Introduction
Nephropathy, which is one of the most common
complications of diabetes, is characterized by proteinuria, renal
dysfunction, and end stage renal disease. However, the precise
mechanisms underlying the progression of diabetes mellitus-induced
renal injury remain unclear. Huang et al (1) reported that the expression of
angiotensin-converting enzyme (ACE) was significantly upregulated
in tubular epithelial cells and infiltrating mononuclear cells in
diabetic kidneys. In addition, diabetic kidneys exhibited
significantly increased chymase expression in mesangial cells and
vascular smooth muscle cells, and increased chymase deposition was
detected in the collagen-rich extracellular matrix (ECM) alongside
diffused and nodular glomerulosclerosis, tubulointerstitial
fibrosis, and vascular sclerosis (1). In a hamster model of unilateral
ureteral obstruction, treatment with a chymase inhibitor
significantly reduced angiotensin (Ang) II levels, significantly
decreased the mRNA expression levels of α-smooth muscle actin, type
I collagen and transforming growth factor (TGF)-β1 in renal tissue,
and appeared to ameliorate tubulointerstitial injury. However,
chymase inhibition did not alter systolic blood pressure, or the
protein levels of renal ACE and Ang II receptor type 1 (2).
As a chymotrypsin-like serine protease, chymase is
synthesized in mast cells, endothelial cells and mesenchymal cells.
Chymase is secreted directly into the interstitium, and is
responsible for the synthesis of ≤80% of Ang II in the human heart
(3). Chymase is inactivated in the
blood immediately after release, thus indicating that chymase is
only active in local tissues (4).
Human and hamster chymases have been reported to activate the
conversion of Ang I to Ang II, and contribute toward TGF-β1
activation (5), whereas rat
chymase activates TGF-β but not Ang II (6). In the present study, a rat model was
selected to determine the role of chymase in diabetes
mellitus-associated renal injury, without the Ang II effects.
Suc-Val-Pro-PheP-(OPh)2 [also
known as (OPh)2] specifically inhibits chymase without
affecting ACE activity, and has a degradation half-life of 20 h in
human plasma (half maximal inhibitory concentration=2.8 nmol/l)
(7). Therefore, (OPh)2
may act as a stable and strong chymase inhibitor in vivo
(8), and was used in the present
study to investigate the role of chymase in diabetic renal
injury.
Materials and methods
Materials
Male Sprague-Dawley rats (180–200 g) were purchased
from Beijing Vital River Laboratory Animal Technology Co., Ltd.
(Beijing, China). The chymase inhibitor (Oph)2 was
generously provided by Dr. Shinji Takai (Department of
Pharmacology, Osaka Medical College, Osaka, Japan). Monoclonal
mouse anti-fibronectin (FN; sc-8422), rabbit anti-type IV collagen
(ColIV; sc-11360) and mouse anti-TGF-β1 (sc-52893) antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
The horseradish peroxidase (HRP)-conjugated secondary antibody from
the EliVision™ Super kit were purchased from Fuzhou Maixin
Biotechnology Development Co., Ltd. (Fuzhou, China). The polyclonal
rabbit anti-vascular endothelial growth factor (VEGF; ab46154)
antibody was purchased from Abcam (Cambridge, UK). The Total RNA
extraction kit (TRIzol) was purchased from BioTeke Corporation
(Beijing, China), oligo-(dT) primers and Moloney Murine Leukemia
Virus (M-MLV) Reverse Transcriptase were purchased from SunBio
Corporation (Dongan-gu, Republic of Korea). The RNase inhibitor was
purchased from Takara Biotechnology Co., Ltd. (Dalian, China), and
the Polymerase Chain Reaction (PCR) Amplification kit (Taq) was
from Sangon Biotech Co., Ltd. (Shanghai, China).
Animal experiment
The rats were housed at 21±2°C at a temperature of
55±2% with a 12 h/12 h light cycle in a specific-pathogen-free
laboratory. The rats received standard rat chow and ad
libitum access to tap water. The rats were randomly divided
into the following groups: The control group (n=7), in which the
rats received an injection of 0.1 M acetate buffer; and the
diabetes (DM) group (n=7), in which the rats received a single
intraperitoneal (i.p.) injection of streptozotocin (STZ; 65 mg/kg
in 0.1 M acetate buffer; pH 4.4; Sigma-Aldrich, St. Louis, MO,
USA), in order to produce a diabetic rat model. A total of 72 h
after the STZ injection, non-fasting blood glucose levels were
measured with a portable glucose meter (Optium Xceed; Abbott
Laboratories Co., Ltd., Shanghai, China) using whole blood samples
obtained from snipped tails, subsequent to anesthesia with 40 mg/kg
intraperitoneal 2% pentobarbital sodium. The rats were considered
diabetic when blood glucose levels reached 16.7 mmol/l. Rats
treated with STZ were then intraperitoneally administered the
chymase inhibitor (OPh)2 (1 mg/kg daily), in order to
generate the DM + Chy-I group (n=10). Rats in the control and DM
groups were treated with the same volume of saline. The whole
experiment lasted 12 weeks. To avoid ketosis and acidosis and
improve long-term survival, non-fasting glycaemia was measured once
a week, and when serum glucose was greater than 25 mmol/l, rats in
the DM and DM + Chy-I groups received 2 U insulin (Humulin NPH;
Lilly Suzhou Pharmaceutical Group Co., Ltd., Suzhou, China)
subcutaneously. All related animal research was approved by the
Ethics Committee of the Peking University First Hospital (Beijing,
China).
At the end of the treatment period, all rats were
housed individually in metabolic cages for 24 h for the collection
of urine, in order to determine protein and creatinine clearance,
which were measured using the Hitachi 7150 Automatic Biochemical
Analyzer (Hitachi, Ltd., Tokyo, Japan). Blood pressure was measured
when the rats were conscious by the tail cuff method. All rats were
sacrificed by cervical dislocation after 12 weeks of
(OPh)2 treatment under 40 mg/kg intraperitoneal 2%
pentobarbital sodium anesthesia. Blood samples were obtained from
the abdominal aorta, and were used for serum biochemical analyses
using the Hitachi 7150 Automatic Biochemical Analyzer. Blood and
urine samples were collected into chilled tubes and were stored at
−80°C until further use. The left kidney was excised, weighed, and
cut into two halves; one half was fixed with 10% formalin and 2.5%
glutaraldehyde, and the other half was prepared for renal
morphological analysis. The right kidney was excised and maintained
at −80°C for mRNA extraction.
Immunohistochemical staining
Paraffin-embedded tissue sections (3 µm for
light microscopy; 4 µm for immunohistochemistry) were
dewaxed with dimethylbenzene (Beijing Yili Fine Chemicals Co.,
Ltd., Beijing, China), hydrated with ethanol, treated with 5%
H2O2, and were incubated in a microwave in
0.01 M citrate buffer/pepsin for antigen retrieval for 10 min.
Subsequently, the sections were rehydrated in phosphate-buffered
saline (PBS) for 15 min, and were blocked with goat serum (AR 0009;
Boster Biological Technology Co., Ltd., Pleasanton, CA, USA) at
room temperature for 20 min to reduce nonspecific binding. The
sections were then incubated with the following primary antibodies:
Mouse monoclonal anti-FN (1:300), mouse monoclonal anti-ColIV
(1:300), rabbit polyclonal anti-VEGF (1:200) and mouse monoclonal
anti-TGF-β1 (1:200), overnight at 4°C. After rinsing, the sections
were incubated with anti-mouse/rabbit HRP-conjugated secondary
antibodies from the EliVision™ Super kit for 30 min at room
temperature, followed by visualization with 0.05% diaminobenzidine.
The negative control sections underwent the same protocol; however,
primary antibodies were replaced with PBS. Images of the renal
tissue sections were digitally captured and quantitatively assessed
using a microscope (Leica DM1000; Leica Microsystems, Wetzlar,
Germany) with Leica QWin Pro V2.8 (Leica Microsystems), and
computer-assisted image analysis software (Image-Pro Plus software,
version 5.02; Media Cybernetics, Inc., Rockville, MD, USA). The
sections were coded (allocated a number) by an investigator;
however, the images were digitally captured and analyzed by a
different investigator who was unaware of the coding. Images of
each section were captured using the Leica DM1000 stereomicroscope
with a digital camera. The areas positive for the target proteins
were automatically calculated by the software, and the affected
areas were divided by the total area of microscopic fields. A total
of 20 glomeruli were examined for each rat, and the average
percentage of positive lesions was obtained for each animal. The
tubulointerstitial area did not include the area of the
glomeruli.
Reverse transcription PCR (RT-PCR)
The mRNA expression levels of FN, ColIV, TGF-β1,
VEGF and rat mast cell proteinase-1 (RMCP-1) were detected by
RT-PCR. β-actin was used as an internal standard. The expression
levels of the target genes were normalized to the levels of β-actin
in each sample. Subsequent to homogenization of the tissues, total
RNA was extracted from the frozen renal tissues using an RNA
extraction kit. Subsequently, the RNA (5 µg) underwent RT to
generate cDNA using Superscript II M-MLV reverse transcriptase (200
U/µl), oligo (dT) 12–18 primers (50 µM; 1 µl;
SunBio Corporation) and RNase-free ddH2O (7.5
µl). The RT reaction was conducted in a mixture containing
5X first-strand M-MLV buffer, 1 mmol/l dNTP and 20 mol/l
dithiothreitol RNase inhibitor (40 U/µl), at 42°C for 50
min. The PCR reaction mixture consisted of 1 µl cDNA, 20
pmol/l primers (SunBio Corporation), 2 µl 10X PCR buffer,
0.4 mmol/l dNTPs, and 2.5 U Taq polymerase. PCR was
performed using a Bioer PCR amplification instrument (GeneQ;
Hangzhou Bioer Technology Co., Ltd., Hangzhou, China) with the
following conditions: Pre-denaturation at 95°C for 5 min; followed
by 30 cycles of amplification at 95°C for 30 sec, annealing at 55°C
for 30 sec and extension at 72°C for 30 sec; and a final extension
step at 72°C for 5 min. The PCR primer sequences were as follows:
β-actin, sense 5′-CTG AACCCTAAGGCCAACC-3′, antisense
5′-CTGAACCCTAAGGCCAACC-3′ (309 bp); FN, sense
5′-CAGTTTGTGGAAGTGACCGA-3′, antisense 5′-TGGAGG TTAGTGGGAGCATA-3′
(272 bp); ColIV, sense 5′-GTGCGGTTTGTGAAGCACCG-3′, antisense
5′-GTTCTTCTC ATGCACACTTC-3′ (363 bp); TGF-β1, sense 5′-TATAGC
AACAATTCCTGGCG3 ′, antisense 5′-CAGAAG TTGGCATGGTAGCC-3′ (446 bp);
RMCP-1, sense 5′-GGA CCAGAACCAAGTGAGA-3′, antisense 5′-TTG
AAAGTGTTGAACCAGGA-3′ (328 bp); and VEGF, sense
5′-CAATTGAGACCCTGGTGGACAT-3′ and antisense
5′-TTGATCCGCATGATCTGCATAG-3′ (169 bp).
The PCR products were subsequently separated by
electrophoresis on 2% agarose gels, stained with ethidium bromide
(Santa Cruz Biotechnology, Inc.) and visualized by ultraviolet (UV)
transillumination (Clinx CUV 20 A; Shanghai Clinx Science
Instruments Co., Ltd., Shanghai, China). The ratio of the 2 DNA
bands (target DNA and β-actin) densities was measured using a Gel
Image Analysis system (AlphaImager; ProteinSimple, San Jose, CA,
USA) under UV light.
Statistical analysis
Statistical analyses were performed using SPSS for
Windows, version 11.5 (SPSS Inc., Chicago, IL, USA). All values are
presented as the mean ± standard deviation. Comparisons of data
between multiple groups were made using one-way analysis of
variance, followed by Student-Newman-Keuls. P<0.05 was
considered to indicate a statistically significant difference.
Results
Biochemical analysis
In the present study, the effects of chymase
inhibition were investigated on diabetic renal injury in an STZ rat
model. Preliminary experiments performed on the STZ-treated rats
determined that 12 weeks was the optimal treatment duration for
renal pathological analyses. No differences in serum creatinine,
blood pressure and triglyceride levels were detected among the
control, DM and DM + Chy-I groups. In the DM group, the kidney
weight/body weight ratio, blood glucose levels, urinary albumin
levels, low-density lipoprotein (LDL) levels and endogenous
creatinine clearance rate (CCr) were significantly increased
compared with in the control group. However, chymase inhibition had
no effect on these factors (Tables
I and II). CCr was elevated
in the 12-week STZ-treated rats, which is considered a symptom of
the glomerular hyperfiltration phase of diabetic nephropathy. The
total serum urinary albumin/creatinine ratio (ACR) and cholesterol
levels were significantly higher in the DM group compared with in
the control group. However, these factors were significantly
reduced in the DM + Chy-I group by 84.42 and 24.64%, respectively
(Tables I and II).
 | Table IComparison of blood pressure and
serum biochemical markers between the groups. |
Table I
Comparison of blood pressure and
serum biochemical markers between the groups.
| Group | Systolic pressure
(mmHg) | Diastolic pressure
(mmHg) | Blood glucose
(mmol/l) | Total
cholesterol(mmol/l) | HDL (mmol/l) | LDL (mmol/l) | Triglyceride
(mmol/l) | Albumin (g/l) |
|---|
| Control | 123.71±13.30 | 100.05±12.27 | 8.44±1.99 | 1.52±0.26 | 1.06±0.16 | 0.25±0.0.08 | 0.70±0.26 | 38.37±1.39 |
| DM | 135.71±11.16 | 93.37±19.95 | 48.40±12.13a | 2.31±0.30a | 1.18±0.12 | 0.40±0.09a | 2.12±1.28 | 34.11±3.00a |
| DM + Chy-I | 136.50±8.92 | 99.35±9.15 | 40.48±16.31 | 1.95±0.31a,b | 1.06±0.23 | 0.31±0.10 | 1.92±1.66 | 35.33±2.83a |
| Table IIComparison of urinary albumin,
urinary ACR and KW/BW between the groups. |
Table II
Comparison of urinary albumin,
urinary ACR and KW/BW between the groups.
| Group | Urinary albumin
(mg/24 h) | Urinary ACR
(mg/g) | Serum creatinine
(µmol/l) | CCr (ml/min) | KW/BW |
|---|
| Control | 1.20±0.97 | 70.21±40.30 | 25.54±2.33 | 0.45±0.36 | 3.67±0.40 |
| DM | 42.57±43.92a |
1,380.67±950.44a | 22.39±3.8 | 1.32±1.10a | 8.03±1.43a |
| DM + Chy-I | 17.88±12.20 |
340.34±150.23a,b | 23.36±4.1 | 1.34±0.59a | 7.7±1.321a |
Effects of chymase inhibition on ECM
deposition and RMCP-1 expression
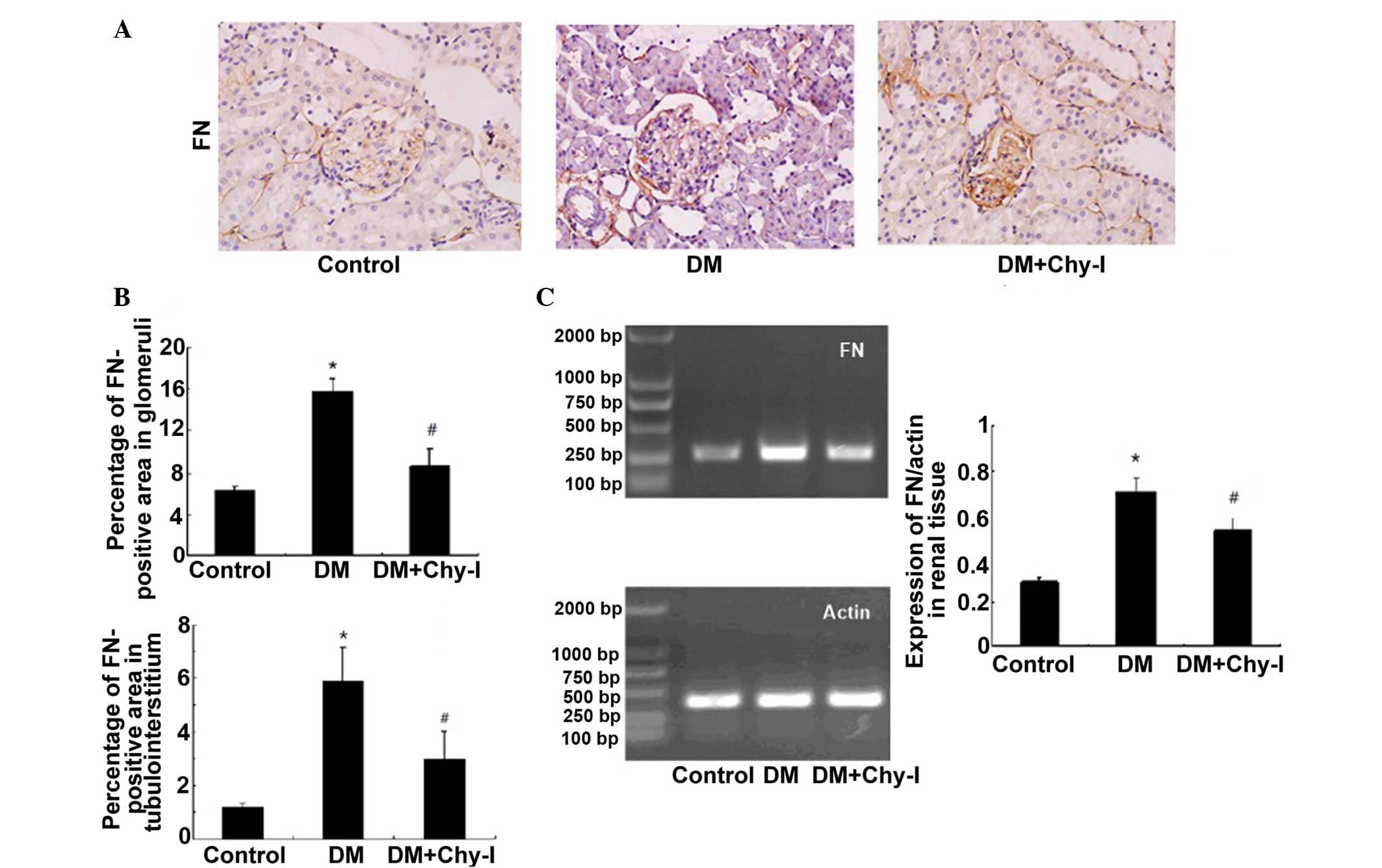
The present study investigated the expression of FN
and ColIV, which are two typical markers of diabetic renal injury.
Immunohistochemical analysis revealed that the protein expression
of FN was significantly increased in glomeruli and at the renal
tubulointerstitium in diabetic rats, as compared with in the
control rats (2.49-fold increase in glomeruli and 4.98-fold
increase in tubulointerstitium). However, in the DM + Chy-I group,
the intensity of FN staining was markedly decreased in these
regions (by 55.15% in glomeruli and 50.59% in tubulointerstitium)
(Fig. 1A and B). RT-PCR was also
performed to detect FN gene expression in the various groups
(Fig. 1C). Compared with in the
normal control group, FN expression exhibited a 2.45-fold increase
in the DM group, whereas chymase inhibition reduced FN expression
by 25.35%. These results indicate that chymase inhibition may limit
the expression of ECM components.
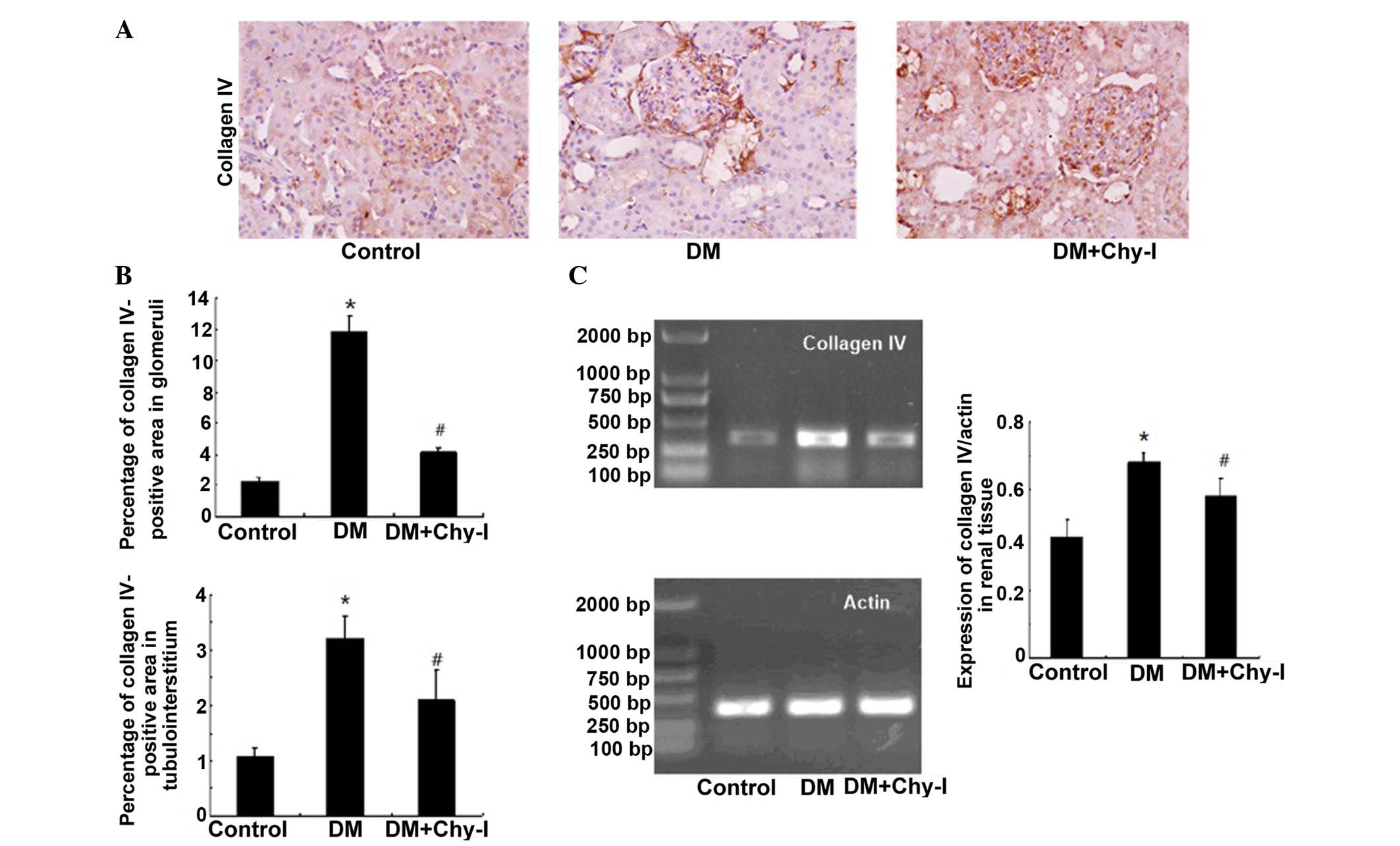
The previously discussed findings were confirmed by
detection of the renal expression of another main component of the
ECM, ColIV. In the DM group, ColIV was localized in the same
regions as in the control group; however, its expression was
significantly increased (5.23-fold increase in the glomerular
mesangial area and 3.00-fold increase in tubulointerstitium).
Treatment with a chymase inhibitor decreased the staining intensity
of ColIV (to 37.75% in the glomerular mesangial area and to 65.27%
in tubulointerstitium) (Fig. 2A and
B). RT-PCR analysis revealed that the expression levels of
ColIV were increased in the diabetic rat renal tissues by 1.61-fold
compared with in the control group, whereas chymase inhibition
downregulated the expression of ColIV by 17.24% (Fig. 2C). Quantitative analysis confirmed
that chymase inhibition reversed the deposition of ECM
components.
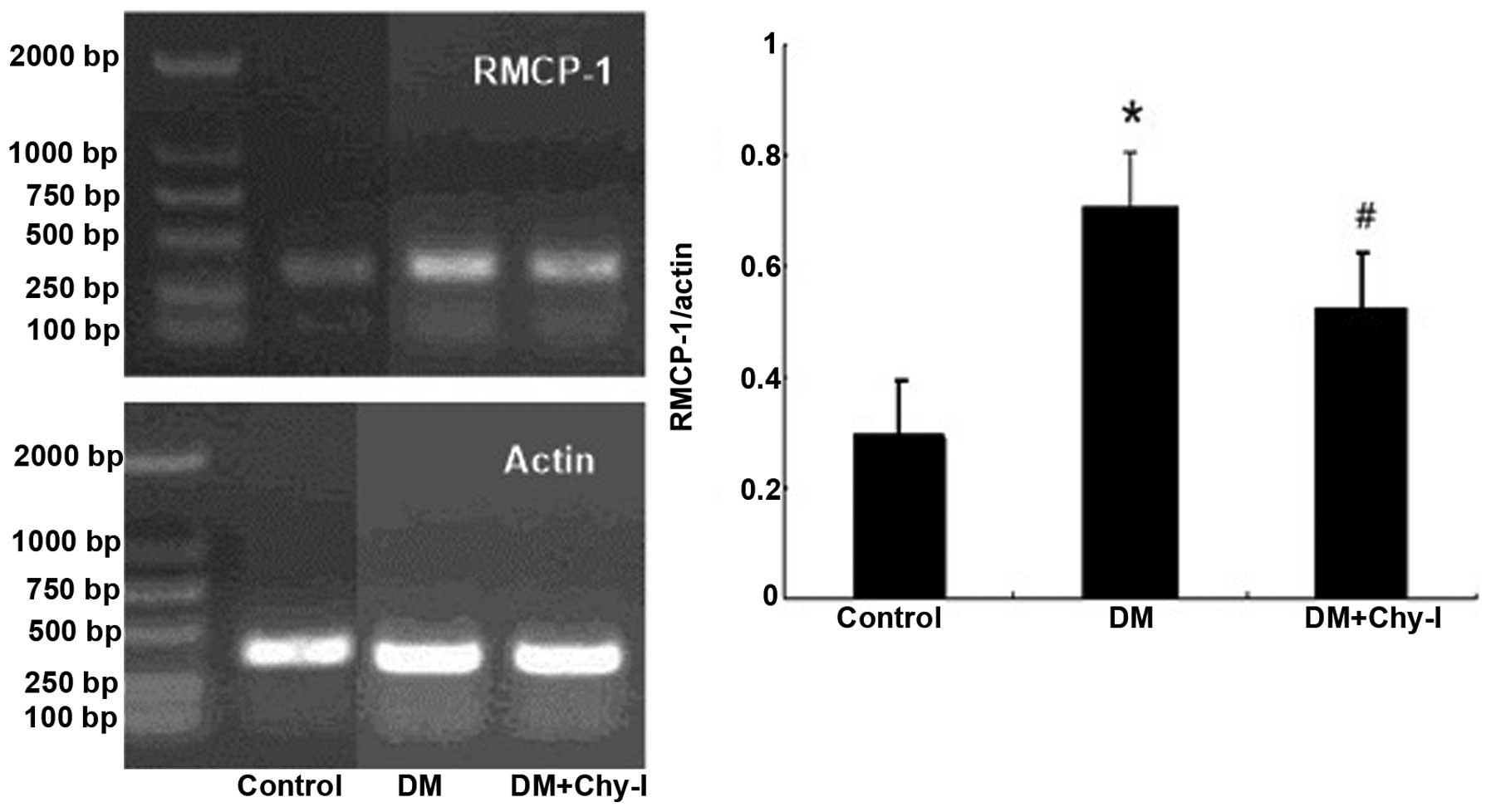
The expression levels of RMCP-1 were significantly
higher in the diabetic renal tissues compared with in the normal
renal tissues, as revealed by RT-PCR (0.288±0.026 vs. 0.709±0.054;
P<0.01). Furthermore, treatment with a chymase inhibitor reduced
the expression levels of RMCP-1 (Fig.
3).
Effects of chymase inhibition on the
expression of VEGF
The expression of VEGF was concentrated in renal
tubules. While, in normal glomeruli, VEGF was expressed
sporadically in epithelial cells. Immunostaining revealed increased
expression of VEGF protein in the kidneys of the DM group compared
with in the control group (10.52-fold increase in glomeruli and
4.03-fold increase in tubules). However, this overexpression was
inhibited following treatment with the chymase inhibitor
(OPh)2 (to 63.65% in glomeruli and to 38.35% in tubules)
(Fig. 4A and B). In addition, in
the DM group, the mRNA expression levels of VEGF were significantly
increased by 2.33-fold compared with in the control group.
Treatment with the chymase inhibitor (OPh)2 reversed the
increases in VEGF mRNA expression (Fig. 4C).
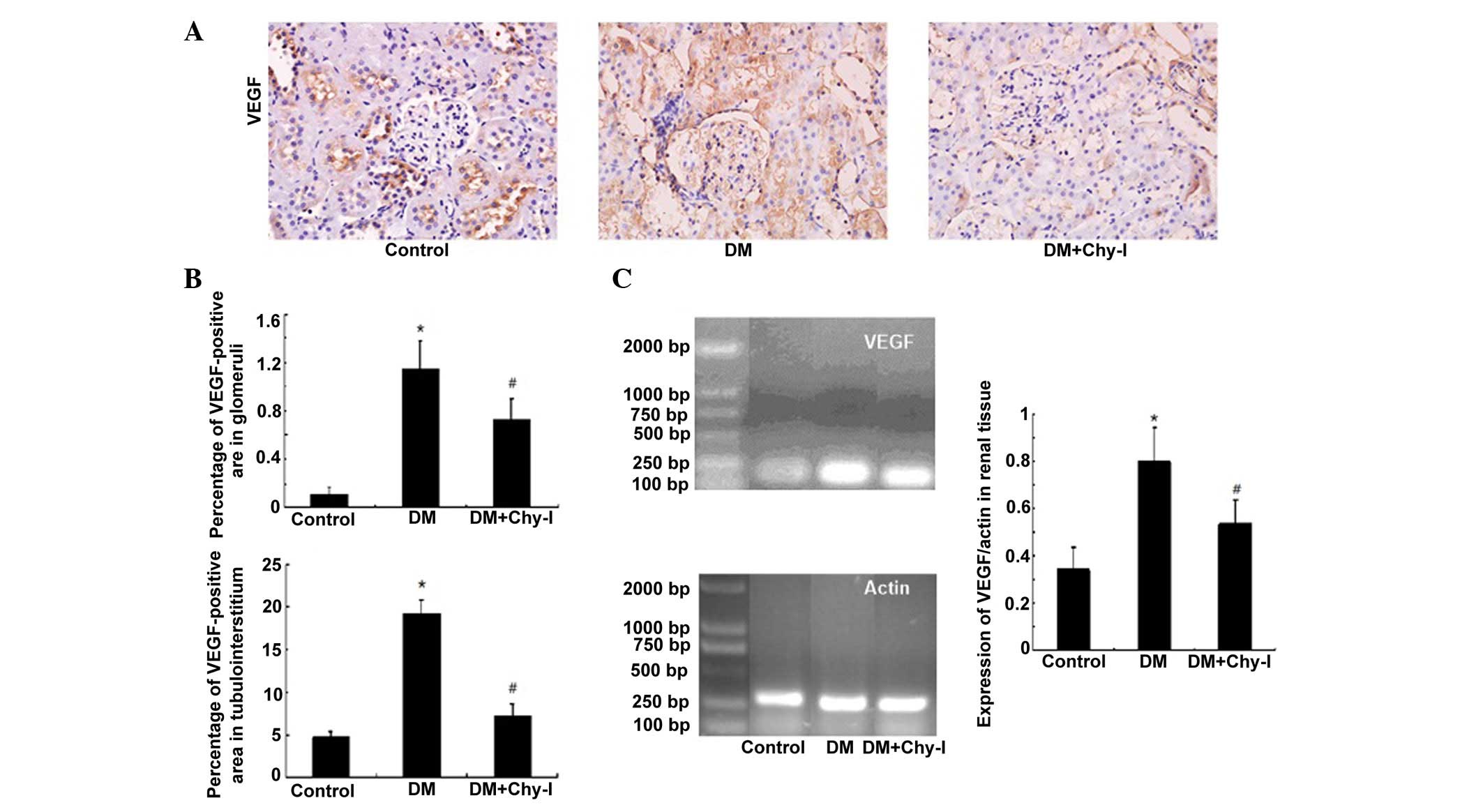 | Figure 4Chymase inhibition downregulated the
expression of VEGF in diabetic rat renal tissues. (A) Diabetic
kidneys exhibited increased VEGF immunostaining, which was reduced
by treatment with the chymase inhibitor (OPh)2
(magnification, ×400). (B) Percentage VEGF-positive area in the
glomeruli and tubulointerstitium of the various groups. Data are
presented as the mean ± standard deviation. *P<0.01,
vs. the control group; #P<0.01, vs. the DM group. (C)
Chymase inhibition downregulated the expression of VEGF, as
detected by reverse transcription polymerase chain reaction. Data
are presented as the mean ± standard deviation.
*P<0.01, vs. the control group;
#P<0.01, vs. the DM group. VEGF, vascular endothelial
growth factor; DM, diabetes group; DM + Chy-I, diabetes + chymase
inhibitor group. |
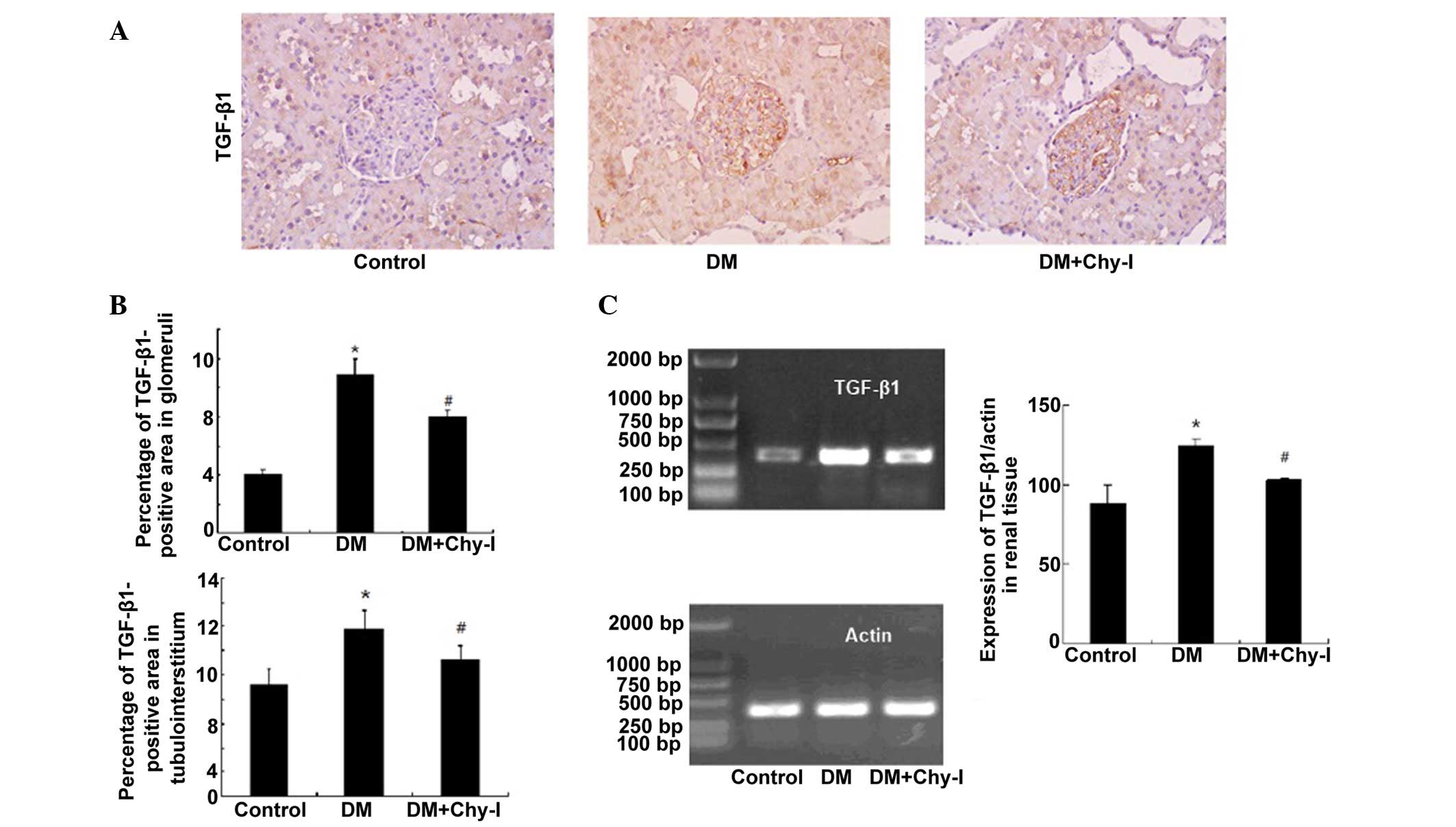
Effects of chymase inhibition on the
expression of TGF-β1
Immunohistochemical analysis revealed that TGF-β1
was upregulated in the glomerular mesangium and tubular cells of
the diabetic rats (2.69-fold increase in glomeruli and 1.51-fold
increase in tubulointerstitium) (Fig.
5A and B). Conversely, chymase inhibition reversed the
overexpression of TGF-β1 in diabetic rats (to 73.43% in glomeruli
and to 81.36% in tubulointerstitium). RT-PCR analysis revealed that
the renal tissues from the DM group exhibited increased TGF-β1 mRNA
expression, as compared with the control rats (82.22±11.35 vs.
124.15±4.32; P<0.05), whereas chymase inhibition reduced the
expression of TGF-β1 in diabetic renal tissues (124.15±4.32 vs.
103.07± 6.30; P<0.05) (Fig.
5C).
Discussion
The present study established an in vivo
model to explore the potential preventive effects of the chymase
inhibitor (OPh)2 on renal lesions in diabetic rats. In a
previous study, almost all renal mast cells in patients with
diabetic nephropathy were shown to produce chymase (9). The present study determined the
expression of chymase in rat renal tissues. The expression levels
of the chymase encoding gene, RMCP-1, were markedly enhanced in
diabetic kidneys. A previous study demonstrated that the mRNA
expression levels of chymase were upregulated in glucose-stimulated
mesangial cells (10). Mast cells
are thought to be the main source of chymase; however, the present
study failed to identify abundant mast cells in renal tissues by
toluidine blue staining (data not shown). This result is consistent
with the findings of a previous study, in which Cristovam et
al (10) demonstrated that
mast cells were absent in both control and diabetic kidneys, thus
suggesting that chymase is synthesized by resident renal cells.
Disordered lipid metabolism is a common feature of
diabetes mellitus. In the present study, the chymase inhibitor
(OPh)2 lowered cholesterol levels in diabetic rats via
an unknown mechanism. Consistent with these findings, it has been
previously reported that arterial chymase is activated when
cholesterol levels are elevated in animals fed a high-lipid diet
(11). Mast cell chymase actively
degrades high-density lipoprotein and thus generates defective
particles that are unable to initiate cholesterol efflux from the
arterial wall (12).
The majority of patients with diabetic nephropathy
develop hypertension, alongside proteinuria. In the present study,
no obvious differences were observed in blood pressure between the
rat groups. (OPh)2 lowered urinary ACR in diabetic rats
but did not affect blood pressure or blood glucose levels, thus
indicating that this chymase inhibitor exerts its effects on renal
lesions independent of hypertension and hyperglycemia.
FN and ColIV are the main components of the ECM. ECM
deposition occurs in the early stage of renal disease. An
indication of ECM overaccumulation is abnormally increased FN
levels, which is a main feature of glomerular sclerosis and
interstitial fibrosis. In addition, an imbalance between ColIV
synthesis and degradation directly affects ECM deposition, and
abnormally high levels of ColIV in patients with renal disease have
been closely associated with the severity of diabetic nephropathy
(13).
In vitro, rat and human chymases have been
reported to degrade FN, which has an important role in the
attachment of cultured vascular smooth muscle cells (14). Conversely, chymase inhibition has
been reported to downregulate the expression of FN and ColIV in
animal models of ischemia and fibrosis (15,16).
These results, together with the findings of the present study,
suggested that chymase may participate in ECM deposition in
diabetes-induced renal fibrosis, and that chymase inhibition could
attenuate the progression of diabetic nephropathy. These findings
may also explain the reduction in urinary protein secretion induced
by chymase inhibition.
VEGF has important roles in blood vessel growth,
endothelial survival and microvasculature maintenance. VEGF is
abundantly expressed in glomerular epithelial cells, also known as
podocytes, and tubular epithelial cells, whereas the glomerular and
peritubular capillary endothelial cells express VEGF receptors
(17). VEGF renal expression is
decreased in several animal models of kidney disease, and VEGF
administration is protective (18). However, plasma VEGF levels in
patients with diabetic nephropathy are increased, and suppressing
VEGF has been shown to improve diabetic nephropathy in animal
models (19–22). In diabetic models, VEGF secretion
and the expression of its receptors are increased by high glucose
concentrations in the kidneys (23). In addition, patients with diabetes
and overt proteinuria express higher levels of VEGF, and excretion
of urinary VEGF is increased in those with a higher degree of
proteinuria (24). In the present
study, diabetic rats exhibited higher urinary ACR and increased
VEGF expression in renal tissues, which is consistent with the
findings of previous animal studies and clinical observations
(23,24). Chymase inhibition reduced urinary
protein excretion and the mRNA expression levels of VEGF in
diabetic rats, thus suggesting that the chymase inhibitor may
decrease urinary protein excretion and renal lesions via its
inhibition of VEGF. It has previously been reported that the
chymase-Ang II-VEGF pathway may operate in granulation tissues as
the primary mediator of angiogenesis (25). However, the results of the present
study did not indicate the involvement of this pathway in VEGF
regulation of chymase. Chymase may induce VEGF via the VEGF
paracrine loop in glomeruli, as reported previously in
podocyte-specific VEGF-deficient mice (26). VEGF itself stimulates collagen and
FN expression in mesangial cells and, in turn, enhances
TGF-β1-induced ECM accumulation (27). TGF-β1 and VEGF interact with each
other, and increased secretion of VEGF, induced by TGF-β1, may be a
possible explanation for the role of TGF-β1 in the development of
albuminuria (28).
Chymase strongly promotes accumulation of
inflammatory cells, and also converts TGF-β and matrix
metalloproteinase-9 precursors to their active forms. These
multiple functions of chymase may have an important role in the
development and promotion of various types of disease (29). TGF-β1 is a critical cytokine in
cell proliferation, ECM deposition and fibrosis. It stimulates
glomerular mesangial cells and tubular epithelial cells, and
enhances the expression of ColI, III, IV and other non-collagen
glycoproteins (30,31). In the present study, chymase
inhibition depressed the upregulation of TGF-β1 in diabetic rats,
alongside urinary albumin secretion. These results indicated that
TGF-β1 may have an important role in chymase-induced renal damage.
It has previously been reported that chymase converts the TGF-β1
precursor to its active form (15). Autocrine TGF-β1 may also have a
critical role in the growth and basal ColIV production of renal
tubular epithelial cells (14,32).
Increased levels of TGF-β1 have been observed in both clinical and
experimental models of diabetes, and the deleterious effects of
high glucose levels are attributed primarily to the autocrine
action of TGF-β1 (33). Autocrine
TGF-β1 signaling in proliferating proximal tubule cells usually
exceeds the levels that are necessary for physiological
regeneration (34), thus
indicating that chymase inhibition may downregulate TGF-β1
expression by depressing its activation, as shown in the present
study.
In conclusion, the results of the present study
indicated that chymase may participate in diabetic renal fibrosis
by inducing excessive ECM deposition, probably via its regulation
of TGF-β1 and VEGF. The chymase inhibitor (OPh)2
reversed the effects of chymase as a fibrogenic factor by
restraining the expression of TGF-β1 and VEGF. In addition, chymase
inhibition resulted in a reduced urinary ACR and alleviated
diabetic renal fibrosis. These results indicated the potential of
chymase inhibitors for the treatment of diabetic nephropathy;
however, the mechanisms underlying the regulation of VEGF and
TGF-β1 by chymase require further investigation.
Acknowledgments
The present study was supported by the Basic
Clinical Cooperation Foundation of Capital Medical University
(grant no. 11JL41).
References
|
1
|
Huang XR, Chen WY, Truong LD and Lan HY:
Chymase is upregulated in diabetic nephropathy: Implications for an
alternative pathway of angiotensin II-mediated diabetic renal and
vascular disease. J Am Soc Nephrol. 14:1738–1747. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan YY, Nishiyama A, Fujisawa Y, Kobori H,
Nakano D, Matsuura J, Hase N, Hitomi H, Kiyomoto H, Urata H and
Kohno M: Contribution of chymase-dependent angiotensin II formation
to the progression of tubulointerstitial fibrosis in obstructed
kidneys in hamsters. J Pharmacol Sci. 111:82–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Urata H, Kinoshita A, Misono KS, Bumpus FM
and Husain A: Identification of a highly specific chymase as the
major angiotensin II-forming enzyme in the human heart. J Biol
Chem. 265:22348–22357. 1990.PubMed/NCBI
|
|
4
|
Miyazaki M and Takai S: Tissue angiotensin
II generating system by angiotensin-converting enzyme and chymase.
J Pharmacol Sci. 100:391–397. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takai S, Sakonjo H, Fukuda K, Jin D,
Sakaguchi M, Kamoshita K, Ishida K, Sukenaga Y and Miyazaki M: A
novel chymase inhibitor,
2-(5-formylamino-6-oxo-2-phenyl-1,6-dihydropyrimidine-1-yl)-N-[[,4-dioxo-1-phenyl-7-(2-pyridyloxy)]
2-heptyl]acetamide (NK3201), suppressed intimal hyperplasia after
balloon injury. J Pharmacol Exp Ther. 304:841–844. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Okamoto Y, Takai S and Miyazaki M: Effect
of chymase-dependent transforming growth factor beta on peritoneal
adhesion formation in a rat model. Surg Today. 34:865–867. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oleksyszyn J and Powers JC: Irreversible
inhibition of serine proteases by peptide derivatives of
(alpha-aminoalkyl) phosphonate diphenyl esters. Biochemistry.
30:485–493. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takai S, Yuda A, Jin D, Nishimoto M,
Sakagichi M, Sasaki S and Miyazaki M: Inhibition of chymase reduces
vascular proliferation in dog grafted veins. FEBS Lett.
467:141–144. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ninichuk V, Khandoga AG, Segerer S,
Loetscher P, Schlapbach A, Revesz L, Feifel R, Khandoga A, Krombach
F, Nelson PJ, et al: The role of interstitial macrophages in
nephropathy of type 2 diabetic db/db mice. Am J Pathol.
170:1267–1276. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cristovam PC, Carmona AK, Arnoni CP,
Maquigussa E, Pereira LG and Boim MA: Role of chymase in diabetic
nephrology. Exp Biol Med (Maywood). 237:985–992. 2012. View Article : Google Scholar
|
|
11
|
Uehara Y, Urata H, Ideishi M, Arakawa K
and Saku K: Chymase inhibition suppresses high-cholesterol
diet-induced lipid accumulation in the hamster aorta. Cardiovasc
Res. 55:870–876. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kovanen PT: Mast cells: Multipotent local
effector cells in atherothrombosis. Immunol Rev. 217:105–122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ruiz-Torres MP, López-Ongil S, Griera M,
Díez-Marqués ML, Rodríguez-Puyol M and Rodríguez-Puyol D: The
accumulation of extracellular matrix in the kidney: Consequences on
cellular function. J Nephrol. 18:334–340. 2005.PubMed/NCBI
|
|
14
|
Leskinen MJ, Lindstedt KA, Wang Y and
Kovanen PT: Mast cell chymase induces smooth muscle cell apoptosis
by a mechanism involving fibronectin degradation and disruption of
focal adhesions. Arterioscler Thromb Vasc Biol. 23:238–243. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanemitsu H, Takai S, Tsuneyoshi H,
Nishina T, Yoshikawa K, Miyazaki M, Ikeda T and Komeda M: Chymase
inhibition prevents cardiac fibrosis and dysfunction after
myocardial infarction in rats. Hypertens Res. 29:57–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maeda Y, Inoguchi T, Takei R, Sawada F,
Sasaki S, Fujii M, Kobayashi K, Urata H, Nishiyama A and Takayanagi
R: Inhibition of chymase protects against diabetes-induced
oxidative stress and renal dysfunction in hamsters. Am J Physiol
Renal Physiol. 299:F1328–F1338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Simon M, Röckl W, Hornig C, Gröne EF,
Theis H, Weich HA, Fuchs E, Yayon A and Gröne HJ: Receptors of
vascular endothelial growth factor/vascular permeability factor
(VEGF/VPF) in fetal and adult human kidney: Localization and
[125I]VEGF binding sites. J Am Soc Nephrol. 9:1032–1044.
1998.PubMed/NCBI
|
|
18
|
Kang DH, Joly AH, Oh SW, Hugo C,
Kerjaschki D, Gordon KL, Mazzali M, Jefferson JA, Hughes J, Madsen
KM, et al: Impaired angiogenesis in the remnant kidney model: I.
Potential role of vascular endothelial growth factor and
thrombospondin-1. J Am Soc Nephrol. 12:1434–1447. 2001.PubMed/NCBI
|
|
19
|
Tsuchida K, Makita Z, Yamagishi S, Atsumi
T, Miyoshi H, Obara S, Ishida M, Ishikawa S, Yasumura K and Koike
T: Suppression of transforming growth factor beta and vascular
endothelial growth factor in diabetic nephropathy in rats by a
novel advanced glycation end product inhibitor, OPB-9195.
Diabetologia. 42:579–588. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cha DR, Kim NH, Yoon JW, Jo SK, Cho WY,
Kim HK and Won NH: Role of vascular endothelial growth factor in
diabetic nephropathy. Kidney Int Suppl. 77:S104–S112. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Vriese AS, Tilton RG, Elger M, Stephan
CC, Kriz W and Lameire NH: Antibodies against vascular endothelial
growth factor improve early renal dysfunction in experimental
diabetes. J Am Soc Nephrol. 12:993–1000. 2001.PubMed/NCBI
|
|
22
|
Schrijvers BF, Flyvbjerg A, Tilton RG,
Lameire NH and De Vriese AS: A neutralizing VEGF antibody prevents
glomerular hypertrophy in a model of obese type 2 diabetes, the
Zucker diabetic fatty rat. Nephrol Dial Transplant. 21:324–329.
2006. View Article : Google Scholar
|
|
23
|
Cooper ME, Vranes D, Youssef S, Stacker
SA, Cox AJ, Rizkalla B, Casley DJ, Bach LA, Kelly DJ and Gilbert
RE: Increased renal expression of vascular endothelial growth
factor (VEGF) and its receptor VEGFR-2 in experimental diabetes.
Diabetes. 48:2229–2239. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hovind P, Tarnow L, Oestergaard PB and
Parving HH: Elevated vascular endothelial growth factor in type 1
diabetic patients with diabetic nephropathy. Kidney Int Suppl.
75:S56–S61. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Katada J, Muramatsu M, Hayashi I, Tsutsumi
M, Konishi Y and Majima M: Significance of vascular endothelial
cell growth factor upregulation mediated via a
chymase-angiotensin-dependent pathway during angiogenesis in
hamster sponge granulomas. J Pharmacol Exp Ther. 302:949–956. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eremina V, Sood M, Haigh J, Nagy A, Lajoie
G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH and Quaggin SE:
Glomerular-specific alterations of VEGF-A expression lead to
distinct congenital and acquired renal diseases. J Clin Invest.
111:707–716. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang L, Kwak JH, Kim SI, He Y and Choi ME:
Transforming growth factor-beta1 stimulates vascular endothelial
growth factor 164 via mitogen-activated protein kinase kinase
3-p38alpha and p38delta mitogen-activated protein kinase-dependent
pathway in murine mesangial cells. J Biol Chem. 279:33213–33219.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li X, Hu J, Zhang Q, Sun X and Li S:
Urocortin 1 improves renal function in rats with
streptozotocin-induced diabetes by inhibiting overproduction of
TGF-beta 1 and VEGF. Br J Pharmacol. 157:994–1003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takai S, Jin D and Miyazaki M: Multiple
mechanisms for the action of chymase inhibitors. J Pharmacol Sci.
118:311–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Khera TK, Martin J, Riley SG, Steadman R
and Phillips AO: Glucose modulates handling of apoptotic cells by
mesangial cells: Involvement of TGF-beta1. Lab Invest. 87:690–701.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Umezono T, Toyoda M, Kato M, Miyauchi M,
Kimura M, Maruyama M, Honma M, Yagame M and Suzuki D: Glomerular
expression of CTGF, TGF-beta 1 and type IV collagen in diabetic
nephropathy. J Nephrol. 19:751–757. 2006.PubMed/NCBI
|
|
32
|
Grande JP, Warner GM, Walker HJ, Yusufi
AN, Cheng J, Gray CE, Kopp JB and Nath KA: TGF-beta1 is an
autocrine mediator of renal tubular epithelial cell growth and
collagen IV production. Exp Biol Med (Maywood). 227:171–181.
2002.
|
|
33
|
Shankland SJ, Scholey JW, Ly H and Thai K:
Expression of transforming growth factor-beta 1 during diabetic
renal hypertrophy. Kidney Int. 46:430–442. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Geng H, Lan R, Wang G, Siddiqi AR, Naski
MC, Brooks AI, Barnes JL, Saikumar P, Weinberg JM and Venkatachalam
MA: Inhibition of autoregulated TGF-beta signaling simultaneously
enhances proliferation and differentiation of kidney epithelium and
promotes repair following renal ischemia. Am J Pathol.
174:1291–1308. 2009. View Article : Google Scholar : PubMed/NCBI
|