Bode AM,
Wang L, Flavell RA and Dong Z: Suppression of skin tumorigenesis in
c-Jun NH2-terminal kinase-2-deficient mice. Cancer Res.
61:3908–3912. 2001.PubMed/NCBI
|
|
19
|
Wu K, Liu BH, Zhao DY and Zhao Y: Effect
of vitamin E succinate on expression of TGF-beta1, c-Jun and JNK1
in human gastric cancer SGC-7901 cells. World J Gastroenterol.
7:83–87. 2001. View Article : Google Scholar
|
|
20
|
Zhao Y, Zhao X, Yang B, Neuzil J and Wu K:
α-Tocopheryl succinate-induced apoptosis in human gastric cancer
cells is modulated by ERK1/2 and c-Jun N-terminal kinase in a
biphasic manner. Cancer Lett. 247:345–352. 2007. View Article : Google Scholar
|
|
21
|
Xia HH, He H, De Wang J, Gu Q, Lin MC, Zou
B, Yu LF, Sun YW, Chan AO, Kung HF and Wong BC: Induction of
apoptosis and cell cycle arrest by a specific c-Jun NH2-terminal
kinase (JNK) inhibitor, SP-600125, in gastrointestinal cancers.
Cancer Lett. 241:268–274. 2006. View Article : Google Scholar
|
|
22
|
Qi X, Pramanik R, Wang J, Schultz RM,
Maitra RK, Han J, DeLuca HF and Chen G: The p38 and JNK pathways
cooperate to trans-activate vitamin D receptor via c-Jun/AP-1 and
sensitize human breast cancer cells to vitamin D(3)-induced growth
inhibition. J Biol Chem. 277:25884–25892. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schroeter H, Spencer J, Rice-Evans C and
Williams R: Flavonoids protect neurons from oxidized
low-density-lipoprotein-induced apoptosis involving c-Jun
N-terminal kinase (JNK), c-Jun and caspase-3. Biochem J.
358:547–557. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu K, Zhao Y, Li GC and Yu WP: c-Jun
N-terminal kinase is required for vitamin E succinate-induced
apoptosis in human gastric cancer cells. World J Gastroenterol.
10:1110–1114. 2004.PubMed/NCBI
|