Introduction
Atrial fibrillation (AF) is the most common cardiac
arrhythmia (1,2). The majority of patients with AF have
underlying cardiovascular diseases, including valvular heart
disease, coronary artery disease, cardiomyopathy and diabetes
mellitus (3). A number of studies
have indicated that inflammation serves an important role in the
pathogenesis of AF (4–6). Patients with AF have been identified
to have an elevated level of serum inflammatory markers including
C-reactive protein (CRP), interleukin 6 (IL-6), tumor necrosis
factor α (TNF-α) and monocyte chemoattractant protein-1 (MCP-1)
(5,7,8).
Histological studies have demonstrated that inflammatory cell
infiltration increases in the atrial myocardium of patients with AF
(6,9). Treatment with anti-inflammatory
agents has been demonstrated to decrease the recurrence and
perpetuation of AF (10,11). In addition, alterations in the
expression of connexins and remodeling is involved in the
pathogenesis of AF (12). Connexin
43 (Cx43) is one of the major connexin isotypes in the atrial
myocardium (13). Previous studies
have indicated that there is a link between Cx43-mediated
gap-junction coupling and atrial arrhythmias (14–16).
Under physiological conditions, Cx43 is localized in intercalated
disks between atrial myocytes. Lateralization of Cx43 has been
observed in patients with AF (14)
and in the chronic pressure overload-induced AF animal model
(17). However, it remains unclear
whether Cx43 is involved in inflammation-induced atrial
fibrillation.
There is an increasing body of evidence that
demonstrates that there may be cross-talk between α-adrenergic
receptor (α-AR) signaling and the immune system during
inflammation. Flierl et al (18) reported that phagocytes are capable
of de novo production of catecholamines when phagocytes are
exposed to lipopolysaccharides (LPS), producing a blockade of
α2-AR, which suppressed lung inflammation. A significant increase
in α1-AR expression has been observed in human periodontal ligament
fibroblasts following LPS pretreatment and blocking α1-AR signaling
prevents the upregulation of inflammatory-associated cytokines
(19). In high altitude native
rats, blockage of the α-AR also induced a complete decrease in
inflammatory mediators (20).
Therefore, it was hypothesized that α1-adrenergic activation may be
involved in inflammation-induced AF.
In the present study, dogs were administrated a low
dose of LPS for 2 weeks to mimic the chronic low-grade system of
inflammation. The roles of Cx43 and α1-AR in inflammation-induced
AF were then investigated.
Materials and methods
Animals
A total of 20 healthy male beagles (weight, 10–12
kg; age, 12–15 months old) from the Experimental Animal Center of
Hangzhou Normal University (Hangzhou, Zhejiang, China), with no
prior symptoms of inflammation or AF, were used. The dogs were
housed in individual cages in a controlled room (18 to 24°C with a
12/12 h light/dark cycle) for 2 weeks prior to the experiment and
were given free access to food and water. The investigation
conformed to the Guide for the Care and Use of Laboratory Animals
published by the US National Institutes of Health (21). All experimental protocols were
approved by the Ethics Committee on Experimental Animal Center of
Hangzhou Normal University (Hangzhou, China).
Drugs and solutions
LPS (derived from Escherichia coli 055:B5)
and doxazosin were purchased from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). Sodium pentobarbital was purchased from
Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Cx43 (cat.
no. #3512), nuclear factor κB (NF-κB) p65 (cat. no. #8242), histone
H3 (cat. no. #9717) and β-actin (cat. no. #4970) antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Toll-like receptor 4 (TLR4) antibody (cat. no. LS-C44226) was
obtained from LifeSpan BioSciences, Inc. (Seattle, WA, USA). The
enzyme-linked immunosorbent assay (ELISA) kits for TNF-α (cat. no.
CATA00) and IL-6 (cat. no. CA6000) were from R&D Systems, Inc.
(Minneapolis, MN, USA).
Experimental protocol
The dogs were divided into four groups of 5 dogs
each. Dogs in the control group were injected with the same volume
of vehicle [0.9% NaCl, 0.2 ml/kg, intraperitoneal (i.p.)] and fed
empty capsules once a day for 2 weeks. Dogs in the LPS group
received LPS (0.1 µg/kg in 0.9% NaCl, i.p.) and fed empty capsules
once a day for 2 weeks. In the LPS + doxazosin group, the dogs were
fed α1-AR antagonist doxazosin (0.2 mg/kg) in the form of a capsule
5 min following LPS injection. Dogs in the doxazosin group were fed
a capsule containing doxazosin (0.2 mg/kg) 5 min following an i.p.
injection of 0.9% NaCl.
Vital signs monitoring
Vital signs (i.e., rectal temperature, heart rates
and blood pressure) were observed and recorded at time 0 h (prior
to the experiment) and 3, 6, 12 and 24 h following each injection
of LPS. For noninvasive measurement of blood pressure, a cuff was
placed on the right femoral region and connected to a commonly used
noninvasive blood pressure monitor. Heart rate was measured by
palpations of the femoral pulse.
Measurement of systemic TNF-α and IL-6
levels
Blood (2 ml) was collected in EDTA-coated tubes at 3
h and then 1, 4, 7 and 14 days post-treatment with the first
injection of LPS. The plasma was separated by centrifugation at
2,000 × g for 10 min at room temperature, and then stored at −80°C
until analysis. The systemic TNF-α and IL-6 levels were determined
by Canine TNF-α Quantikine ELISA kit (cat. no. CATA00) and Canine
IL-6 Quantikine ELISA kit (cat. no. CA6000) according to the
manufacturer's instructions (R&D Systems, Inc.).
Animal preparation
Following 2 weeks of the different treatment, the
dogs were fasted for ≥10 to 12 h and then anesthetized with sodium
pentobarbital (30 mg/kg, i.p.). A multi-electrode catheter (Cordis
Webster, Inc.; Biosense Webster, Inc.; Johnson & Johnson, New
Brunswick, NJ, USA) was introduced from the right external jugular
vein and was placed in the right atrial appendage to record right
atrial electrograms and for atrial pacing. The pacing and recording
leads were connected to a cardiac electrophysiology stimulator
(model DF-5A; Suzhou Dongfang Electronic Instrument Factory,
Shuzhou, China) and a multichannel electrophysiological recording
system (model TOP-2001; Shanghai Hongtong Industrial Co., Shanghai,
China). ECGs were recorded with the use of bipolar percutaneous
electrodes placed in each of the dogs' four limbs. Body temperature
was maintained at 36.5±1.5°C using a heating pad situated under the
dog. Anesthesia was maintained with 6 mg/kg of sodium pentobarbital
i.p. administration hourly.
Programmed stimulation
Programmed stimulation was used to determine the
atrial effective refractory periods (ERP) and window of
vulnerability (WOV). The right atrium was paced at an atrial pacing
cycle length of 300 msec; electrical pacing was repeated every 300
msec with each pace lasting 0.5 msec in duration. The ERP at 2x, 4x
and 10x diastolic threshold was determined by programmed
stimulation of the right atrial appendage, which consisted of 8
consecutive stimuli (S1S1=300 msec) followed by a premature
stimulus (S1S2). The S1S2 intervals were decreased from 200 msec to
refractoriness by decrements of 2 msec. As the S1S2 intervals
approached the ERP, decrements were reduced to 1 msec. The atrial
ERP was defined as the longest S1-S2 interval that failed to induce
atrial depolarization (22).
The WOV was used as a quantitative measurement of AF
inducibility. AF was defined as irregular atrial rates faster than
500 bpm associated with irregular atrioventricular conduction
lasting longer than 5 sec. During ERP measurements, if AF was
induced by decremental S1S2 stimulation, the difference between the
longest and shortest S1-S2 interval (in msec) at which AF was
induced was defined as the WOV. The cumulative WOV was the sum of
the individual WOVs determined at 2x, 4x and 10x threshold levels
in each dog (23).
The ability of the atria to develop sustained AF was
also analyzed by burst pacing at 10x threshold (S1S1=100 msec and
50 msec) for 120 sec. Sustained AF was defined as a fast irregular
rhythm that lasted for >60 sec following cessation of burst
pacing (24).
Cardiac TNF-α and IL-6 content
analyses
Following the measurements for AF inducibility,
right atrial tissues from all 20 dogs were harvested and
homogenized thoroughly on ice in a lysis buffer (50 mM Tris-HCl,
0.1 mM EDTA-2Na, 1 mM sucrose, 0.8% sodium chloride, pH 7.4). The
homogenates were centrifuged at 12,000 × g and 4°C for 10 min, and
the supernatant was collected and stored at −80°C until analysis.
The TNF-α and IL-6 levels in the supernatant were measured using
the Canine TNF-α Quantikine ELISA kit (cat. no. CATA00) and Canine
IL-6 Quantikine ELISA kit (cat. no. CA6000) according to the
manufacturer's instruction (R&D Systems, Inc.). Protein
concentration of the supernatant was determined using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology, Haimen, China) according to the manufacturer's
instructions. The cardiac TNF-α and IL-6 contents were expressed as
picograms per milligram of protein.
Western blot analysis
Total proteins were obtained from right atrial
myocardium by homogenization in ice-cold radioimmunoprecipitation
lysis solution (Cell Signaling Technology, Inc.) containing 1%
Triton X-100, phosphatase, protease inhibitors and PMSF. Nuclear
protein extracts were obtained using a Nuclear and Cytoplasmic
Protein Extraction kit and quantified using a bicinchoninic kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Equal amounts of protein (20 µg) from
each sample were separated by 10% SDS-PAGE and transferred to a
nitrocellulose membrane (EMD Millipore, Billerica, MA, USA). The
membranes were blocked for 1 h with 5% bovine serum albumin (BSA)
at room temperature, then incubated overnight at 4°C with the
primary antibodies, including anti-Cx43 (1:1,000), anti-TLR4
(1:500), anti-NF-κB p65 (1:1,000), anti-β-actin (1:1,000) and
anti-histone H3 (1:1,000). Following washing using TBS-T
(Tris-buffered saline with 0.1% Tween-20), the membrane was
incubated with a horseradish peroxidase-conjugated secondary
antibody [1:1,000; goat anti-rabbit IgG (cat. no. #7074) or horse
anti-mouse IgG (cat. no. #7076); Cell Signaling Technology, Inc.]
for 45 min at room temperature. All reactions were detected using
an enhanced chemiluminescence kit (Beyotime Institute of
Biotechnology) according to manufacturer's instructions. The
experiment was repeated three times. The band intensities were
analyzed with Quality One software (version 4.6.2; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Immunohistochemistry
Immunohistochemistry staining was used to determine
the localization of Cx43 in the atria. Each tissue section (5-µm
thick) was deparaffinized, rehydrated and blocked with 5% BSA for 1
h at room temperature. Then, the sections were incubated with the
anti-Cx43 antibody (dilution, 1:500) overnight at 4°C, followed by
incubation with the biotin-conjugated secondary antibody (dilution,
1:200; cat. no. A0277; Beyotime Institute of Biotechnology) for 1 h
at room temperature. Nuclei were counterstained with hematoxylin
staining solution (cat. no. C0107; Beyotime Institute of
Biotechnology) for 5 min at room temperature. Staining was
visualized using 3,3-diaminobenzidine (DAB). The sections were then
observed and photographed under the BX51 microscope (Olympus
Corporation, Tokyo, Japan). To quantify staining for Cx43 in the
analyzed regions, integrated optical density was calculated as the
product of staining area and intensity using image analysis
software (Image-Pro Plus version 6.0.0.26; Media Cybernetics, Inc.,
Rockville, MD, USA).
Statistical analysis
Data are expressed as the mean ± standard error and
were analyzed by one-way analysis of the variance (ANOVA) with
Newman-Keuls' post hoc tests, or two-way ANOVA with Bonferroni post
hoc tests as required using Prism v6.0 (GraphPad Software, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of LPS on vital signs
The dogs in the control and doxazosin only groups
had no visible symptoms including, fever, lethargy, vomiting,
diarrhea or increased heart rate. In the LPS only group, all dogs
treated with LPS (0.1 mg/kg; n=5) developed fever and lethargy
within 3 h. Signs of increased gastrointestinal motility, such as
vomiting (1/5) and diarrhea (4/5), were observed within 6 h
following the first LPS administration. However, these symptoms
disappeared within 24 h and did not recur throughout the study. The
dogs in the LPS + doxazosin group developed the same symptoms
following the first LPS treatment and all symptoms disappeared
within 24 h. Heart rate increased within 6 h and peaked at 12 h,
then returned to normal in both the LPS and LPS + doxazosin groups.
Treatment with LPS and/or doxazosin for 2 weeks did not alter body
weight, the mean arterial blood pressure or heart rate. All dogs
completed the study without mortality. Data are not shown.
Effect of LPS on inflammatory factor
and inducibility of AF
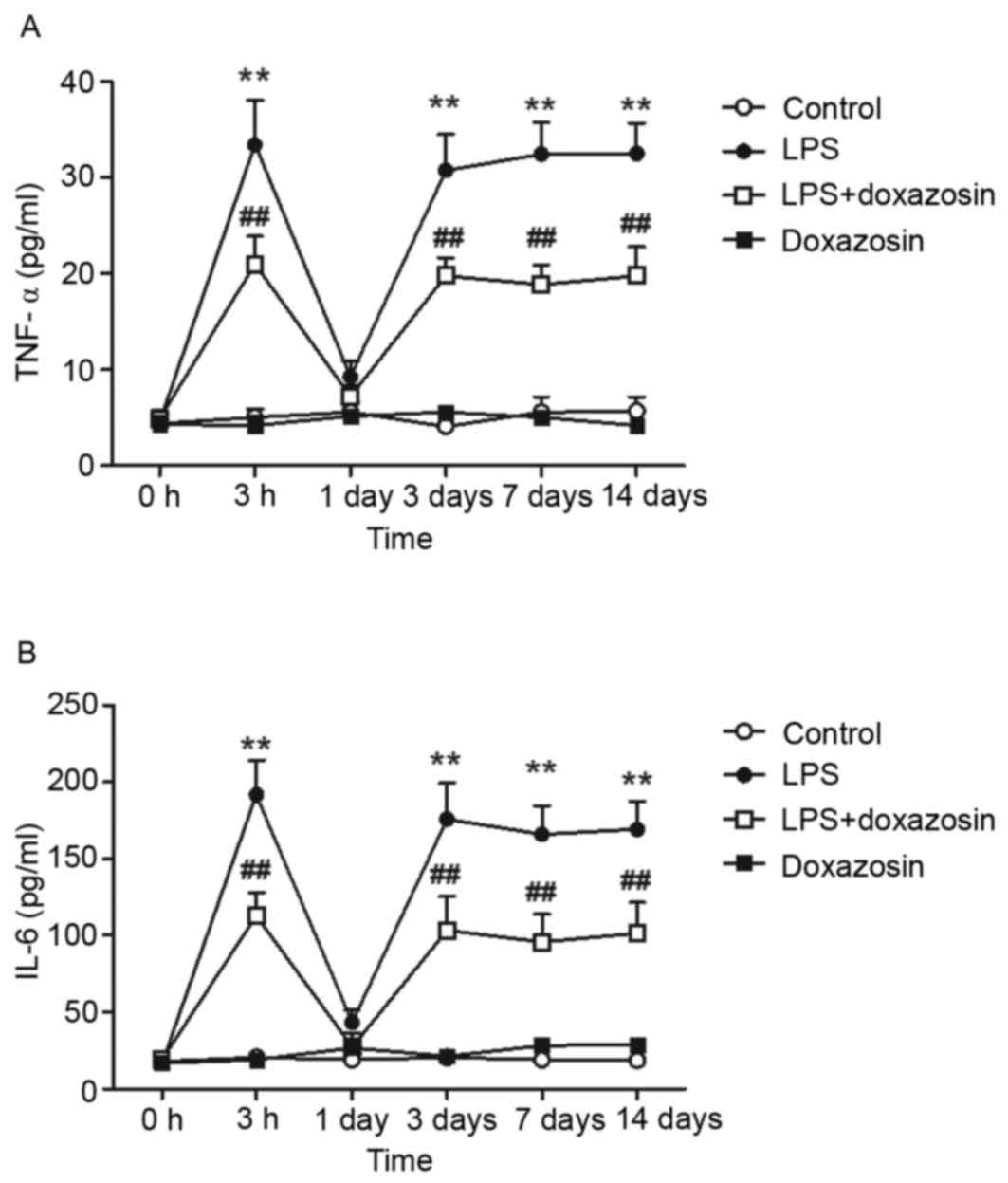
When compared with the control group, plasma TNF-α
and IL-6 levels increased at 3 h following treatment with LPS, then
returned to normal levels at 24 h following treatment. However,
administration of LPS for 3 to 14 days increased plasma TNF-α
concentration over 6-fold. Similarly, the level of plasma IL-6
increased by ~10-fold following 3–14 days of LPS injection
(P<0.01; Fig. 1). The TNF-α and
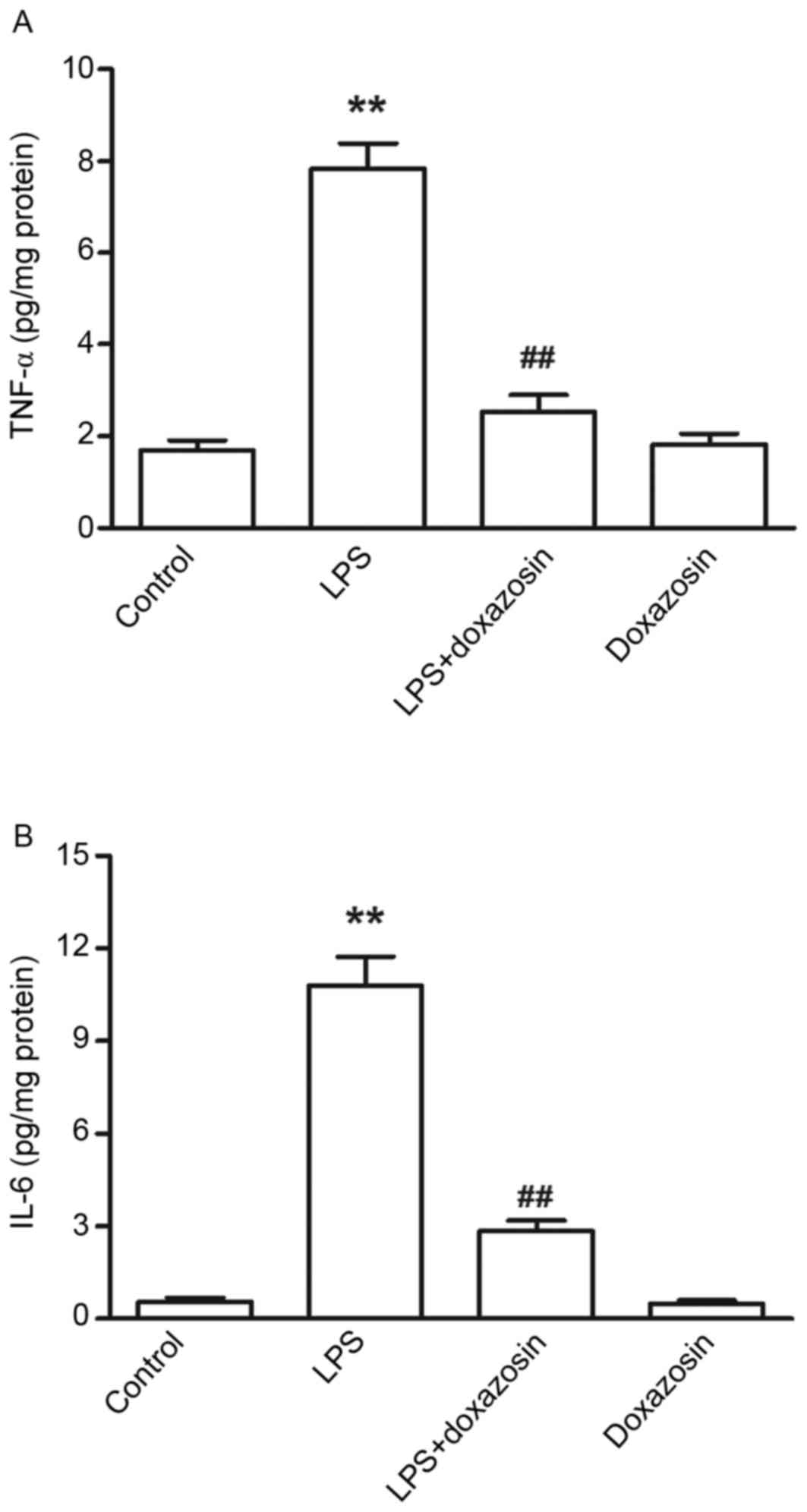
IL-6 contents in the atrial tissue were also significantly
increased in the LPS group when compared with the control
(P<0.01; Fig. 2). The
concentrations of TNF-α and IL-6 were not altered in the doxazosin
only group when compared with the control group (Figs. 1 and 2). When compared with the control group,
the levels of TNF-α and IL-6 were higher in the LPS + doxazosin
group. However, the TNF-α content and level of IL-6 was
significantly lower in the LPS + doxazosin group when compared with
the LPS only group (P<0.01; Figs.
1 and 2).
Following treatment with LPS (0.1 µg/kg, daily) for
2 weeks, ERP was significantly shortened at either 2x, 4x or 10x
threshold and the cumulative WOV was significantly widened in the
LPS group (P<0.01 vs. control; Fig.
3). Burst pacing at 10x threshold failed to develop sustained
AF in the control group, however, it did induce the occurrence of
sustained AF in 3/5 dogs treated with LPS. Doxazosin alone did not
influence the ERP and cumulative WOV (P>0.05; vs. control;
Fig. 3). When compared with the
LPS only group, doxazosin prevented the LPS-induced decrease in ERP
and increase in WOV (P<0.05; doxazosin + LPS vs. LPS; Fig. 3). In addition, AF was reported in
1/5 dogs in the LPS + doxazosin group (P<0.05 vs. LPS; data not
shown).
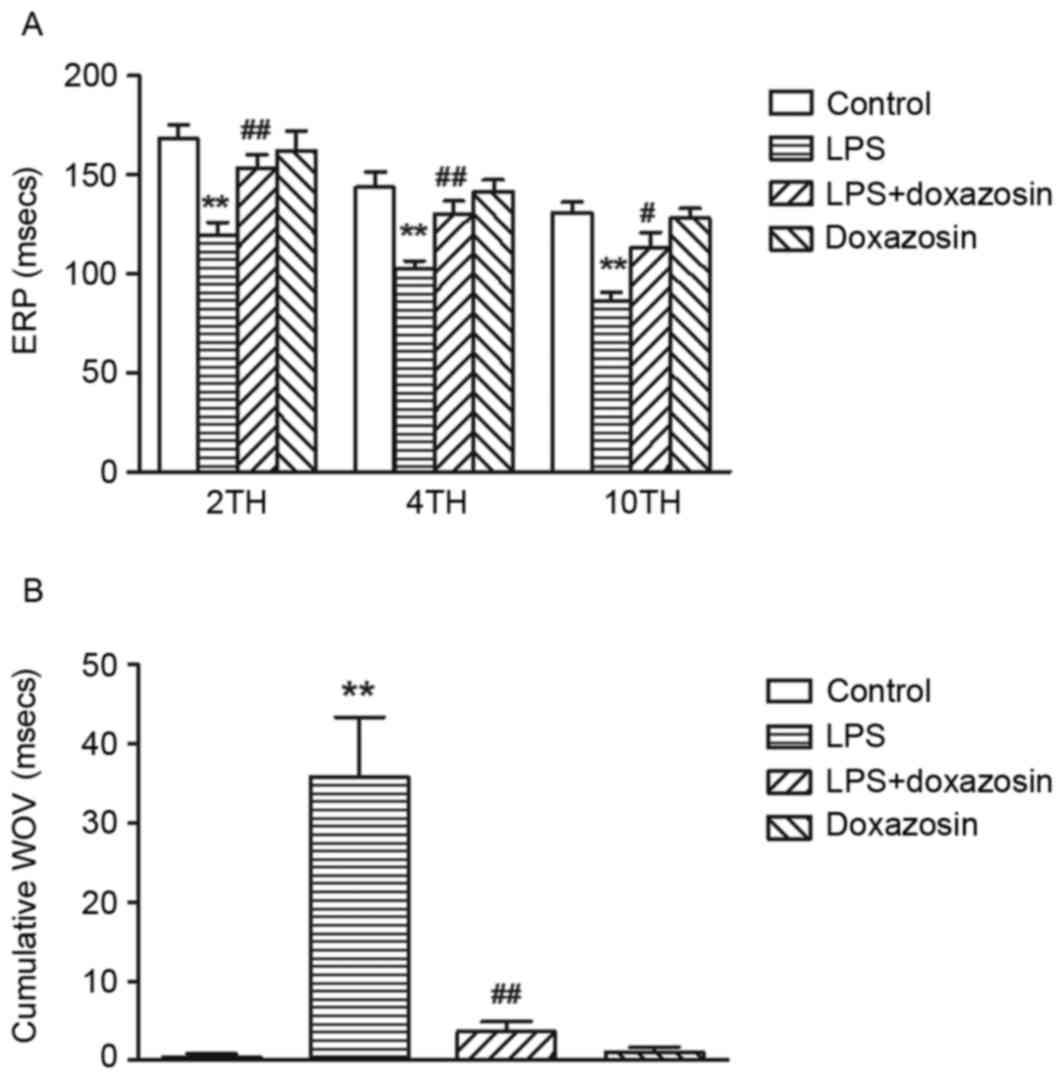 | Figure 3.Effect of LPS on atrial ERP and WOV
in canines. (A) ERP and (B) WOV were measured in canines from the
control (vehicle), LPS (0.1µg/kg LPS), LPS + doxazosin (0.1 µg/kg
LPS + 0.2 mg/kg doxazosin) and doxazosin only (0.2 mg/kg doxazosin)
groups, at the 2x, 4x and 10x threshold. Data are expressed as the
mean ± standard error (n=5/group). **P<0.01 vs. control;
#P<0.05 and ##P<0.01 vs. LPS. LPS,
lipopolysaccharide; ERP, effective refractory periods; 2TH, 2x
threshold; 4TH, 4x threshold; 10TH, 10x threshold; WOV, cumulative
window of vulnerability. |
Effect of LPS on Cx43, TLR4 and NF-kB
protein expression
Results from western blot analysis demonstrated that
the expression of Cx43 protein significantly increased in the LPS
treated group when compared with the control group (P<0.01;
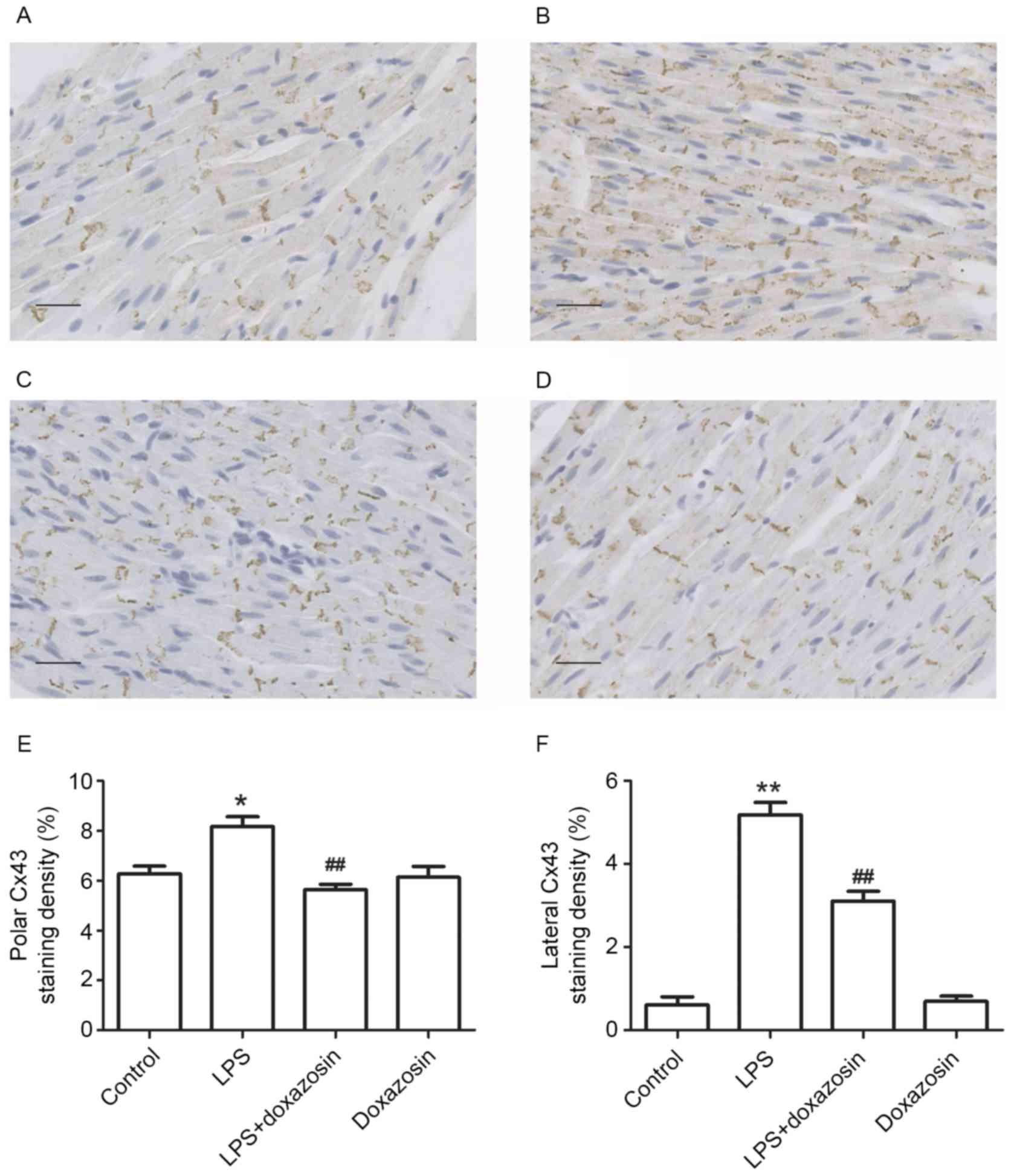
Fig. 4). Immunohistochemistry
analysis demonstrated that Cx43 was localized to a large extent in
the intercalated disk in the canine atria of the control group.
Following treatment with LPS, the amount of Cx43 protein in the
area of the intercalated disk increased, and a heterogeneous
distribution pattern of Cx43 was identified with a high-density in
lateral connection (P<0.05; Fig.
5). Doxazosin only treatment did not influence the expression
and distribution of Cx43 (P>0.05 vs. control; Figs. 4 and 5). However, in the LPS + doxazosin group,
doxazosin inhibited the LPS-induced increase in Cx43 protein and
the heterogeneous distribution, when compared with LPS only
(P<0.01; Figs. 4 and 5).
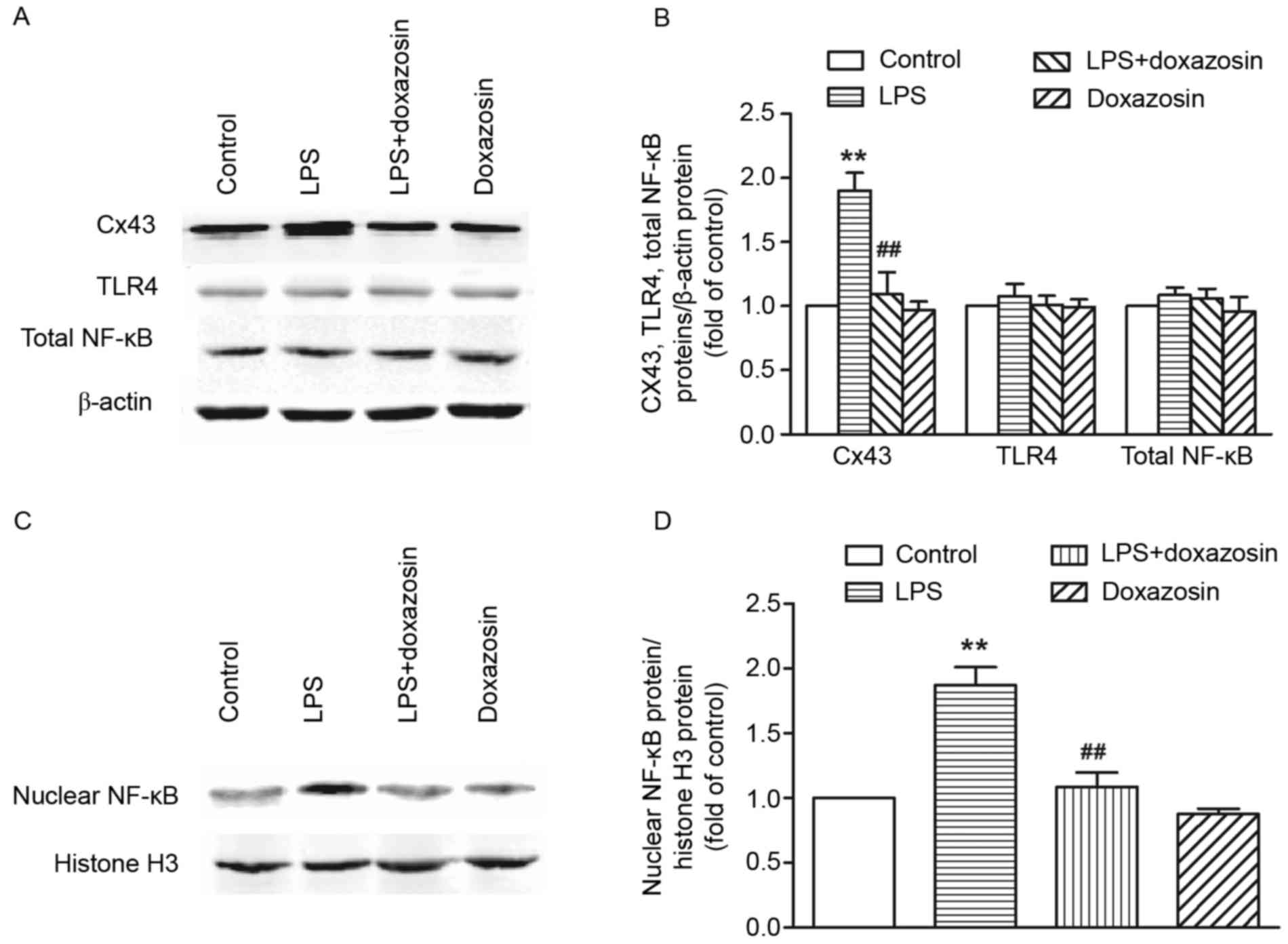 | Figure 4.Western blot analysis of Cx43, TLR4
and NF-κB protein expression in the right atrial tissue. Protein
expression was measured in canines from the control (vehicle), LPS
(0.1 µg/kg LPS), LPS + doxazosin (0.1 µg/kg LPS + 0.2 mg/kg
doxazosin) and doxazosin only (0.2 mg/kg doxazosin) groups. (A)
Representative blots of Cx43, TLR4, total NF-κB and β-actin protein
expression. (B) Densitometric quantification of Cx43, TLR4 and
total NF-κB protein expression normalized to the β-actin level.
Data are expressed as the mean ± standard error of three
experiments and expressed as the fold increase relative to the
control β-actin value. (C) Representative blots of nuclear NF-κB
and histone H3 protein (nuclear internal protein) expression in
nuclear extraction. (D) Densitometric quantification of nuclear
NF-κB protein expression normalized by histone H3 level. Data are
expressed as the mean ± standard error of three experiments and
expressed as the fold increase relative to the control histone H3
value. **P<0.01 vs. control group; ##P<0.01 vs.
LPS. LPS, lipopolysaccharide; Cx43, connexin 43; TLR4, Toll-like
receptor 4; TNF-α, tumor necrosis factor α; NF-κB, nuclear factor
κB. |
TLR4 protein expression in the canine atrium did not
increase following 2 weeks of LPS only, LPS + doxazosin or
doxazosin only administration. Although LPS treatment did not alter
the amount of total NF-κB protein expression, it enhanced the
nuclear NF-κB level in the canine atrial myocardium (P<0.01;
Fig. 4), suggesting that LPS may
induce the activation of NF-κB and promote the nuclear
translocation of NF-κB in the canine atrium. Administration with
doxazosin only did not alter the total and nuclear levels of NF-κB
when compared with control group. However, the level of nuclear
NF-κB in the LPS + doxazosin group was significantly lower when
compared with the LPS only group (P<0.01; Figs. 4 and 5).
Discussion
AF has traditionally been regarded as a sporadic and
acquired disease, however, a large population-based cohort study
and animal experimental data have suggested that local and systemic
inflammation serve an important role in the initiation and
perpetuation of AF (25,26). In 2007, Boos et al (27) reported that an LPS challenge as an
intravenous bolus of low dose induced a significant increase in
acute inflammatory indexes, however, it did not lead to the
development of acute new-onset AF in a large cohort of healthy
LPS-challenged subjects. However, it has been previously determined
that increased risk of AF is mainly associated with low-grade
chronic inflammation (28).
Low-grade inflammation is induced by a number of systemic diseases
including obesity, hypertension and coronary artery disease
(4,26,29).
Transgenic mice overexpressing TNF-α in the heart have been
demonstrated to develop atrial arrhythmias (30). Perfusion with IL-6 >20 min
induced an increase in the duration of the action potential in
isolated rat atrial tissue and led to the appearance of atrial
fibrillation (31). However,
whether the subacute treatment of LPS could directly affect cardiac
rhythms or induce atrial fibrillation is unclear. In the present
study, a chronic low-grade system inflammation model was
established by administration of 0.1 µg/kg of LPS once a day for 2
weeks. This dosage of LPS (from Escherichia coli 055:B5) can
cause detectable inflammation although no severe clinical symptoms
or death (32,33); however, a previous study has
demonstrated that using LPS from Escherichia coli 0111:B4
with the same dosage induces severe fever, vomiting, diarrhea and
even death in dogs (34). A
low-grade subacute or chronic system inflammation model could be
established by continuous subcutaneous infusion or repeated daily
injection of low dose LPS for ~5 to 28 days (35,36).
Therefore in the present study, LPS from Escherichia coli
055:B5 was used, which was induced only transient fever,
vomiting and diarrhea. Although repeated administration of LPS
developed an adaptive response in a set of behaviors (such as the
febrile response), which was similar to the results of previous
studies (36,37), it did induce a system inflammatory
response and increase in the inducibility of AF.
In the present study, total Cx43 protein expression
increased in the LPS group dogs, and treatment with LPS increased
the expression of the Cx43 protein in the intercalated disk and
also produced a high density distribution of Cx43 at the lateral
borders of atrial myocytes. Cx43 serves a crucial role in the
normal function of the cardiovascular system, and acts as a crucial
factor to generate arrhythmias. An increase in Cx43 was identified
in patients with lone AF or AF in mitral valve repair when compared
with patients in sinus rhythm (38). It has also been reported that
atrial Cx43 expression increased in the canine model of
pacing-induced sustained atrial fibrillation (39,40).
Somatic genetic defects of Cx43 have been reported as a potential
cause of AF in patients with sporadic, nonfamilial lone AF
(41). Abnormal distribution of
connexins seems to serve a crucial role in the initiation and
perpetuation of AF (42). Fastened
side-to-side conduction velocity due to an increase in the lateral
side of the gap junction may predispose the atrium to reentry
(43).
The molecular mechanism of altered Cx43 protein
expression and distribution during low-grade systemic
inflammation-induced AF is unclear. TNF-α has been suggested as a
main mediator of the inflammatory response, contributing to the
development of a number of cardiovascular diseases such as AF
(30,44). TNF-α may change the intracellular
distribution of Cx43 in mouse cardiomyocytes (45,46).
The activation of the TLR4 receptor has been known as a classical
pathway to increase the production of proinflammatory cytokines
(such as TNF-α) through the phosphorylation of inhibitor of κB and
activation of NF-κB (47). TLR4
expression has been detected in the atrium (48). In the present study, administration
of low-dose LPS for 2 weeks led to the activation of NF-κB and
significantly increased the levels of TNF-α and IL-6 in the atria,
however, it did not alter TLR4 expression.
An increasing body of evidence has demonstrated that
there is a cross-talk between α1-AR signaling and cytokine
production. Catecholamines have been reported to be produced by
phagocytes and enhance acute inflammatory injuries (18). In human monocytes and macrophages,
α1-AR positively regulates the LPS-induced cytokine production
(IL-1β) production (49).
Stimulation of α2-AR augments the production of TNF-α in
vitro from alveolar macrophage (50). Panama et al (51) identified that activation of α1-AR
regulates the fast transient outward K+ current in rat
ventricular myocytes via the NF-κB-dependent signaling pathway.
Neuronally induced atrial arrhythmias can be modified if a α-AR
blockade targets the intrinsic cardiac local circuit of neurons
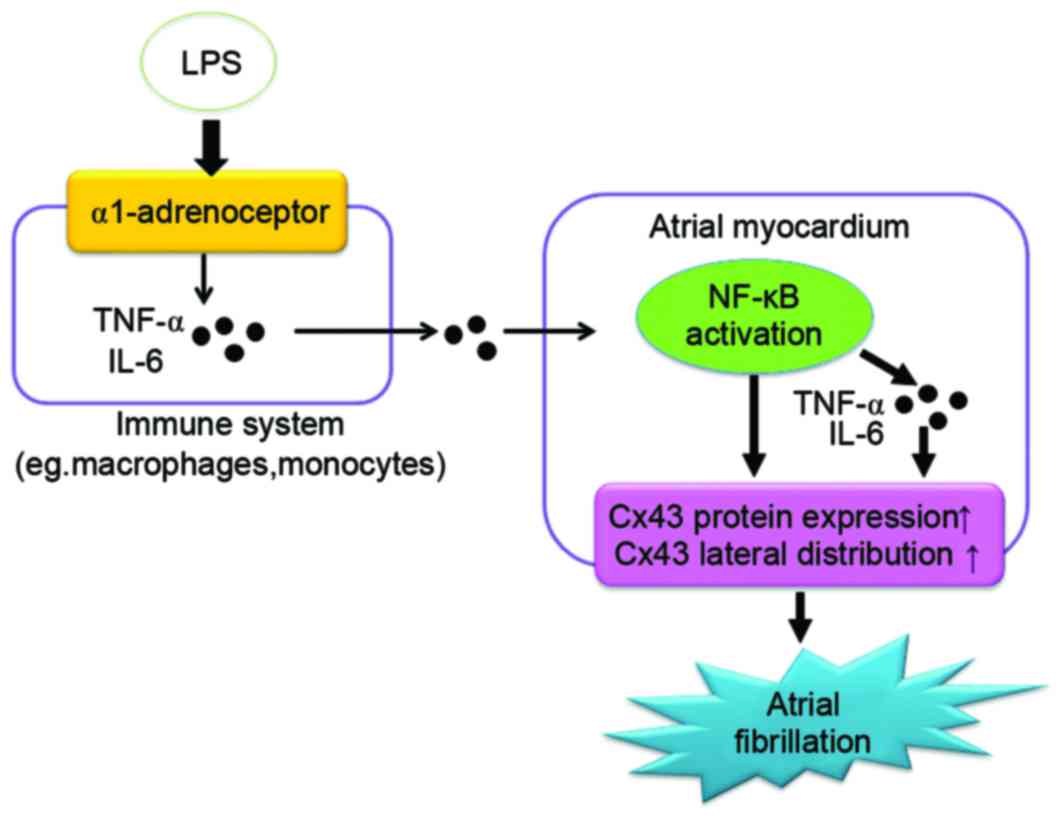
(52). In the present study,
blockage of α1-AR inhibited NF-κB activation, and the production of
TNF-α and IL-6, suggesting that α1-AR may participate in the
LPS-induced NF-κB activation in canine hearts. The LPS-induced
decrease in Cx43 and increase in inducibility of AF was also
inhibited by an α1-AR antagonist, indicating that α1-adrenergic
activation may be involved in the low-grade system of
inflammation-induced AF (Fig.
6).
One of the limitations of the present study was that
the effect of infiltrated monocytes and activated endothelium,
which may also contribute to the mechanism of the low-grade system
of inflammation-induced AF, cannot be excluded. Recruitment of
immune cells across the atrial endocardium has been observed in
human AF (53), and immune cells
(such as monocytes and macrophages) are the stronger regulator in
the production of LPS-induced inflammatory cytokines (49). It is possible that infiltrated
monocytes and activated endothelium are also involved in
LPS-induced cytokine generation in the heart. Besides Cx43, Cx40 is
another major composition of gap junctions expressed in the atrium
(54). It will be of interest to
examine whether the changes in Cx40 expression or distribution are
also involved in this canine model of inflammation-induced AF.
In conclusion, administration of a low-dose of LPS
for 2 weeks generated a system inflammatory response and increased
the inducibility of AF in the canine model. The mechanism may be
involved in the LPS-induced activation of NF-κB, and the increase
in Cx43 expression and lateral distribution via a α1-AR-dependent
signaling pathway. The present study will help to further elucidate
the pathogenesis of the system inflammation-induced atrial
fibrillation, and provide novel ideas for the occurrence of atrial
fibrillation.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81170167,
81,270,002 and 81471837) and the Science and Technology Department
of Zhejiang Province (grant no. 2015C37129).
References
|
1
|
Desai NR and Giugliano RP: Can we predict
outcomes in atrial fibrillation? Clin Cardiol. 35(Suppl 1):
S10–S14. 2012. View Article : Google Scholar
|
|
2
|
Zoni-Berisso M, Lercari F, Carazza T and
Domenicucci S: Epidemiology of atrial fibrillation: European
perspective. Clin Epidemiol. 6:213–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leonardi M and Bissett J: Prevention of
atrial fibrillation. Curr Opin Cardiol. 20:417–423. 2005.PubMed/NCBI
|
|
4
|
Harada M, Van Wagoner DR and Nattel S:
Role of inflammation in atrial fibrillation pathophysiology and
management. Circ J. 79:495–502. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pellegrino PL, Brunetti ND, De Gennaro L,
Ziccardi L, Grimaldi M and Biase MD: Inflammatory activation in an
unselected population of subjects with atrial fibrillation: Links
with structural heart disease, atrial remodeling and recent onset.
Intern Emerg Med. 8:123–128. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen MC, Chang JP, Liu WH, Yang CH, Chen
YL, Tsai TH, Wang YH and Pan KL: Increased inflammatory cell
infiltration in the atrial myocardium of patients with atrial
fibrillation. Am J Cardiol. 102:861–865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marcus GM, Smith LM, Ordovas K, Scheinman
MM, Kim AM, Badhwar N, Lee RJ, Tseng ZH, Lee BK and Olgin JE:
Intracardiac and extracardiac markers of inflammation during atrial
fibrillation. Heart Rhythm. 7:149–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li J, Solus J, Chen Q, Rho YH, Milne G,
Stein CM and Darbar D: Role of inflammation and oxidative stress in
atrial fibrillation. Heart Rhythm. 7:438–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Friedrichs K, Adam M, Remane L,
Mollenhauer M, Rudolph V, Rudolph TK, Andrié RP, Stöckigt F,
Schrickel JW, Ravekes T, et al: Induction of atrial fibrillation by
neutrophils critically depends on CD11b/CD18 integrins. PLoS One.
9:e893072014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pinho-Gomes AC, Reilly S, Brandes RP and
Casadei B: Targeting inflammation and oxidative stress in atrial
fibrillation: Role of 3-hydroxy-3-methylglutaryl-coenzyme a
reductase inhibition with statins. Antioxid Redox Signal.
20:1268–1285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ozaydin M, Icli A, Yucel H, Akcay S, Peker
O, Erdogan D, Varol E, Dogan A and Okutan H: Metoprolol vs.
carvedilol or carvedilol plus N-acetyl cysteine on post-operative
atrial fibrillation: A randomized, double-blind, placebo-controlled
study. Eur Heart J. 34:597–604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thomsen MB: Strengthening intercellular
communication to prevent atrial fibrillation. Cardiovasc Res.
92:187–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gros DB and Jongsma HJ: Connexins in
mammalian heart function. Bioessays. 18:719–730. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rothe S, Busch A, Bittner H, Kostelka M,
Dohmen PM, Mohr FW and Dhein S: Body mass index affects connexin43
remodeling in patients with atrial fibrillation. Thorac Cardiovasc
Surg. 62:547–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bikou O, Thomas D, Trappe K, Lugenbiel P,
Kelemen K, Koch M, Soucek R, Voss F, Becker R, Katus HA and Bauer
A: Connexin 43 gene therapy prevents persistent atrial fibrillation
in a porcine model. Cardiovasc Res. 92:218–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan J, Kong W, Zhang Q, Beyer EC, Walcott
G, Fast VG and Ai X: c-Jun N-terminal kinase activation contributes
to reduced connexin43 and development of atrial arrhythmias.
Cardiovasc Res. 97:589–597. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shin SY, Jo WM, Min TJ, Kim BK, Song DH,
Hyeon SH, Kwon JE, Lee WS, Lee KJ, Kim SW, et al: Gap junction
remodelling by chronic pressure overload is related to the
increased susceptibility to atrial fibrillation in rat heart.
Europace. 17:655–663. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Flierl MA, Rittirsch D, Nadeau BA, Chen
AJ, Sarma JV, Zetoune FS, McGuire SR, List RP, Day DE, Hoesel LM,
et al: Phagocyte-derived catecholamines enhance acute inflammatory
injury. Nature. 449:721–725. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu H, Xu M, Wang F, Liu S, Gu J and Lin S:
Chronic stress enhances progression of periodontitis via
α1-adrenergic signaling: A potential target for periodontal disease
therapy. Exp Mol Med. 46:e1182014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Hashem FH, Assiri AS, Shatoor AS,
Elrefaey HM, Alessa RM and Alkhateeb MA: Increased systemic
low-grade inflammation in high altitude native rats mediated by
adrenergic receptors. Saudi Med J. 35:538–546. 2014.PubMed/NCBI
|
|
21
|
The National Academies Collection: Reports
funded by National Institutes of Health, . National Research
Council (US) Committee for the Update of the Guide for the Care and
Use of Laboratory Animals. Guide for the Care and Use of Laboratory
Animals. 8th. National Academies Press (US); Washington (DC): pp.
1–197. 2011
|
|
22
|
Lu Z, Scherlag BJ, Lin J, Niu G, Fung KM,
Zhao L, Ghias M, Jackman WM, Lazzara R, Jiang H and Po SS: Atrial
fibrillation begets atrial fibrillation: Autonomic mechanism for
atrial electrical remodeling induced by short-term rapid atrial
pacing. Circ Arrhythm Electrophysiol. 1:184–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rashid A, Hines M, Scherlag BJ, Yamanashi
WS and Lovallo W: The effects of caffeine on the inducibility of
atrial fibrillation. J Electrocardiol. 39:421–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bode F, Katchman A, Woosley RL and Franz
MR: Gadolinium decreases stretch-induced vulnerability to atrial
fibrillation. Circulation. 101:2200–2205. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aviles RJ, Martin DO, Apperson-Hansen C,
Houghtaling PL, Rautaharju P, Kronmal RA, Tracy RP, Van Wagoner DR,
Psaty BM, Lauer MS and Chung MK: Inflammation as a risk factor for
atrial fibrillation. Circulation. 108:3006–3010. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu YF, Chen YJ, Lin YJ and Chen SA:
Inflammation and the pathogenesis of atrial fibrillation. Nat Rev
Cardiol. 12:230–243. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boos CJ, Lip GY and Jilma B: Endotoxemia,
inflammation, and atrial fibrillation. Am J Cardiol. 100:986–988.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liuba I, Ahlmroth H, Jonasson L, Englund
A, Jönsson A, Säfström K and Walfridsson H: Source of inflammatory
markers in patients with atrial fibrillation. Europace. 10:848–853.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goudis CA, Ntalas IV and Ketikoglou DG:
Atrial fibrillation in athletes. Cardiol Rev. 23:247–251. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saba S, Janczewski AM, Baker LC,
Shusterman V, Gursoy EC, Feldman AM, Salama G, McTiernan CF and
London B: Atrial contractile dysfunction, fibrosis, and arrhythmias
in a mouse model of cardiomyopathy secondary to cardiac-specific
overexpression of tumor necrosis factor-{alpha}. Am J Physiol Heart
Circ Physiol. 289:H1456–H1467. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mitrokhin VM, Mladenov MI and Kamkin AG:
Effects of interleukin-6 on the bio-electric activity of rat atrial
tissue under normal conditions and during gradual stretching.
Immunobiology. 220:1107–1112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Flatland B, Fry MM, LeBlanc CJ and
Rohrbach BW: Leukocyte and platelet changes following low-dose
lipopolysaccharide administration in five dogs. Res Vet Sci.
90:89–94. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
LeBlanc CJ, Horohov DW, Bauer JE, Hosgood
G and Mauldin GE: Effects of dietary supplementation with fish oil
on in vivo production of inflammatory mediators in clinically
normal dogs. Am J Vet Res. 69:486–493. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
LeMay DR, LeMay LG, Kluger MJ and D'Alecy
LG: Plasma profiles of IL-6 and TNF with fever-inducing doses of
lipopolysaccharide in dogs. Am J Physiol. 259:R126–R132.
1990.PubMed/NCBI
|
|
35
|
Cani PD, Amar J, Iglesias MA, Poggi M,
Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et
al: Metabolic endotoxemia initiates obesity and insulin resistance.
Diabetes. 56:1761–1772. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sparkman NL, Martin LA, Calvert WS and
Boehm GW: Effects of intraperitoneal lipopolysaccharide on Morris
maze performance in year-old and 2-month-old female C57BL/6J mice.
Behav Brain Res. 159:145–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Marais M, Maloney SK and Gray DA: The
development of endotoxin tolerance, and the role of
hypothalamo-pituitary-adrenal function and glucocorticoids in Pekin
ducks. J Exp Biol. 214:3378–3385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wetzel U, Boldt A, Lauschke J, Weigl J,
Schirdewahn P, Dorszewski A, Doll N, Hindricks G, Dhein S and
Kottkamp H: Expression of connexins 40 and 43 in human left atrium
in atrial fibrillation of different aetiologies. Heart. 91:166–170.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sakabe M, Fujiki A, Nishida K, Sugao M,
Nagasawa H, Tsuneda T, Mizumaki K and Inoue H: Enalapril prevents
perpetuation of atrial fibrillation by suppressing atrial fibrosis
and over-expression of connexin43 in a canine model of atrial
pacing-induced left ventricular dysfunction. J Cardiovasc
Pharmacol. 43:851–859. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Elvan A, Huang XD, Pressler ML and Zipes
DP: Radiofrequency catheter ablation of the atria eliminates
pacing-induced sustained atrial fibrillation and reduces connexin
43 in dogs. Circulation. 96:1675–1685. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thibodeau IL, Xu J, Li Q, Liu G, Lam K,
Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, et al: Paradigm
of genetic mosaicism and lone atrial fibrillation: Physiological
characterization of a connexin 43-deletion mutant identified from
atrial tissue. Circulation. 122:236–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Duffy HS and Wit AL: Is there a role for
remodeled connexins in AF? No simple answers. J Mol Cell Cardiol.
44:4–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Verheule S, Wilson EE, Arora R, Engle SK,
Scott LR and Olgin JE: Tissue structure and connexin expression of
canine pulmonary veins. Cardiovasc Res. 55:727–738. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ren M, Li X, Hao L and Zhong J: Role of
tumor necrosis factor alpha in the pathogenesis of atrial
fibrillation: A novel potential therapeutic target? Ann Med.
47:316–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liew R, Khairunnisa K, Gu Y, Tee N, Yin
NO, Naylynn TM and Moe KT: Role of tumor necrosis factor-α in the
pathogenesis of atrial fibrosis and development of an
arrhythmogenic substrate. Circ J. 77:1171–1179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sawaya SE, Rajawat YS, Rami TG, Szalai G,
Price RL, Sivasubramanian N, Mann DL and Khoury DS: Downregulation
of connexin40 and increased prevalence of atrial arrhythmias in
transgenic mice with cardiac-restricted overexpression of tumor
necrosis factor. Am J Physiol Heart Circ Physiol. 292:H1561–H1567.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Baumgarten G, Knuefermann P, Nozaki N,
Sivasubramanian N, Mann DL and Vallejo JG: In vivo expression of
proinflammatory mediators in the adult heart after endotoxin
administration: The role of toll-like receptor-4. J Infect Dis.
183:1617–1624. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Katoh S, Honda S, Watanabe T, Suzuki S,
Ishino M, Kitahara T, Funayama A, Netsu S, Sasaki T, Shishido T, et
al: Atrial endothelial impairment through Toll-like receptor 4
signaling causes atrial thrombogenesis. Heart Vessels. 29:263–272.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Grisanti LA, Woster AP, Dahlman J, Sauter
ER, Combs CK and Porter JE: α1-adrenergic receptors positively
regulate Toll-like receptor cytokine production from human
monocytes and macrophages. J Pharmacol Exp Ther. 338:648–657. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Spengler RN, Allen RM, Remick DG, Strieter
RM and Kunkel SL: Stimulation of alpha-adrenergic receptor augments
the production of macrophage-derived tumor necrosis factor. J
Immunol. 145:1430–1434. 1990.PubMed/NCBI
|
|
51
|
Panama BK, Latour-Villamil D, Farman GP,
Zhao D, Bolz SS, Kirshenbaum LA and Backx PH: Nuclear factor kappaB
downregulates the transient outward potassium current I(to,f)
through control of KChIP2 expression. Circ Res. 108:537–543. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Richer LP, Vinet A, Kus T, Cardinal R,
Ardell JL and Armour JA: Alpha-adrenoceptor blockade modifies
neurally induced atrial arrhythmias. Am J Physiol Regul Integr Comp
Physiol. 295:R1175–R1180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yamashita T, Sekiguchi A, Iwasaki YK, Date
T, Sagara K, Tanabe H, Suma H, Sawada H and Aizawa T: Recruitment
of immune cells across atrial endocardium in human atrial
fibrillation. Circ J. 74:262–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Verheule S, Van Kempen MJ, te Welscher PH,
Kwak BR and Jongsma HJ: Characterization of gap junction channels
in adult rabbit atrial and ventricular myocardium. Circ Res.
80:673–681. 1997. View Article : Google Scholar : PubMed/NCBI
|