Introduction
Thoracic aortic aneurysm and dissection (TAAD) is
associated with marked cardiovascular morbidity and mortality. The
incidence of acute aortic dissection has been suggested to be 3–6
cases/100,000 individuals/year, with an increasing incidence in
recent decades (1,2). The etiology of TAAD is complex and
heterogeneous. Hypertension and male sex are the most common risk
factors for TAAD in older patients, and other risk factors include
a family history of aortic diseases, pre-existing aortic or aortic
valve disease, a history of cardiac surgery and cigarette smoking
(2,3). However, genetic disease may be a
consideration in early-onset patients without the usual risk
factors (4,5). Genetic predisposition to TAAD may
occur in individuals with syndromic features (<5% of all cases
of TAAD), including in Marfan syndrome (MFS), or in the absence of
syndromic features. Of the total cases of non-syndromic TAAD, ~20%
are familial (FTAAD) and the rest are sporadic TAAD (6–8).
MFS, as an autosomal dominant disease, is the most
frequent heritable connective tissue disorder, associated with
mutations in the fibrillin 1 (FBN1) gene (9). The incidence of MFS is estimated to
be 2–3 per 10,000 individuals and patients are generally young; the
disease frequently manifests by the third decade of life (10,11).
MFS, a multisystem disorder, typically involves the skeletal,
cardiovascular and ocular systems (10). A number of connective tissue
disorders present overlapping clinical phenotypic features with
MFS, including Loeys-Dietz syndrome, Ehlers-Danlos syndrome,
Shprintzene-Goldberg syndrome and familial TAAD (12). With an improved understanding of
MFS, the revised Ghent Nosology gives greater weight to aortic root
aneurysm/dissection and FBN1 mutation in the diagnosis of MFS
(13). TAAD, which frequently
follows a period of progressive dilatation of the ascending aorta,
remains the most life-threatening manifestation of MFS and occurs
on average 20 years earlier in these patients compared with
patients without MFS (14). The
majority of patients suffer premature mortality from acute type A
aortic dissection or rupture without surgical aortic root
replacement. However, the incidence of type A aortic dissection has
decreased in individuals with MFS due to prophylactic aortic root
and ascending aorta replacement (15). Additionally, with the improvement
in life expectancy of patients with MFS, type B aortic dissection,
not involving the ascending aorta, occurs more frequently (16). FBN1 has been recognized to be the
causal gene of MFS and mutations in FBN1 increase the concentration
of transforming growth factor (TGF)-β1, which contributes notably
to the pathology of MFS. To date, 1,847 different mutations and
1,096 protein variants have been identified (http://www.umd.be/FBN1), and affected individuals
exhibit a broad phenotype although they may share the same mutation
within a family (17). Therefore,
the timely and accurate diagnosis of MFS remains a challenge.
Whole exome sequencing (WES) is an efficient tool to
identify the underlying genetic cause in TAAD (18,19).
In contrast with routine Sanger sequencing, WES is more efficient,
sensitive and cost-effective due to its high-throughput property.
In the present study, two patients were assessed who presented with
acute type B aortic dissection, and were subsequently identified to
harbor FBN1 pathogenic mutations by WES analysis. According to the
revised Ghent Nosology, the patients were diagnosed with MFS. The
two patients would have been diagnosed with sporadic TAAD without
the results of the WES analysis. It was hypothesized that WES may
contribute to molecular diagnosis for patients without the typical
features of MFS, thus providing genetic counseling and timely
intervention for affected individuals.
Patients and methods
Patients and clinical evaluation
A total of two male patients, not related by birth,
were respectively admitted in Marth and May 2015 to the department
of Vascular Surgery, Nanjing Drum Tower Hospital, Nanjing, China.
The two individuals, designated T287 and T267, complained of acute
severe chest and back pain. They were reported to be in good health
prior to this acute incidence. Regular physical examination and
clinical testing was performed on the two patients, including
electrocardiography, computed tomography angiography (CTA) and
echocardiography. Z-score was used to estimate the degree of aortic
root dilatation (http://www.marfan.org/dx/zscore). Genomic DNA was
extracted from peripheral blood samples. The present study was
approved by the Institutional Research Ethics Committee of Nanjing
Drum Tower Hospital and informed written consent was obtained. A
total of 100 ethnically-matched unrelated subjects (aged 29.5±6.5
years) were recruited between June and August 2016 as controls.
Peripheral blood was drawn and DNA were subsequent extracted.
WES and data analysis
Genomic DNA was extracted from peripheral whole
blood samples from the subjects using the QIAamp DNA Blood Mini kit
(Qiagen GmbH, Hilden, Germany), according to the manufacturer's
protocol. WES analysis was performed by Beijing Novogene
Bioinformatics Technology, Co., Ltd. (Beijing, China). Target
enrichment was performed to construct the exome library using the
Agilent SureSelect Human All Exon V5 kit (Agilent Technologies,
Inc., Santa Clara, CA, USA), according to manufacturer's protocol,
and sequenced on the Illumina HiSeq 2000 platform (Illumina, Inc.,
San Diego, CA). The clean reads from the Illumina Genome HiSeq 2000
were aligned to the human genome reference (University of
California Santa Cruz hg19; genome.ucsc.edu) using the Burrows-Wheeler Alignment
tool. The Sequence Alignment/Map tools were used to identify single
nucleotide polymorphisms (SNPs) and insertions/deletions (INDELs).
Picard (http://sourceforge.net/projects/picard/) was employed
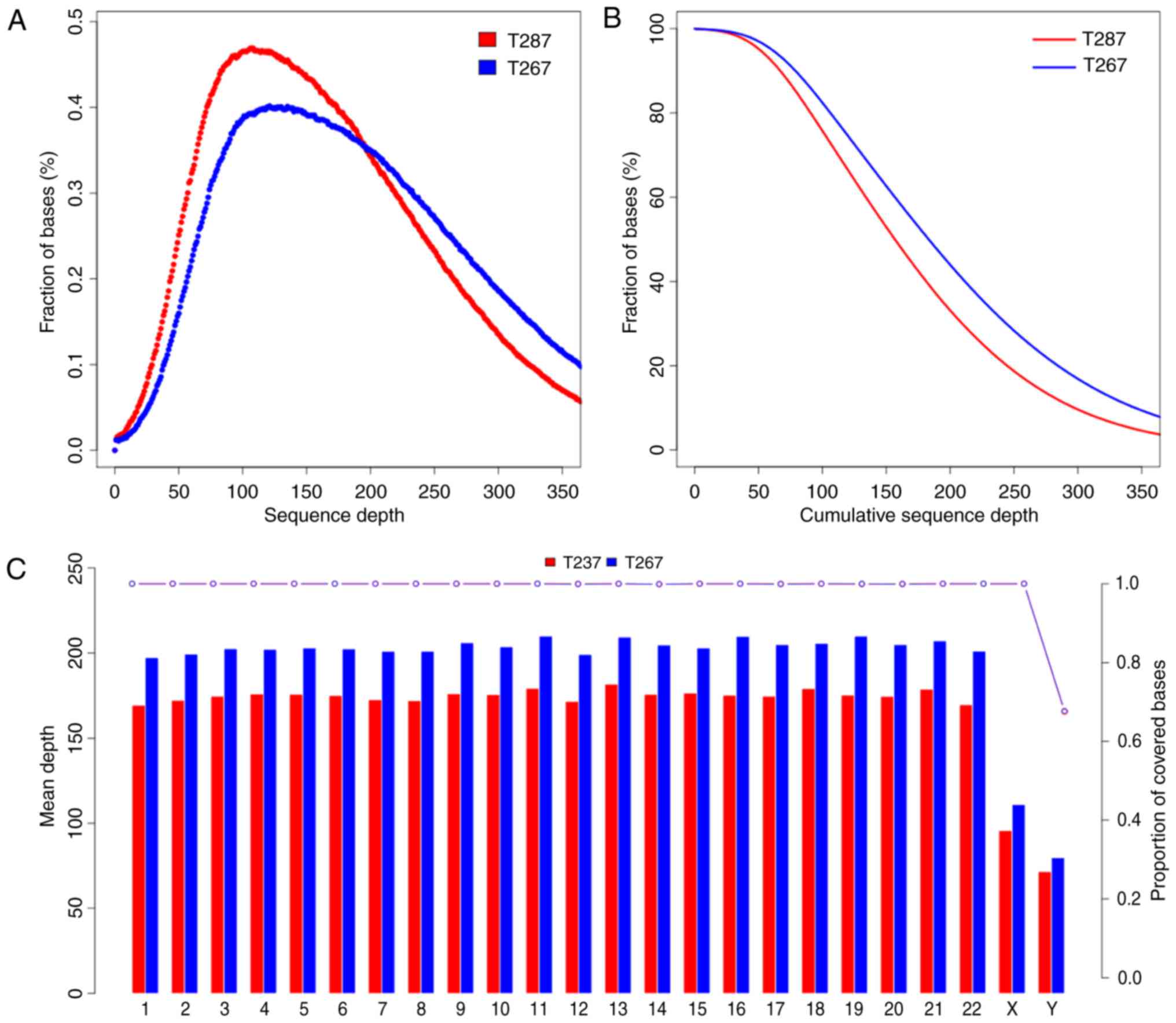
to mark duplicate reads. An average sequencing depth of 171.32× was
achieved and >99.4% of targeted variants were covered at least
by 20× for T287, while a 199.80× average sequencing depth and
>99.6% of targeted variants covered at least by 20× was achieved
for T267.
All annotated variants were screened with databases
including the SNP database (dbSNP142, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi),
1000 Genomes Project (version 2014 October, http://www.1000genomes.org/), and National Heart,
Lung, and Blood Institute Exome Sequencing Project (ESP) 6500
(http://evs.gs.washington.edu/EVS/).
Functional prediction was assessed by sorts intolerant from
tolerant (SIFT; http://sift.jcvi.org/) and
polymorphism phenotyping version 2 (Polyphen-2; http://genetics.bwh.harvard.edu/pph2/).
Candidate SNPs or INDELs were annotated using ANNOVAR software
(http://annovar.openbioinformatics.org).
Sanger sequencing confirmation
For Sanger sequencing verification of variants
detected by WES, oligonucleotide primers were designed using the
Primer3 program (primer3.ut.ee). The regions containing the
suspected variants were amplified by standard polymerase chain
reaction (PCR) analysis using GoTaq polymerase (Promega). Primers
used to amplify the mutant sequence were FBN1-Ex39-F
(5′-AACTTACTTCAGACGGGCAGAG-3′), FBN1-Ex39-R
(5′-TAGCTCCTGGCACTCATCAATA-3′), FBN1-Ex57-F
(5′-ATGTGAGAGAGGGAAGGAAGGT-3′), FBN1-Ex39-R
(5′-GTCAATACGGCATCTCCAAAAT-3′). The amplification reaction mixture
(50 µl) was subjected to denaturation at 98°C for 3 min followed by
35 cycles at 95°C for 30 sec, annealing temperature 60°C for 30
sec, 72°C for 30 sec and by a final extension at 72°C for 15 min.
PCR products were purified and sequenced on the ABI PRISM3730
automated sequencer (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) using the BigDye terminator v3.1 cycle
sequencing kit (Thermo Fisher Scientific, Inc.).
Results
Clinical findings
The two patients complained of acute episodes of
chest and back pain. T267 is a 35-year-old man, 182 cm in height
and 60 kg in weight. He was diagnosed with descending aortic
dissection by CTA. Echocardiography of T267 demonstrated that the
diameter of the aortic sinus was 4.3 cm, and identified hypertrophy
of the left ventricle. T287 is a 32-year-old male, 178 cm in height
and 95 kg in weight. A CTA scan revealed descending aortic
dissection and ascending aortic dilatation. Echocardiography of
T287 demonstrated that the diameter of the aortic sinus was 4.5 cm,
in addition to identifying mild aortic valve regurgitation, and
mild bicuspid and tricuspid valve regurgitation. Additionally, the
grandfather of T287 died suddenly of an unknown cause at age of 40.
Clinical manifestations of other systems, including the skeletal
and ocular systems, were absent. The clinical data of the two
patients are presented in Table
I.
 | Table I.Clinical characteristics of the two
individuals affected with acute type B aortic dissection. |
Table I.
Clinical characteristics of the two
individuals affected with acute type B aortic dissection.
|
|
|
|
|
|
|
| Cardiovascular |
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|---|
| ID | Age, years | Sex | Height, cm | Weight, kg | Diameter of aortic
sinus, cma | Z-Scoreb | Aorta | Other | Skeletal | Ocular |
|---|
| T267 | 35 | M | 182 | 60 | 4.3 | 3.86 | TAD | Hypertension; left
ventricular hypertrophy | None | None |
| T287 | 32 | M | 178 | 95 | 4.5 | 4.12 | TAD | Hypertension;
dilatation of ascending aorta; mild valve regurgitation | None | None |
WES
The present study generated a total of 58,189,201
and 50,222,226 raw reads of 180–280 bp paired-end read sequences
from the patients T267 and T287, respectively. The raw depth of
T267 and T287 was 346.49× and 299.06×, respectively; and the
Phred-like Q20 (that is, a 99% accuracy of the base call) was 95.02
and 94.26%. The average depth of targeted sequences was 199.80× and
171.32× for T267 and T287, respectively. The sequence depth and
proportion covered bases of every sequence and chromosome of T267
and T287 are presented in Fig. 1.
Variant genes were filtered with dbSNP142, the 1000 Genomes
Project, and ESP6500. Subsequently, variant genes were identified
in either the exonic regions or the splice sites, and synonymous
SNPs or INDELs were excluded. SIFT and PolyPhen-2 were used to
excluded synonymous mutations and to predict the functions of the
mutations.
In order to identify pathogenic mutations in the two
patients, candidate genes were filtered with reported
disease-causing genes for TAAD (data for the two patients are
presented in Table II). Applying
the above strategy, two heterozygous variants in the FBN1 gene were
observed. One was a nonsense mutation, c.C4786T (p.R1596X), and was
deleterious due to nonsense-mediated RNA decay. The other was a
missense mutation, c.G6953A (p.C2318Y), which was reported to
disrupt disulfide bond formation, and thus may be disease-causing
(20).
| Table II.Number of variants present in the
patients at different stages of the filtering process. |
Table II.
Number of variants present in the
patients at different stages of the filtering process.
|
| Number |
|---|
|
|
|
|---|
| Filtering
criteria | T267 | T287 |
|---|
| Total number of
variants | 221,478 | 198,478 |
| Variants not in
dbSNP, MAF<1% | 22,723 | 19,655 |
| Filtering using 1000
Genomes and ESP | 22,529 | 17,494 |
| Variants in exonic or
splicing | 1,082 | 1,021 |
| Non-synonymous and
frameshift variants | 950 | 917 |
| Functional analysis
by SIFT and Polyphen-2 | 776 | 753 |
| Genes associated with
TAAD | 1 | 1 |
Mutation conformation
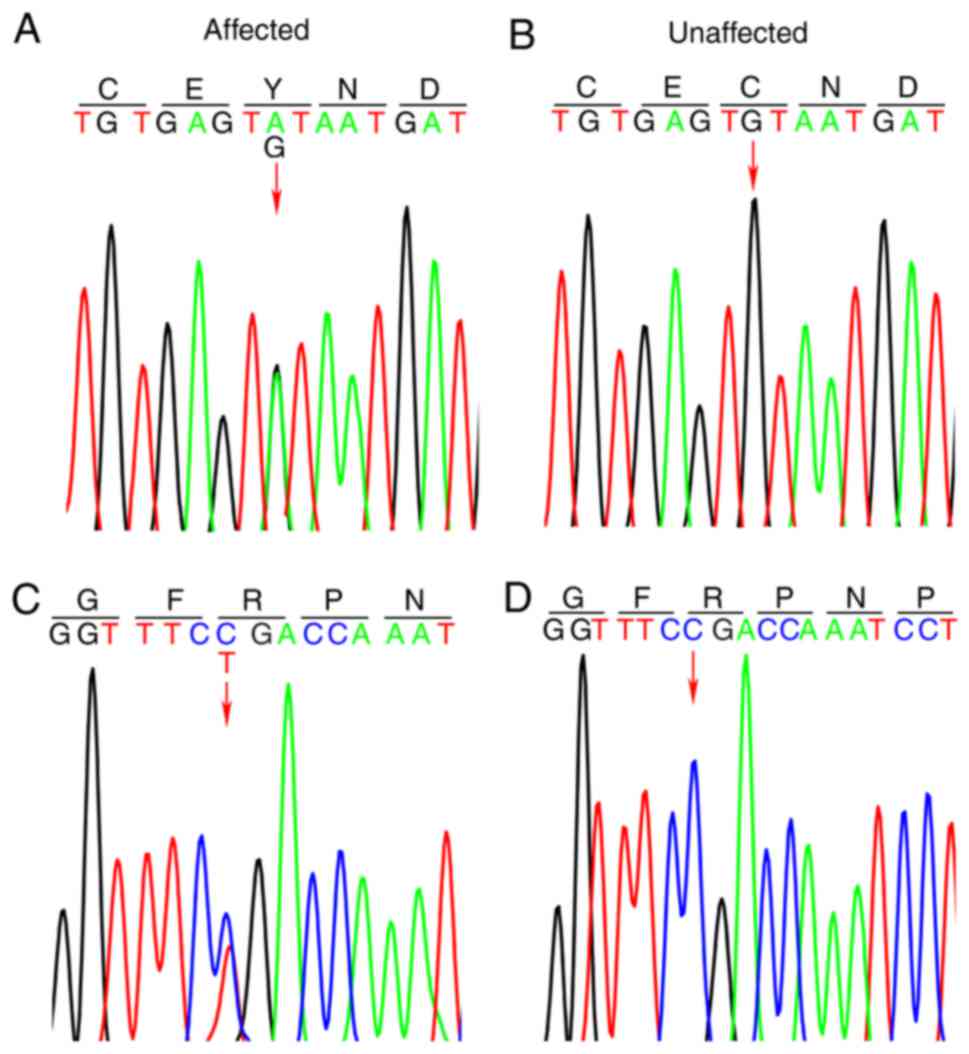
The two variants were confirmed by Sanger sequencing
(Fig. 2). The variants were absent
in 100 ethnically-matched unrelated subjects.
Discussion
MFS is a common multisystem connective tissue
disorder, inherited in an autosomal dominant manner, caused by
mutations in the FBN1 gene on chromosome 15q21.1 (9). FBN1, the major component of
elastin-associated microfibrils, is a large gene (>200 kb),
highly fragmented into 65 exons, and ~1,847 different mutations
have been described to date (http://www.umd.be/FBN1). FBN1 mutations may affect
elastic fiber deposition and cytokine-regulatory functions via
regulation of the TGF-β signaling pathway (21). The principal clinical features of
MFS involve the skeletal system (arachnodactyly, bone overgrowth
and joint laxity), cardiovascular system (particularly valve
regurgitation and TAAD), and the ocular system (ectopia lentis)
(13). Due to phenotypic overlap
and the genetic heterogeneity of multiple Marfan-like disorders, it
is challenging for clinicians to make an accurate diagnosis of MFS
(17). The revised Ghent Nosology
highlights the role of the Z-score of the aortic sinus, ectopia
lentis and identification of a bona fide FBN1 mutation to establish
the diagnosis of MFS (13).
Considering the phenotypic overlap and genetic
heterogeneity of diseases featuring aortopathy, molecular genetic
testing is often required for the timely and accurate diagnosis of
affected individuals. Traditional Sanger sequencing is costly and
laborious due to the size of the FBN1 gene. The present study
identified one missense mutation (c.G6953A:p.C2318Y in exon 57) and
one nonsense mutation (c.C4786T:p.R1596X in exon 39) in the FBN1
gene by WES in two early-onset affected individuals with type B
aortic dissection. The two patients lacked a family history of TAAD
or MFS. For T287, the diameter of the aortic sinus was measured and
the Z-score was calculated to be 4.12, using the Z-score calculator
at www.marfan.org. According to the revised Ghent
Nosology (2010), T287 was diagnosed with MFS with manifestation of
type B aortic dissection. Similarly, the Z-score of T267 was 3.86
and this individual was diagnosed with MFS subsequently. Without
identification of the FBN1 mutation, the two patients may have been
diagnosed with sporadic TAAD. Therefore, using the technique of
WES, patients with MFS may benefit from a timely and accurate
diagnosis of MFS, which aids in periodic surveillance, prophylactic
surgical measures and genetic counseling for family members.
Clinical manifestations vary between TAAD caused by
MFS and non-syndromic TAAD. In previous decades, without the use of
surgical aortic root replacement, the incidence of type A aortic
dissection was increased compared with type B aortic dissection in
patients with MFS (10). Den
Hartog et al (15) observed
that the rate of type B aortic dissection was 9% in 600 patients
with MFS without previous TAAD, during a median follow-up period of
6 years, and that dissection generally occurred in the mildly
dilated proximal descending aorta. Hypertension is considered to be
the most important risk factor in non-syndromic TAAD. However, TAAD
caused by MFS, as the most life-threatening clinical manifestation
of the cardiovascular system, is necessarily associated with the
mutations in the FBN1 gene (22).
Patients with MFS frequently suffer acute TAAD at a younger age
compared with patients with non-syndromic TAAD. At present,
patients with MFS who have undergone prophylactic surgical aortic
root replacement are associated with an increased risk of acute
type B aortic dissection, which frequently occurs without prior
significant aortic dilatation (15,16).
In the present study, the two patients were admitted
to the Department of Vascular Surgery with acute type B aortic
dissection in their thirties. It was inferred that the symptoms
were likely to be caused by a genetic defect. Subsequent molecular
analysis confirmed this hypothesis. It is widely accepted that
endovascular stent grafting may not be suitable for the treatment
of type B dissection, particularly in young patients with MFS, due
to weakening of the aortic walls and the lack of long-term
durability of stent grafting (23–25);
however, the majority of these previous studies used
first-generation stent grafts. Considering the hemodynamic
instability, increased surgical risk and absence of definitive
diagnostic criteria for MFS, thoracic endovascular aortic repair
was performed on the two patients recruited for the present study.
Angiotensin-II type-1 receptor blockers have been demonstrated to
reverse vascular complications in a fibrillin-deficient mouse model
of MFS and losartan was recommended for used in medical therapy
(26,27). With advances in thoracic
endovascular stent grafting and improved recognition of MFS,
improved treatment options may be identified for young patients
with MFS.
In conclusion, the present study identified one
missense mutation and one nonsense mutation via WES in two patients
with early-onset type B aortic dissection without typical syndromic
features, who were subsequently diagnosed with MFS. the present
study emphasized the necessity of genetic testing for young
patients with type B aortic dissection. WES is a timely, robust and
inexpensive technique for genetic sequencing, particularly for TAAD
which is caused by numerous genes. Genetic diagnosis of MFS may
facilitate periodic surveillance, prophylactic surgical measures
and genetic counseling.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Jiangsu Province, China (grant no.
BK20140103) and the Natural Science Foundation of China (grant no.
81270396).
References
|
1
|
Nienaber CA and Powell JT: Management of
acute aortic syndromes. Eur Heart J. 33:26–35b. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Olsson C, Thelin S, Ståhle E, Ekbom A and
Granath F: Thoracic aortic aneurysm and dissection: Increasing
prevalence and improved outcomes reported in a nationwide
population-based study of more than 14,000 cases from 1987 to 2002.
Circulation. 114:2611–2618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Eusanio M, Trimarchi S, Patel HJ,
Hutchison S, Suzuki T, Peterson MD, Di Bartolomeo R, Folesani G,
Pyeritz RE, Braverman AC, et al: Clinical presentation, management,
and short-term outcome of patients with type A acute dissection
complicated by mesenteric malperfusion: Observations from the
International registry of acute aortic dissection. J Thorac
Cardiovasc Surg. 145:385–390.e1. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Backer J, Campens L and De Paepe A:
Genes in thoracic aortic aneurysms/dissections - do they matter?
Ann Cardiothorac Surg. 2:73–82. 2013.PubMed/NCBI
|
|
5
|
Campens L, Callewaert B, Muiño Mosquera L,
Renard M, Symoens S, De Paepe A, Coucke P and De Backer J: Gene
panel sequencing in heritable thoracic aortic disorders and related
entities results of comprehensive testing in a cohort of 264
patients. Orphanet J Rare Dis. 10:92015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pyeritz RE: Heritable thoracic aortic
disorders. Curr Opin Cardiol. 29:97–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pannu H, Avidan N, Tran-Fadulu V and
Milewicz DM: Genetic basis of thoracic aortic aneurysms and
dissections: Potential relevance to abdominal aortic aneurysms. Ann
N Y Acad Sci. 1085:242–255. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Milewicz DM, Chen H, Park ES, Petty EM,
Zaghi H, Shashidhar G, Willing M and Patel V: Reduced penetrance
and variable expressivity of familial thoracic aortic
aneurysms/dissections. Am J Cardiol. 82:474–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robinson PN, Arteaga-Solis E, Baldock C,
Collod-Béroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA,
Judge DP, et al: The molecular genetics of Marfan syndrome and
related disorders. J Med Genet. 43:769–787. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Judge DP and Dietz HC: Marfan's syndrome.
Lancet. 366:1965–1976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murdoch JL, Walker BA, Halpern BL, Kuzma
JW and McKusick VA: Life expectancy and causes of death in the
Marfan syndrome. N Engl J Med. 286:804–808. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wooderchak-Donahue W, VanSant-Webb C,
Tvrdik T, Plant P, Lewis T, Stocks J, Raney JA, Meyers L, Berg A,
Rope AF, et al: Clinical utility of a next generation sequencing
panel assay for Marfan and Marfan-like syndromes featuring
aortopathy. Am J Med Genet A. 167A:1–1757. 2015.PubMed/NCBI
|
|
13
|
Loeys BL, Dietz HC, Braverman AC,
Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y,
Jondeau G, Faivre L, Milewicz DM, et al: The revised Ghent nosology
for the Marfan syndrome. J Med Genet. 47:476–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rylski B, Hoffmann I, Beyersdorf F,
Suedkamp M, Siepe M, Nitsch B, Blettner M, Borger MA and Weigang E:
Multicenter Prospective Observational Study: Acute aortic
dissection type A: Age-related management and outcomes reported in
the German registry for acute aortic dissection type A (GERAADA) of
over patients. Ann Surg. 259:598–604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
den Hartog AW, Franken R, Zwinderman AH,
Timmermans J, Scholte AJ, van den Berg MP, de Waard V, Pals G,
Mulder BJ and Groenink M: The risk for type B aortic dissection in
Marfan syndrome. J Am Coll Cardiol. 65:246–254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Engelfriet PM, Boersma E, Tijssen JG,
Bouma BJ and Mulder BJ: Beyond the root: Dilatation of the distal
aorta in Marfan's syndrome. Heart. 92:1238–1243. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Loeys B, Nuytinck L, Delvaux I, De Bie S
and De Paepe A: Genotype and phenotype analysis of 171 patients
referred for molecular study of the fibrillin-1 gene FBN1 because
of suspected Marfan syndrome. Arch Intern Med. 161:2447–2454. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Poninska JK, Bilinska ZT, Franaszczyk M,
Michalak E, Rydzanicz M, Szpakowski E, Pollak A, Milanowska B,
Truszkowska G, Chmielewski P, et al: Next-generation sequencing for
diagnosis of thoracic aortic aneurysms and dissections: Diagnostic
yield, novel mutations and genotype phenotype correlations. J
Transl Med. 14:1152016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ziganshin BA, Bailey AE, Coons C, Dykas D,
Charilaou P, Tanriverdi LH, Liu L, Tranquilli M, Bale AE and
Elefteriades JA: Routine genetic testing for thoracic aortic
aneurysm and dissection in a clinical setting. Ann Thorac Surg.
100:1604–1611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stheneur C, Collod-Béroud G, Faivre L,
Buyck JF, Gouya L, Le Parc JM, Moura B, Muti C, Grandchamp B,
Sultan G, et al: Identification of the minimal combination of
clinical features in probands for efficient mutation detection in
the FBN1 gene. Eur J Hum Genet. 17:1121–1128. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Neptune ER, Frischmeyer PA, Arking DE,
Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY and Dietz HC:
Dysregulation of TGF-beta activation contributes to pathogenesis in
Marfan syndrome. Nat Genet. 33:407–411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brautbar A, LeMaire SA, Franco LM, Coselli
JS, Milewicz DM and Belmont JW: FBN1 mutations in patients with
descending thoracic aortic dissections. Am J Med Genet A.
152A:1–416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pacini D, Parolari A, Berretta P, Di
Bartolomeo R, Alamanni F and Bavaria J: Endovascular treatment for
type B dissection in Marfan syndrome: Is it worthwhile? Ann Thorac
Surg. 95:737–749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ptaszek LM, Kim K, Spooner AE,
MacGillivray TE, Cambria RP, Lindsay ME and Isselbacher EM: Marfan
syndrome is associated with recurrent dissection of the dissected
aorta. Ann Thorac Surg. 99:1616–1623. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Demers P, Miller DC, Mitchell RS, Kee ST,
Sze D, Razavi MK and Dake MD: Midterm results of endovascular
repair of descending thoracic aortic aneurysms with
first-generation stent grafts. J Thorac Cardiovasc Surg.
127:664–673. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Habashi JP, Judge DP, Holm TM, Cohn RD,
Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al:
Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse
model of Marfan syndrome. Science. 312:117–121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Erbel R, Aboyans V, Boileau C, Bossone E,
Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H,
Gaemperli O, et al: 2014 ESC Guidelines on the diagnosis and
treatment of aortic diseases: Document covering acute and chronic
aortic diseases of the thoracic and abdominal aorta of the adult.
The task force for the diagnosis and treatment of aortic diseases
of the European Society of Cardiology (ESC). Eur Heart J.
35:2873–2926. 2014. View Article : Google Scholar : PubMed/NCBI
|