Introduction
Nonalcoholic fatty liver disease (NAFLD) is one of
the most common forms of chronic liver disease (1). Nonalcoholic steatohepatitis, which is
intimately associated with the innate immune response, can be
severe and lead to hepatic fibrosis and cirrhosis (2). Toll-like receptors (TLRs) are pattern
recognition receptors, which are important in innate immunity.
Increasing evidence has shown that TLRs are involved in the
pathogenesis and progression of various liver diseases, including
alcoholic liver disease, viral hepatitis and autoimmune liver
disease (2–4). However, the mechanisms underlying the
involvement of TLRs in NAFLD remain to be fully elucidated. Certain
TLRs, particularly TLR4, are widely expressed on hepatic stellate
cells (HSCs) and Kuppffer cells, and may be involved in liver
fibrosis (5).
Peroxisome proliferator-associated receptor γ
(PPARγ) is a member of the nuclear receptor superfamily of
transcription factors (6). Our
previous investigations demonstrated that rosglitazone, a specific
PPARγ agonist, has a protective role in the progression of
nutritional fibrotic steatohepatitis, through the modulation of
lipid homeostasis and the inflammatory response, and maintenance of
HSC quiescence in mice (7–9). It has also been reported that
pioglitazone, another specific PPARγ agonist, alleviates steatosis
and steatohepatitis induced by consumption of a methioninecholine
deficient (MCD) diet (10,11).
In the present study, we aimed to clarify whether
the TLR4-dependent signaling pathway is involved in the development
of fibrotic steatohepatitis induced by an MCD diet, and to further
elucidate whether pioglitazone can prevent liver injury by
modulation of the TLR4 pathway and associated genes. The present
study may provide an effective therapeutic target for the treatment
of patients with nutritional fibrotic steatohepatitis.
Materials and methods
Animals and treatment
Male C57BL/6J mice (8-week-old; 20–25 g) were
obtained from the Experimental Animals Center of the Chinese
Academy of Medical Sciences (Beijing, China), and were bred in a
temperature-controlled animal facility with a 12 h light-dark
cycle. The mice had free access to water, and were fed with a
standard rodent diet ad libitum for 1 week prior to the
experiment to enable adaptation to the food and environment. The
mice were randomly divided into four groups (n=6 per group): i)
Control group, fed an MCD diet supplemented with choline bitartrate
(2 g/kg) and DL-methionine (3 g/kg; both ICN, Aurora, OH, USA); ii)
MCD group, fed an MCD diet (ICN); iii) MCD+pioglitazone (PIO)
group, fed an MCD diet with pioglitazone (50 mg/kg/day; chow;
GlaxoSmithKline Co., Ltd., Tianjin, China); iv) MCD+GW9662 group,
fed an MCD diet and administered intraperitoneally with
2-chloro-5-nitrobenzani-liden (GW9662; 1 mg/kg; three times per
week; Alexis Biochemicals, Lausen, Switzerland). The duration of
the experiment lasted up to 8 weeks. Following overnight fasting at
the end of the experiments, all animals were sacrificed via
exposure to 0.08 ml/l aether anesthesia (1.9%), which was presented
on a cotton ball inside a conical tube for 5–10 min. Blood samples
were collected from the femoral arteries for biochemical analysis.
The livers were weighed and either fixed in 10% formalin for
histological analysis, or snap-frozen in lipid nitrogen and stored
at −80°C until required. All protocols and procedures were
performed in accordance with the the guidelines of the Hebei
Committee for Care and Use of Laboratory Animals (Hebei, China) and
were approved by the Animal Experimentation Ethics Committee of
Hebei Medical University (Shijiazhuang, China).
Biochemical analysis
Prior to euthanasia, 0.5 ml blood was harvested from
each mouse via an orbital sinus puncture and the serum was
separated by centrifugation at 1,500 × g for 15 min at 4°C. The
levels of alanine aminotransferase (ALT) and aspartate
aminotransferase (AST) were measured using an the enzymatic kinetic
method with an automatic biochemical analyzer (UA2700; Olympus
Corporation, Tokyo, Japan), according to the manufacturer's
protocol.
Histological analysis
Liver tissues were cut into 5 µm-thick
sections, paraffin-embedded and fixed for 16 h in 4%
phosphate-buffered formalin at 4°C prior to hematoxylin and eosin
and Masson's trichromatism staining. The liver sections were
subsequently scored for hepatic steatosis, inflammation and
fibrosis using a Leica DM 2000 microscope (Leica Microsystems,
Inc., Buffalo Grove, IL), as described previously, in accordance
with Brunt's criteria (12) and
the histological scoring system for NAFLD issued by the Pathology
Committee of the Nonalcoholic Steatohepatitis Clinical Research
Network (13). The histological
scoring system for NAFLD was as follows: Steatosis (0–3), lobular
inflammation (0–2), hepatocellular ballooning (0–2), and fibrosis
(0–4).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis of hepatic messenger
RNA (mRNA) expression levels
Liver tissue samples were homogenized using 1 ml
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) per
50–100 mg tissue using a glass homogenizer. A total of 0.2 ml
chloroform was subsequently added per 1 ml TRIzol reagent and the
samples were vigorously vortexed for 15 sec prior to incubation at
room temperature for 2 min. Following this, samples were
centrifuged at 12,000 × g for 15 min at 4°C. The aqueous phase of
the sample was transferred to a new tube and supplemented 0.5 ml
100% isopropanol prior to incubation at room temperature for 10 min
and centrifugation at 12,000 × g for 10 min at 4°C. Total RNA was
isolated from the frozen liver tissues using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's protocol. Subsequently, 1 µg
total RNA was reverse transcribed into cDNA using a cDNA synthesis
kit (Thermo Fisher Scientific Inc.), according to the
manufacturer's instructions The hepatic mRNA levels of PPARγ, TLR4,
myeloid differentiation primary response gene 88 (MyD88), inhibitor
of κB kinase-β (IKK-β), nuclear factor-κB (NF-κB); c-Jun N-terminal
protein kinase 1 (JNK1), activator protein-1 (AP-1), bone
morphogenetic protein and activin membrane-bound inhibitor (BAMBI),
transforming growth factor-β1 (TGF-β1), α-smooth muscle actin
(α-SMA), collagen type I (Col-1), tumor-necrosis factor-α (TNF-α),
interleukin-1β (IL-1β), monocyte chemoattractant protein-1 (MCP-1),
macrophage inflammatory protein-1α (MIP-1α), were determined by
RT-qPCR using an ABI PRISM 7500 sequence detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). RT-qPCR was performed
at a final reaction volume of 50 µl containing 2 µl
cDNA, 5 pmol specific primers, 25 µl SYBR Green reagent
(CWBio, Beijing, China) and 23 µl water. Thermal cycling was
completed as follows: 95°C for 5 min, followed by 20 cycles of 95°C
for 30 sec and 60°C for 30 sec. Expression levels of the target
genes were normalized against an endogenous reference gene,
glyceraldehydes 3-phosphate dehydrogenase (GAPDH), and were
calculated using the 2−ΔΔCq method (14) using ABI PRISM 7500 Sequence
Detection software. The specific primers for PPARγ, TLR4, MyD88,
IKK-β, NF-κB, JNK1, AP-1, TNF-α, IL-1β, MCP-1, MIP-1α, BAMBI,
TGF-β1, α-SMA, Col-1 and GAPDH were designed using Primer Premier
5.0 (Premier Biosoft, Palo Alto, CA, USA), and the sequences are
listed in Table I. All data were
obtained using Sequence Detector software.
 | Table IPrimers used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Primers used for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Product length
(bp) | Primer sequence |
|---|
| PPARγ | 150 | F:
5′-CACTCGCATTCCTTTGACATC-3′ |
| | R:
5′-CGCACTTTGGTATTCTTGGAG-3′ |
| TLR4 | 166 | F:
5′-GAGCCGTTGGTGTATCTTTGA-3′ |
| | R:
5′-CTCCCATTCCAGGTAGGTATT-3′ |
| MyD88 | 150 | F:
5′-CACTCGCATTCCTTTGACATC-3′ |
| | R:
5′-CGCACTTTGGTATTCTTGGAG-3′ |
| IKK-β | 308 | F:
5′-GGACTTCTTCAAAACCAGCATC-3′ |
| | R:
5′-CACCTTCTGTCCTTTGGTCTCT-3′ |
| NF-κB | 98 | F:
5′-GTAGAGGATTTGCTGAGGGTG-3′ |
| | R:
5′-ATTCTGTCGTGTCCTTCTTTGG-3′ |
| JNK-1 | 166 | F:
5′-TCAAGCACCTTCACTCTG-3′ |
| | R:
5′-CAAACCATTTCTCCCATA-3′ |
| AP-1 | 308 | F:
5′-CGGACCGTTCTATGACTGC-3′ |
| | R:
5′-AGCGTGTTCTGGCTATGC-3′ |
| TNF-α | 98 | F:
5′-CTGTGAAGGGAATGGGTGTT-3′ |
| | R:
5′-CAGGGAAGAATCTGGAAAGGTC-3′ |
| IL-1β | 233 | F:
5′-AGGCTCCGAGATGAACAA-3′ |
| | R:
5′-AAGGCATTAGAAACAGTCC-3′ |
| MCP-1 | 150 | F:
5′-TTCCACGCTCTTATCCTA-3′ |
| | R:
5′-CATCTCGTTGCTACCTCC-3′ |
| MIP-1α | 166 | F:
5′-CTGCCCTTGCTGTTCTTC-3′ |
| | R:
5′-GTTCCAGGTCAGTGATGTA-3′ |
| Col-1 | 308 | F:
5′-GGGCGAGTGCTGTGCCTTT-3′ |
| | R:
5′-GAGCCATTGGACCTGAACC-3′ |
| α-SMA | 98 | F:
5′-CTGACAGAGGCACCACTGAA-3′ |
| | R:
5′-CATCTCCAGAGTCCAGCACA-3′ |
| TGF-β1 | 233 | F:
5′-GGCGGTGCTCGCTTTGTA-3′ |
| | R:
5′-AGCCACTCAGGCGTATCAG-3′ |
| BAMBI | 308 | F:
5′-AGTGACTAGCAGGGAAAT-3′ |
| | R:
5′-AAGGAGCAGATAGAGGAG-3′ |
| GAPDH | 233 | F:
5′-GGTGAAGGTCGGTGTGAACG-3′ |
| | R:
5′-CTCGCTCCTGGAAGATGGTG-3′ |
Western blot analysis of hepatic protein
expression levels
Total hepatic proteins were extracted using
radioimmunoprecipitation assay lysis buffer (Thermo Fisher
Scientific, Inc.), and the protein concentration was measured using
the Bradford method (DC protein assay; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Loading buffer (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) was added to each lysate,
which contained equal quantities of protein (100 µg/well).
The lysate was boiled for 5 min and then electrophoresed on 10%
SDS-polyacrylamide gels. The proteins were then transferred onto
equilibrated polyvinylidene difluoride membranes (EMD Millipore,
Billerica, MA, USA) by electroblotting. The membranes were
incubated, respectively, with the following primary antibodies
overnight at 4°C: Mouse polyclonal anti-PPARγ (1:400; sc-122729;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA); rabbit
polyclonal anti-TLR4 (1:1,000; ab13556; Abcam, Cambridge, MA, USA);
rabbit monoclonal anti-MyD88 (1:,1000; 4283; Cell Signaling
Technology, Inc., Danvers, MA, USA); rabbit polyclonal anti-IKK-β
(ab55404; Abcam); and mouse monoclonal anti-NF-κB (1:400; sc-8414;
Santa Cruz Biotechnology, Inc.). And the following rabbit
polyclonal primary antibodies against: JNK1 (bs-10562R); AP-1
(bs-0670R); TNF-α (bs-2081R); IL-1β (bs-0812R); MCP-1 (bs-0562R);
MIP-1α (bs-1045R); BAMBI (bs-12418R); TGF-β1 (bs-0086R); α-SMA
(bs-0819R); Col-1 (bs-7158R); and β-actin (bs-0061R; all 1:400; all
Biosynthesis Biotechnology, Co., Ltd., Beijing, China). The
membranes were then incubated with appropriate
peroxidase-conjugated secondary antibodies at room temperature for
1 h. Bands were visualized using enhanced chemiluminescence (GE
Healthcare Bio-Sciences, Pittsburgh, PA, USA), followed by exposure
to an X-ray film (Kodak, Rochester, NY, USA). The protein
expression levels were normalized to that of β-actin in the same
sample, and the expression levels were quantified by scanning
densitometry using Quantity One V4.62 software (Bio-Rad
Laboratories, Inc.).
Statistical analysis
All data are expressed as the mean ± standard
deviation. Statistical analyses of the data were performed using
one-way analysis of variance (15)
or a Kruskal-Wallis H-test, with a least significant
difference t-test or Mann-Whitney U test for post-hoc comparison
using SPSS 17.0 (SPSS Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Pioglitazone activates PPARγ, and
ameliorates hepatic inflammation and fibrosis in mice fed an MCD
diet
The MCD diet induced steatohepatitis and liver
fibrosis in the mice, as shown in Fig.
1, which was in accordance with the effects observed in our
previous study (16). Compared
with the control group, the MCD group showed disordered lobule
structure, macrosteatosis in Zone 3, spot or focal hepatocyte
necrosis, inflammatory infiltration (Fig. 1A), and portal and persinusoidal
fibrosis (Fig. 1B) in the liver
sections, which were accompanied with significantly downregulated
mRNA and protein expression levels of hepatic PPARγ, which were
decreased further by GW9662 administration (P<0.01).
Pioglitazone administration markedly ameliorated hepatic steatosis,
inflammation (Fig. 1A) and
progressive fibrosis (Fig. 1B),
which were associated with marked decreases in serum ALT (1) and AST (Fig. 1C and D; P<0.01). However, the
opposite effect was observed in the MCD+GW9662 group, which
exhibited aggravated steatosis, inflammation and fibrosis (Fig. 1A and B), and further increase in
the serum aminotransferase levels (Fig. 1C and D).
 | Figure 1Histopathological changes and serum
levels of ALT and AST of mice under various treatment conditions.
(A) Hematoxylin and eosin-stained and (B) Masson's trichome-stained
liver sections from the mice (Original magnification, ×200), (a)
Control, (b) MCD, (c) MCD+PIO, (d) MCD+GW9662; (C) Serum levels of
ALT; (D) serum levels of AST (n=6 per group). Data are expressed as
the mean ± standard deviation. **P<0.01, compared
with the Control group; #P<0.05, compared with the
MCD group; ##P<0.01, compared with the MCD group.
ALT, alanine aminotransferase; AST, aspartate aminotransferase;
MCD, methionine-choline deficient; PIO, pioglitazone. |
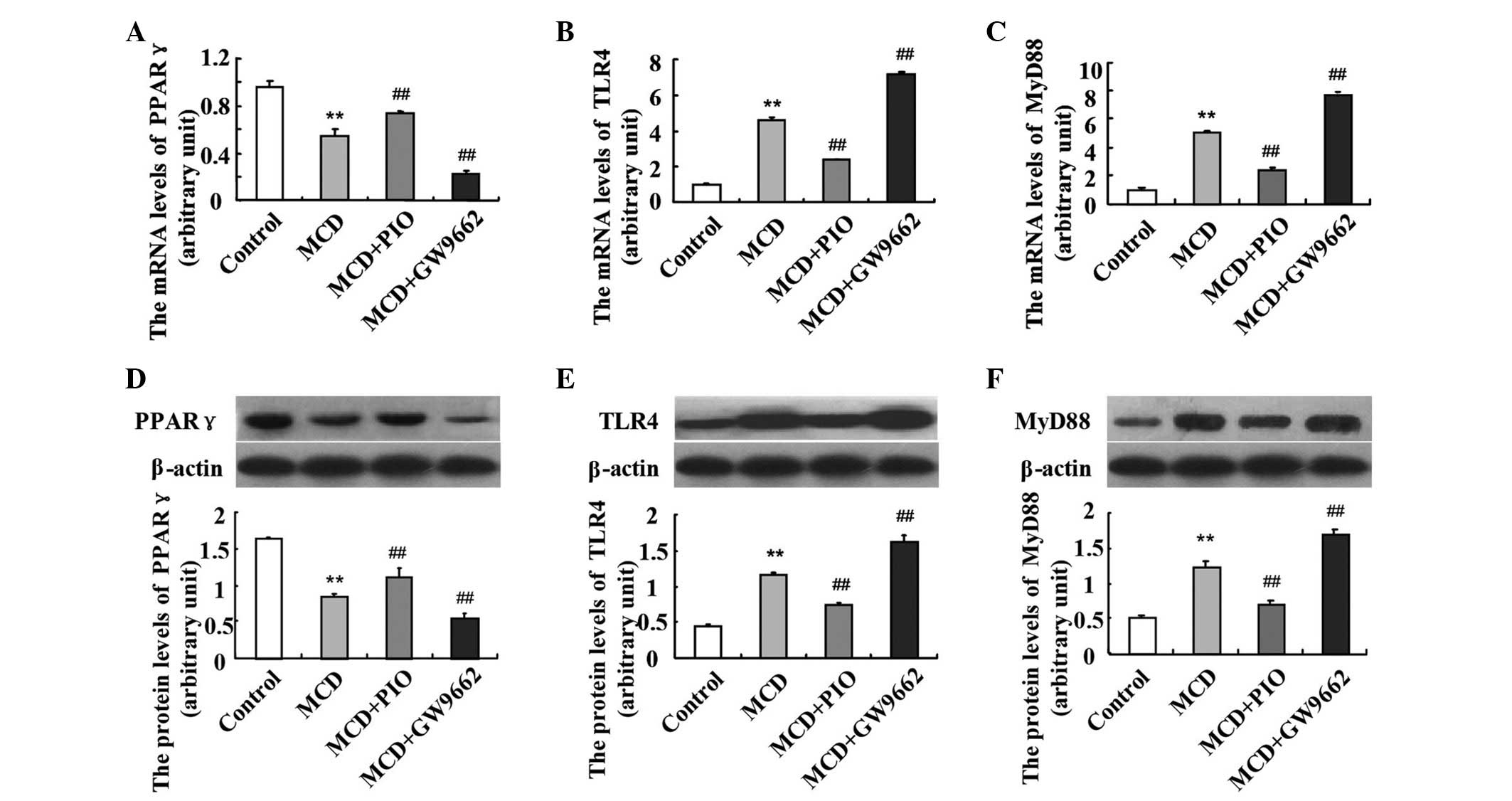
Pioglitazone downregulates the hepatic
expression levels of TLR4 and MyD88
The hepatic expression levels of TLR4 and MyD88 were
significantly higher in the MCD-fed mice, compared with those in
the control mice (P<0.01). Pioglitazone administration
significantly upregulated the mRNA expression of PPARγ (Fig. 2). Following the induction of PPARγ
by pioglitazone, the mRNA expression levels of TLR4 and MyD88 were
downregulated, compared with those in the MCD group. However, the
administration of GW9662 led to further upregulation in the mRNA
levels of TLR4 and MyD88 (Fig. 2B and
C; P<0.01). The same results were observed on examination of
the protein expression levels of PPARγ (Fig. 2D), TLR4 (Fig. 2E) and MyD88 (Fig. 2F).
Pioglitazone suppresses hepatic gene
expression levels down-stream of TLR4
To further investigate the TLR4-mediated
inflammatory response and determine the anti-inflammatory effects
of pioglitazone, the present study investigated hepatic mRNA and
protein expression levels of the MyD88-dependent signaling pathway
and downstream genes, IKK-β, NF-κB, JNK1 and AP-1. Compared with
the controls, the hepatic mRNA and protein expression levels of
IKK-β, NF-κB, JNK1 and AP-1 were markedly increased in the MCD-fed
mice (P<0.01), and these levels were significantly reduced by
pioglitazone (P<0.01), as shown in Fig. 3. By contrast, the hepatic mRNA and
protein expression levels of IKK-β, NF-κB, JNK1 and AP-1 were
further upregulated by GW9662 treatment, compared with the MCD
group (Fig. 3; P<0.01).
 | Figure 3Effect of pioglitazone on the
expression levels of IKK-β, NF-κB, JNK1 and AP-1 in nutritional
fibrotic steatohepatitis-induced mice. The mRNA expression levels
of (A) IKK-β, (B) NF-κB, (C) JNK1 and (D) AP-1 were examined using
reverse transcription-quantitative polymerase chain
reactionanalysis. The protein expression levels of (E) IKK-β, (F)
NF-κB, (G) JNK1 and (H) AP-1 were measured using Western blotting.
n=6 per group. Data are expressed as the mean ± standard deviation.
**P<0.01, compared with the Control group;
##P<0.01, compared with the MCD group. MCD,
methionine-choline deficient; PIO, pioglitazone; IKK-β, inhibitor
of κB kinase-β; NF-κB, nuclear factor κB; JNK1, c-Jun N-terminal
protein kinase-1, AP-1, activator protein-1. |
Pioglitazone downregulates hepatic
expression levels of proinflammatory cytokines and chemokines
To further examine the mechanism underlying
PPARγ-induced amelioration of liver histology, the present
study examined the hepatic expression levels of inflammatory
cytokines and chemokines. Compared with the control group, the
hepatic expression levels of TNF-α, IL-1β, MCP-1 and MIP-1α were
upregulated in the MCD diet-fed mice (P<0.01; Fig. 4). These induced expression levels
were significantly decreased by pioglitazone treatment. Treatment
with GW9662 led to further increases in the expression levels of
TNF-α, IL-1β, MCP-1 and MIP-1α, compared with the levels in the
mice fed an MCD diet (Fig. 4;
P<0.01).
 | Figure 4Effect of pioglitazone on the
expression of inflammatory cytokines and chemokines in mice. The
mRNA expression levels of (A) TNF-α, (B) IL-1β, (C) MCP-1 and (D)
MIP-1α were examined using reverse transcription-quantitative
polymerase chain reaction analysis. Protein expression levels of
(E) TNF-, (F) IL-1β, (G) MCP-1 and (H) MIP-1α were measured using
Western blotting. n=6 per group. Data are expressed as mean ±
standard deviation. **P<0.01, compared with the
Control group; ##P<0.01, compared with the MCD group.
MCD, methionine-choline deficient; PIO, pioglitazone; TNF-α,
tumor-necrosis factor-α; IL-1β, interleukin-1β; MCP-1, monocyte
chemoattractant protein-1; MIP-1α, macrophage inflammatory
protein-1α. |
Pioglitazone modulates the expression
levels of fibrosis-associated genes
To evaluate the effect and mechanisms of
PPARγ-induced fibrotic steatohepatitis, the present study
assessed the hepatic expression levels of fibrosis-associated
genes. The mice fed an MCD diet showed enhanced hepatic mRNA and
protein expression levels of TGF-β1, α-SMA and Col-1 (P<0.01),
which were depressed by pioglitazone administration. By contrast,
the mRNA and protein expression levels of BAMBI were significantly
lower in the MCD-fed mice, compared with the control mice, and were
markedly increased following treatment of pioglitazone (both
P<0.01). The opposite changes were observed in the GW9662 group,
compared with the MCD group (Fig.
5; P<0.01).
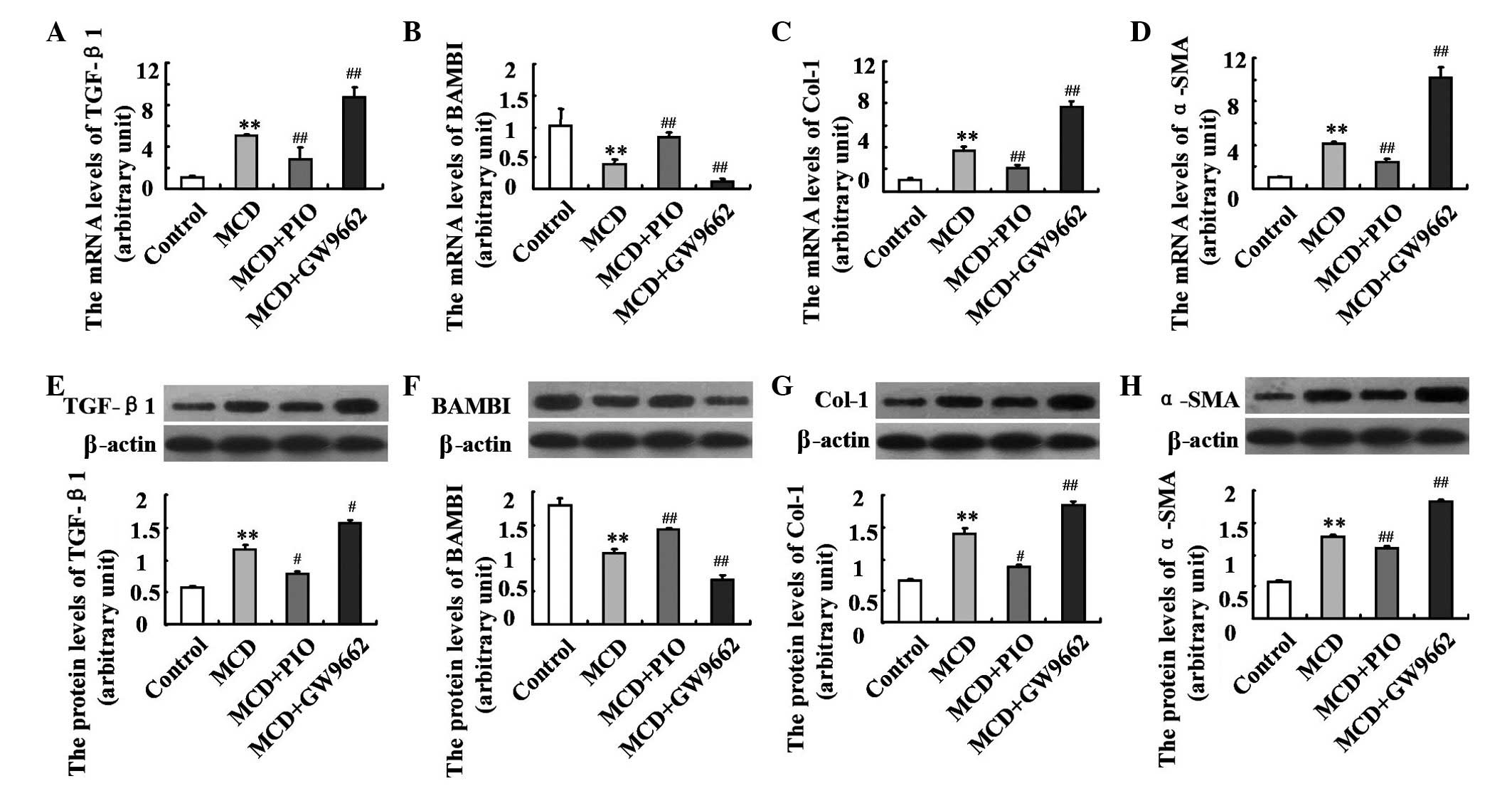 | Figure 5Effect of pioglitazone on the
expression levels of fibrosis-associated genes in nutritional
fibrotic steatohepatitis-induced mice. The mRNA expression levels
of (A) TGF-β1, (B) BAMBI, (C) Col-1 and (D) α-SMA were examined
using reverse transcription quantitative polymerase chain reaction
analysis. The protein expression levels of (E) TGF-β1, (F) BAMBI,
(G) Col-1 and (H) α-SMA were measured using Western blotting. n=6
per group. Data are expressed as the mean ± standard deviation.
*P<0.05 and **P<0.01, compared with the
Control group; #P<0.05 and ##P<0.01,
compared with the MCD group. MCD, methionine-choline deficient;
PIO, pioglitazone; Col-1, collagen type I; α-SMA, α-smooth muscle
actin; TGF-β1, transforming growth factor-β1; BAMBI, basic membrane
protein and activin membrane-bound inhibitor. |
Discussion
Nutritional fibrotic steatohepatitis is a complex
multifactorial disease, and is associated with abnormal lipid
metabolism, oxidative stress, inflammation and the immune response
(17). Investigations have
focussed on the potential mechanisms responsible for the effect of
PPARγ (18–20). The results of the present study
showed that the hepatic expression of PPARγ was significantly lower
in the MCD-fed mice, compared with the control mice, which was
consistent with indicators of liver injury, including increased
levels of serum ALT and AST, hepatic steatosis, necrotic
inflammation and fibrosis. The present study further demonstrated
that the induction of PPARγ by administration of the specific
agonist, pioglitazone, for 8 weeks markedly attenuated liver
injury, as evidenced by decreased levels of serum ALT and AST, a
reduced inflammatory response, reduced collagen deposition and
suppressed HSC activation. These results indicated that PPARγ
modulation had an important protective role in the progression of
NALFD
A novel finding in the present study was that TLR4
was closely involved in the development of liver fibrosis, and the
inhibitory effects of pioglitazone on the inflammatory response and
fibrogenesis in fibrotic steatohepatitis. With the progression of
liver fibrosis in the present study, the hepatic expression of TLR4
increased, suggesting that TLR4 may be involved in the development
of MCD-diet induced liver injury. The downstream proinflammatory
cytokines, MyD88, IKK-β, NF-κB, JNK1 and AP-1, remained consistent
with the change in TLR4 in the mice. The results of the present
study were supported by evidence in a previous study, that the
expression levels of TLR4 and MyD88, the adaptor protein for TLR4,
are significantly increased in fructose or L-amino-acid-defined
diet-induced NAFLD (21–23). Furthermore, the interaction between
TLR4 and MyD88 triggered a downstream signaling cascade, which led
to activation of the IKK-β/NF-κB and JNK1/AP-1 pathways, which then
activated the transcription of proinflammatory genes. These genes
encode proinflammatory molecules, including cytokines, chemokines,
and other effectors of the innate immune response, and induce the
onset and progression of murine diet-induced nonalcoholic
steatohepatitis (24,25). The activation of
TLR4-MyD88-dependent signaling also exacerbates liver inflammation
through stimulating TNF-α and IL-1β. This effect has been
demonstrated by the observations that TLR4-deficient mice show
decreased expression levels of TNF-α and IL-1β in NAFLD models
(26,27). The present study also found that
the hepatic expression of TLR4 was decreased following the
administration of pioglitazone, which was accompanied with
downregulation in the expression levels of MyD88, IKK-β, NF-κB,
JNK1, AP-1, TNF-α and IL-1β. Thus, pioglitazone may alleviate
MCD-induced liver inflammation and fibrosis through downregulation
of the expression levels of TLR4 and its downstream genes.
To further clarify the mechanism by which
pioglitazone alleviates MCD-induced liver fibrosis in mice, the
hepatic expression levels of BAMBI, TGF-β1, α-SMA and Col-1 were
assessed. It has been reported that activated HSCs express TLR4
(28). However, TLR4-signaling
modulates the activation of quiescent HSCs and the transformation
into myofibroblast-like cells, which secrete extracellular matrix
(ECM) and promotes hepatic fibrogenesis in nutritional fibrotic
steatohepatitis. TGF-β1 is considered to be the most potent
mediator of HSC activation (29).
Previously, Li et al reported that TLR4 downregulated the
expression of the TGF-β1 pseudoreceptor, BAMBI, to sensitize HSCs
to TGF-β1-induced signals (30).
In the present study, with the induction of TLR4, the expression
levels of hepatic TGF-β1, α-SMA and Col-1 were significantly
increased, whereas the expression of BAMBI was markedly decreased
in the MCD diet-fed mice, compared with the mice in the control
group, indicating HSC activation and collagen deposition. The
possible mechanisms underlying the TLR4-mediated HSC activation and
hepatic fibrosis in the MCD mice were that TLR4/MyD88 induced the
stimulation of NF-κB, repressed the transcription of BAMBI in HSCs
and increased the production of IL-1β. In addition, the HSC-derived
MCP-1 acted in an autocrine manner to activate the HSCs (31), and MIP-1α enhanced extensive HSC
proliferation (32,33). Activated HSCs secrete excessive ECM
to induce liver fibrosis (34).
Due to its inflammatory action in, pioglitazone reversed the
activation of HSCs and inhibited hepatic nutritional fibrosis in
the mice by modulating the expression levels of PPARγ and TLR4,
which further upregulated the expression of BAMBI and downregulated
the expression levels of TGF-β1, α-SMA and Col-1. This effect may
be enhanced by the efficacy of the PPARγ agonist, pioglitazone, by
modulation of the hepatic expression levels of lipid metabolism-,
oxidative stress- and apoptosis-associated genes. On assessment of
the findings, the present study hypothesized that pioglitazone has
a protective role in nutritional fibrotic steatohepatitis in mice,
which is associated with the targeted activation of PPARγ and
modulation of the TLR4-mediated signaling pathway.
In conclusion, the present study provided insight
into the importance of TLR4-dependent signaling in the development
of nutritional fibrotic steatohepatitis. In addition, the PPARγ
agonist, pioglitazone exerted its anti-inflammatory and
anti-fibrotic effects by modulating the TLR4-dependent signaling
pathways of IKK-β/NF-κB and JNK1/AP-1, and the expression levels of
the fibrogenic genes, BAMBI, TGF-β1, α-SMA and Col-1. These results
indicated the potential role, and underlying molecular mechanism,
of pioglitazone in the prevention and treatment of nutritional
fibrotic steatohepatitis.
Acknowledgments
The study was supported by the National Natural
Science Foundation of China (grant no. 81370536) and the Hebei
Provincial Natural Science Fund (grant no. H2013206276).
Abbreviations:
|
PPARγ
|
peroxisome proliferator-associated
receptor γ
|
|
TLR4
|
Toll-like receptor 4
|
|
MyD88
|
myeloid differentiation primary
response gene 88
|
|
IKK-β
|
inhibitor of κB kinase-β
|
|
NF-κB
|
nuclear factor κB
|
|
JNK1
|
c-Jun N-terminal protein kinase-1
|
|
AP-1
|
activator protein-1
|
|
TNF-α
|
tumor-necrosis factor-α
|
|
IL-1β
|
interleukin-1β
|
|
MCP-1
|
monocyte chemoattractant protein-1
|
|
MIP-1α
|
macrophage inflammatory protein-1α
|
|
Col-1
|
collagen type I
|
|
α-SMA
|
α-smooth muscle actin
|
|
TGF-β1
|
transforming growth factor-β1
|
|
BAMBI
|
basic membrane protein and activin
membrane-bound inhibitor
|
|
GAPDH
|
glyceraldehydes 3-phosphate
dehydrogenase
|
References
|
1
|
Mitchell PS, Parkin RK, Kroh EM, Fritz BR,
Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant
KC, Allen A, et al: Circulating microRNAs as stable blood-based
markers for cancer detection. Proc Natl Acad Sci USA.
105:10513–10518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cohen JC, Horton JD and Hobbs HH: Human
fatty liver disease: Old questions and new insights. Science.
332:1519–1523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kelishadi R and Poursafa P: Obesity and
air pollution: Global risk factors for pediatric non-alcoholic
fatty liver disease. Hepat Mon. 11:794–802. 2011. View Article : Google Scholar
|
|
4
|
Söderberg C, Stål P, Askling J, Glaumann
H, Lindberg G, Marmur J and Hultcrantz R: Decreased survival of
subjects with elevated liver function tests during a 28-year
follow-up. Hepatology. 51:595–602. 2010. View Article : Google Scholar
|
|
5
|
Guo J and Friedman SL: Toll like receptor
4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis
Tissue Repair. 3:212010. View Article : Google Scholar
|
|
6
|
Fuentes E, Guzmán-Jofre L, Moore-Carrasco
R and Palomo I: Role of PPARs in inflammatory processes associated
with metabolic syndrome. Mol Med Rep. 8:1611–1616. 2013.PubMed/NCBI
|
|
7
|
Nan YM, Han F, Kong LB, Zhao SX, Wang RQ,
Wu WJ and Yu J: Adenovirus-mediated peroxisome proliferator
activated receptor gamma overexpression prevents nutritional
fibrotic steatohepatitis in mice. Scand J Gastroenterol.
46:358–369. 2011. View Article : Google Scholar
|
|
8
|
Nan YM, Fu N, Wu WJ, Liang BL, Wang RQ,
Zhao SX, Zhao JM and Yu J: Rosiglitazone prevents nutritional
fibrosis and steatohepatitis in mice. Scand J Gastroenterol.
44:358–365. 2009. View Article : Google Scholar
|
|
9
|
Ji Y, Liu J, Wang Z, Liu N and Gou W:
PPARgama agonist, rosiglitazone, regulates angiotensin II-induced
vascular inflammation through the TLR4-dependent signaling pathway.
Lab Invest. 89:887–902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leclercq IA, Lebrun VA, Stärkel P and
Horsmans YJ: Intrahepatic insulin resistance in a murine model of
steatohepatitis: Effect of PPARgamma agonist pioglitazone. Lab
Invest. 87:56–65. 2007. View Article : Google Scholar
|
|
11
|
Da Silva Morais A, Lebrun VA,
Abarca-Quinones J, Brichard S, Hue L, Guigas B, Viollet B and
Leclercq IA: Prevention of steatohepatitis by pioglitazone:
Implication of adiponectin-dependent inhibition of SREBP-1c and
inflammation. J Hepatol. 50:489–500. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ballestri S, Lonardo A and Loria P:
Nonalcoholic fatty liver disease activity score and Brunt's
pathologic criteria for the diagnosis of nonalcoholic
steatohepatitis: What do they mean and do they agree? Hepatology.
53:2142–2143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alkhouri N, De Vito R, Alisi A, Yerian L,
Lopez R, Feldstein AE and Nobili V: Development and validation of a
new histological score for pediatric non-alcoholic fatty liver
disease. J Hepatol. 57:1312–1318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
15
|
Estep M, Armistead D, Hossain N, Elarainy
H, Goodman Z, Baranova A, Chandhoke V and Younossi ZM: Differential
expression of miRNAsliver disease. Aliment Pharmacol Ther.
32:487–497. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang RQ, Nan YM, Wu WJ, Kong LB, Han F,
Zhao SX, Kong L and Yu J: Induction of heme oxygenase-1 protects
against nutritional fibrosing steatohepatitis in mice. Lipids
Health Dis. 10:312011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Berlanga A, Guiu-Jurado E, Porras JA and
Auguet T: Molecular pathways in non-alcoholic fatty liver disease.
Clin Exp Gastroenterol. 7:221–239. 2014.PubMed/NCBI
|
|
18
|
Hasenfuss SC, Bakiri L, Thomsen MK,
Williams EG, Auwerx J and Wagner EF: Regulation of steatohepatitis
and PPARγ signaling by distinct AP-1 dimers. Cell Metab. 19:84–95.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu E, Forest MP, Schwab M, Avramoglu RK,
St-Amand E, Caron AZ, Bellmann K, Shum M, Voisin G, Paquet M, et
al: Hepatocyte-specific Ptpn6 deletion promotes hepatic lipid
accretion, but reduces NAFLD in diet-induced obesity: Potential
role of PPARγ. Hepatology. 59:1803–1815. 2014. View Article : Google Scholar
|
|
20
|
Villalpando-Arteaga EV, Mendieta-Condado
E, Esquivel-Solís H, Canales-Aguirre AA, Gálvez-Gastélum FJ,
Mateos-Díaz JC, Rodríguez-González JA and Márquez-Aguirre AL:
Hibiscus sabdariffa L. aqueous extract attenuates hepatic steatosis
through down-regulation of PPAR-γ and SREBP-1c in diet-induced
obese mice. Food Funct. 4:618–626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Spruss A, Kanuri G, Wagnerberger S, Haub
S, Bischoff SC and Bergheim I: Toll-like receptor 4 is involved in
the development of frustose-induced hepatic steatosis in mice.
Hepatology. 50:1094–1104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wagnerberger S, Spruss A, Kanuri G,
Volynets V, Stahl C, Bischoff SC and Bergheim I: Toll-like
receptors 1–9 are elevated in livers with fructose-induced hepatic
steatosis. Br J Nutr. 107:1727–1738. 2012. View Article : Google Scholar
|
|
23
|
Shirai Y, Yoshiji H, Noguchi R, Kaji K,
Aihara Y, Douhara A, Moriya K, Namisaki T, Kawaratani H and Fukui
H: Cross talk between toll-like receptor-4 signaling and
angiotensin-II in liver fibrosis development in the rat model of
non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 28:723–730.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo J and Friedman SL: Toll-like receptor
4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis
Tissue Repair. 3:21–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szabo G, Velayudham A, Romics L Jr and
Mandrekar P: Modulation of non-alcoholic steatohepatitis by pattern
recognition receptors in mice: The role of toll-like receptors 2
and 4. Alcohol Clin Exp Res. 29(Suppl 11): S140–S145. 2005.
View Article : Google Scholar
|
|
26
|
Dauphinee SM and Karsan A:
Lipopolysaccharide signaling in endothelial cells. Lab Invest.
86:9–22. 2006. View Article : Google Scholar
|
|
27
|
Csak T, Velayudham A, Hritz I, Petrasek J,
Levin I, Lippai D, Catalano D, Mandrekar P, Dolganiuc A, Kurt-Jones
E and Szabo G: Deficiency in myeloid differentiation factor-2 and
toll-like receptor 4 expression attenuates nonalcoholic
steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest
Liver Physiol. 300:G433–G441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aoyama T, Paik YH and Seki E: Toll-like
receptor signaling and liver fibrosis. Gastroenterol Res Pract.
2010:pii: 192543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baghy K, Iozzo RV and Kovalszky I:
Decorin-TGFβ axis in hepatic fibrosis and cirrhosis. J Histochem
Cytochem. 60:262–268. 2012.PubMed/NCBI
|
|
30
|
Li YS, Ni SY, Meng Y, Shi XL, Zhao XW, Luo
HH and Li X: Angiotensin II facilitates fibrogenic effect of TGF-β1
through enhancing the down-regulation of BAMBI caused by LPS: A new
pro-fibrotic mechanism of angiotensin II. PLoS One. 8:e762892013.
View Article : Google Scholar
|
|
31
|
Saiman Y, Agarwal R, Hickman DA, Fausther
M, El-Shamy A, Dranoff JA, Friedman SL and Bansal MB: CXCL12
induces hepatic stellate cell contraction through a
calcium-independent pathway. 305:G375–G382. 2013.
|
|
32
|
Cynis H, Kehlen A, Haegele M, Hoffmann T,
Heiser U, Fujii M, Shibazaki Y, Yoneyama H, Schilling S and Demuth
HU: Inhibition of glutaminyl cyclases alleviates CCL2-mediated
inflammation of non-alcoholic fatty liver disease in mice. Int J
Exp Pathol. 94:217–225. 2013.PubMed/NCBI
|
|
33
|
Heinrichs D, Berres ML, Nellen A, Fischer
P, Scholten D, Trautwein C, Wasmuth HE and Sahin H: The chemokine
CCL3 promotes experimental liver fibrosis in mice. PLoS One.
8:e661062013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gressner AM: Transdifferentiation of
hepatic stellate cells (Ito cells) to myofibroblasts: A key event
in hepatic fibrogenesis. Kidney Int Suppl. 54:S39–S45.
1996.PubMed/NCBI
|