Introduction
Alagille syndrome (AGS), additionally known as
arteriohepatic dysplasia, syndromic bile duct paucity and
Alagille-Watson syndrome, is a highly variable autosomal dominant
disease which affects the liver, heart and other parts of the body
(1,2). AGS is one of the familial
intrahepatic cholestatic syndromes associated with jagged 1
(JAG1), neurogenic locus notch homolog protein 2
(NOTCH2), uridine phosphorylase glucuronosyltransferase
family 1 member A1 (UGT1A1), adenosine triphosphatase
phospholipid transporting 8B1 (ATP8B1), adenosine
tripshosphate-binding cassette, sub-family B member 11
(ABCB11) and adenosine tripshosphate-binding cassette,
sub-family B member 4 (ABCB4) genes, and is one of the
important causes of intrahepatic cholestasis in infancy (3–5). The
incidence of AGS is ~1 in 70,000 newborns with neonatal jaundice
(6), and the mortality rate is
~17% (7). AGS is the result of
JAG1 mutations; however, the inherited pathogenic variants
only account for 30–50% of all cases, with the remaining 50–70% of
cases being de novo pathogenic variants (8–10).
Traditionally, a diagnosis of AGS requires three of the five
primary clinical features, or two features if the patient has a
positive family history. The five primary clinical features are:
Chronic cholestasis, cardiac disease, skeletal abnormalities,
ocular abnormalities and a characteristic facial phenotype
(11). However, following the
demonstration that mutations in the JAG1 gene cause AGS
(11,12), the diagnostic criteria has been
modified; if an individual carries a harmful mutation in the
JAG1 gene, AGS may be diagnosed even in the absence of
clinical manifestations.
JAG1 and NOTCH2 genes are associated
with AGS. Pathogenic mutations in JAG1 and NOTCH2
genes may impair the Notch signaling pathway, which is an
evolutionarily conserved, intercellular signaling mechanism
essential for healthy embryonic development (13–16).
The majority of AGS cases (~90%) are caused by a detrimental
mutation in the JAG1 gene (17). The JAG1 gene encodes a
ligand of the notch receptor which is a key signaling molecule on
the cell surface, and a component of the highly conserved Notch
signaling pathway (14). Mutations
located in almost all regions of the 26 exons of the JAG1
gene have been identified, and the majority are pathogenic
variants. It has been reported that the pathogenic mutations of
JAG1 include missense mutations (11%), nonsense and
frame-shift mutations (69%), pathogenic splice site mutations
(16%), and deletion of the entire JAG1 gene (4%) (8,9,11,12,18–26).
A few reported cases (<1%) have been caused by a mutation in the
NOTCH2 gene (17,27). The NOTCH2 gene encodes a
member of the Notch family, and the Notch receptors (Notch1, 2, 3
and 4 in humans) share structural characteristics including an
intracellular domain with seven ankyrin (ANK) repeats and an
extracellular domain composed of multiple epidermal growth
factor-like (EGF) repeats. To date, ten pathogenic variants
associated with AGS in the NOTCH2 gene have been identified.
Of these, seven are localized in the EGF-like repeats and ANK
repeats domains in NOTCH2 protein (27). The remaining three pathogenic
variants are splice site mutations, frame-shift variants and
nonsense variants (28,29).
Materials and methods
Ethics statement
The present study was approved by the Institutional
Review Board on Bioethics and Biosafety of The Third People's
Hospital of Shenzhen (Shenzhen, China). Written informed consent
was obtained from all participants. All of samples were obtained
and analyzed according to appropriate ethical approvals.
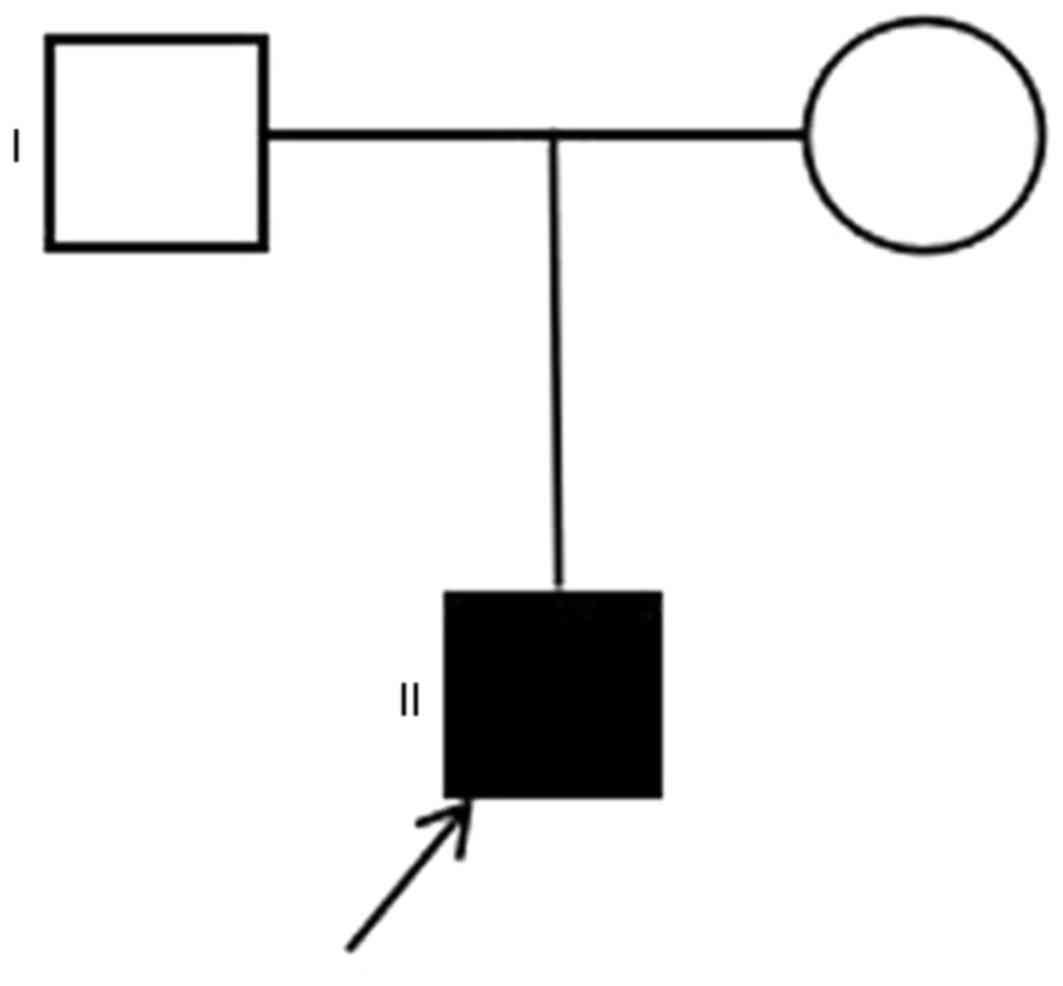
Pedigree and subjects
Samples were obtained from two generations of a
Chinese family. The proband (II:1; Fig. 1) was an 11 year-old boy diagnosed
with chronic intrahepatic cholestasis resulting from AGS, who had a
9-year history of pruritus and a 7-year history of jaundice. He was
the first full-term child of this family, and in 2005 presented
with cutaneous pruritus with no clear cause, particularly on the
lower limbs, accompanied with yellow urine. In 2007, the patient
experienced loss of strength and appetite, jaundice, and occasional
vomiting. In March 2015, liver function tests revealed that levels
of total bilirubin (TBIL, 30.9 µmol/l), direct bilirubin (DBIL,
15.9 µmol/l), total bile acid (TBA, 49.6 µmol/l), alanine
transaminase (ALT, 204 U/l), aspartate aminotransferase (AST, 128
U/l), γ-glutamyl transferase (γ-GGT, 517 U/l), total cholesterol
(TC, 10.77 mmol/l) and free triiodothyronine (FT3, 7.04 pmol/l) in
the patients were greater compared with healthy levels. Normal
ranges are as follows: TBIL, 5.1–19.0 µmol/l; DBIL, 0.1–5.1 µmol/l;
TBA 0–10 µmol/l; ALT, 0–40 U/l; AST, 0–40 U/l; γ-GGT, 7–32 U/l; TC,
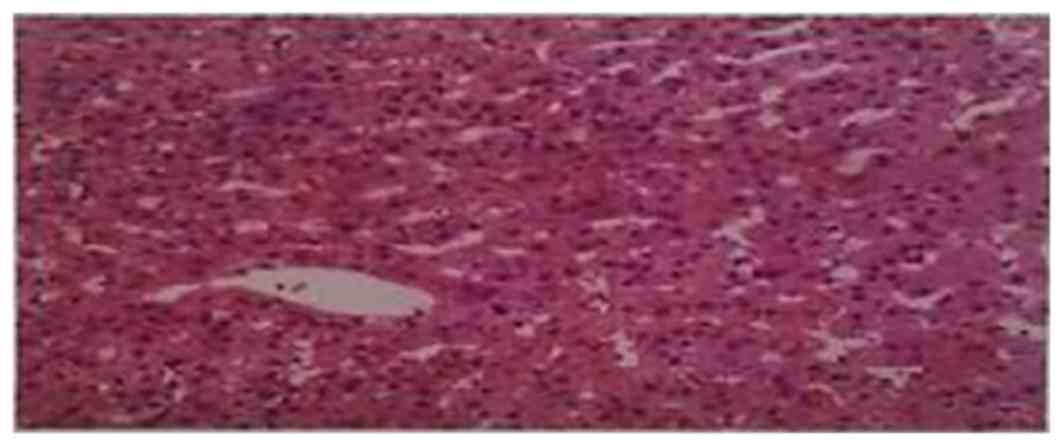
3.35–6.45 mmol/l; FT3, 3.10–6.80 pmol/l. The right lobe of his
liver was biopsied (length, 14 mm; diameter, 1 mm) and was observed
to have minor lesions in the incomplete portal area analyzed
(Fig. 2), suggesting that the
proband had hepatobiliary hypoplasia. A B-scan ultrasound revealed
that the liver, bladder, spleen and pancreas were healthy. The
patient did not exhibit any other clear symptoms.
Preparation of DNA
Peripheral blood was obtained from the proband and
his healthy parents. Genomic DNA was extracted from the blood using
a blood DNA extraction kit (QIAamp DNA Blood Midi kit; Qiagen GmbH,
Hilden, Germany) according to the manufacturer's protocol.
Targeted region capture
sequencing
To construct the capture library, DNA was sheared
randomly by sonication using an LE220 Focused-Ultrasonicator
(Covaris, Inc., Woburn, MA, USA); fragments of 200 to 250 bps were
retained and subsequently purified by Ampure Beads (Beckman
Coulter, Inc., Brea, CA, USA) according to the manufacturer's
protocol. Following this, the two ends of the purified fragments
were repaired, bound to A base and ligated with adapters. DNA
fragments were subsequently amplified by ligation-mediated
polymerase chain reaction (LM-PCR) using Platinum Pfx DNA
polymerase (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) according to the manufacturer's protocol. Cycling
conditions were as follows: An initial predenaturation step at 95°C
for 5 min, followed by 30 cycles of denaturation at 95°C for 30
sec, annealing 58°C for 30 sec, extension at 72°C for 30 sec, and a
final extension step at 72°C for 7 min. PCR products were purified
and hybridized to a customized gene-trapping chip (Roche Applied
Science, Madison, WI, USA) for enrichment. Non-hybridized fragments
were washed out using a NimbleGen Wash and Elution kit (Roche
NimbleGen, Inc., Madison, WI, USA) as per the manufacture's
protocol. A 2100 Bioanalyzer system (Agilent Technologies, Inc.,
Santa Clara, CA, USA) and an ABI StepOne Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) were used to
estimate the magnitude of the enrichment, and the size and the
concentration of the fragments in the two libraries. Subsequently,
the library enriched for target regions was analyzed using a HiSeq
2500 Sequencing system (Illumina, Inc., San Diego, CA, USA) by
paired-end sequencing to obtain raw reads, which were subsequently
analyzed using Illumina Genome Analyzer Pipeline software version
1.3.4 (Illumina, Inc.).
Read mapping and variant analysis
The first step of the analysis was to assess the
quality of the raw data and to remove contaminated or low quality
reads. The reads were mapped to the reference hg19 (build 37.1)
using Burrows Wheeler Aligner software version 0.7.12 (sourceforge.net/projects/bio-bwa/files) and estimated
the effect capturing at the same time. Subsequently, Short
Oligonucleotide Analysis Package single nucleotide polymorphism
(SNP) software version 1.03 (soap.genomics.org.cn/soapsnp.html) and SAMtools
software version 1.2 (samtools.sourceforge.net) were used to detect single
nucleotide variants, and insertions and deletions (indels),
respectively. The last step was to annotate and screen the
suspected mutations. The coverage of targeted region and sequencing
depth calculation were based on all mapped reads. All alterations
were compared and filtered against exome data from the SNP database
(dbSNP build 137; www.ncbi.nlm.nih.gov/snp), the 1000 human genome
dataset (http://www.internationalgenome.org/), HapMap
(http://www.ncbi.nlm.nih.gov/probe/docs/projhapmap/)
and an in-house database of 100 Chinese healthy adults. Sorting
Intolerant From Tolerant software version 1.03 (sift.jcvi.org) was used to predict if amino acid
substitutions, insertions or deletions affected protein function
(30).
Sanger sequencing for validation
The samples of the three individuals in this
pedigree were used for validation. The primers were designed using
Primer 3 software version 4.0.0 (primer3.ut.ee). PCR amplification
and Sanger sequencing were conducted to validate the candidate
mutation (human JAG1; locus, NM_000214; c.3254_3255insT) using
standard protocols on an ABI 3730XL sequencer (Applied Biosystems).
The forward primer for JAG1 was 5′-TTGGTGGTGTTGTCCTCAGA-3′ and the
reverse primer was 5′-AGGGATAAAGGGCAGGAGAA-3′ (product size, 244
bp). PCR amplification was performed using Ex Taq™ DNA
polymerase (Takara Bio, Inc., Otsu, Japan) with an initial
predenaturation step at 95°C for 5 min, followed by 33 cycles of
denaturation at 95°C for 30 sec, annealing at 58°C for 30 sec,
extension at 72°C for 30 sec and a final extension at 72°C for 7
min. PCR products were pooled and subsequently purified with an
AxyPrep DNA Gel Extraction kit (Axygen Biosciences, Inc., Union
City, CA, USA) according to the manufacturer's protocol.
Results
Mutation screening
Targeted region capture sequencing of the proband in
a Chinese family with chronic intrahepatic cholestasis linked with
JAG1, NOTCH2, UGT1A1, ATP8B1, ABCB11 and
ABCB4 genes was performed. The reads were aligned with the
human genome reference. The average depth of the target region was
186.55-fold and the coverage of the target region was 99.67%. A
total of 10,277 genetic variants were revealed, including 10,132
SNPs and 1435 indels. Of the variants, there were 758 missense, 3
nonsense, 1087 synonymous, 248 splice site, 7047 intron and 2188 3′
untranslated region (UTR) mutations, 311 5′UTR and 8 frame-shift
coding indels that were more likely to be pathogenic compared with
the other variants. Priority was given to frame-shift,
non-synonymous and splice site mutations. Using a preliminary
screening process, ten candidate sites were revealed (Table I). Considering the frequency in the
public and in-house databases, nine other candidate sites were
excluded. A novel insertion mutation, c.3254_3255insT
(p.Leu1085PhefsX24), was identified in exon 26 of the JAG1
gene in the proband. The mutation in the JAG1 gene was
selected for further validation, as it is known to be the
disease-causing gene.
 | Table I.Targeted sequence capture
sequencing. |
Table I.
Targeted sequence capture
sequencing.
| Gene | Transcript | Nucleotide
change | AA change | Gene subregion | Hom/Het | Location | RS number | Frequency (public
database) | Frequency (in-house
database) |
|---|
| ATP8B1 | NM_005603 | c.3454G>A | p.Ala1152Thr | EX27 | Hom | chr18:55317676 | rs222581 | 0.9991 | C |
| ATP8B1 | NM_005603 | c.811A>C | p.Arg271Arg | EX10 | Hom | chr18:55362532 | rs319443 | 0.9908 | C |
| ATP8B1 | NM_005603 | c.696T>C | p.Asp232Asp | EX8 | Hom | chr18:55364852 | rs319438 | 0.9908 | C |
| NOTCH2 | NM_024408 | c.7341T>A | p.Gly2447Gly | EX34 | Hom | chr1:120458004 | rs6685892 | 0.0495 | B |
| JAG1 | NM_000214 | c.765C>T | p.Tyr255Tyr | EX6 | Hom | chr20:10633237 | rs1131695 | 0.1767 | C |
| JAG1 | NM_000214 | c.588C>T | p.Cys196Cys | EX4 | Het | chr20:10639222 | rs1801138 | 0.2637 | C |
| ABCB11 | NM_003742 | c.3084A>G | p.Ala1028Ala | EX24 | Het | chr2:169789016 | rs497692 | 0.4103 | C |
| ABCB11 | NM_003742 | c.1331T>C | p.Val444Ala | EX13 | Hom | chr2:169830328 | rs2287622 | 0.3526 | C |
| ABCB4 | NM_018849 | c.504C>T | p.Asn168Asn | EX6 | Het | chr7:87082292 | rs1202283 | 0.3782 | C |
| JAG1 | NM_000214 |
c.3254_3255insT |
p.Leu1085PhefsX24 | EX26 | Het |
chr20:10620548–10620549 | – | 0 | A |
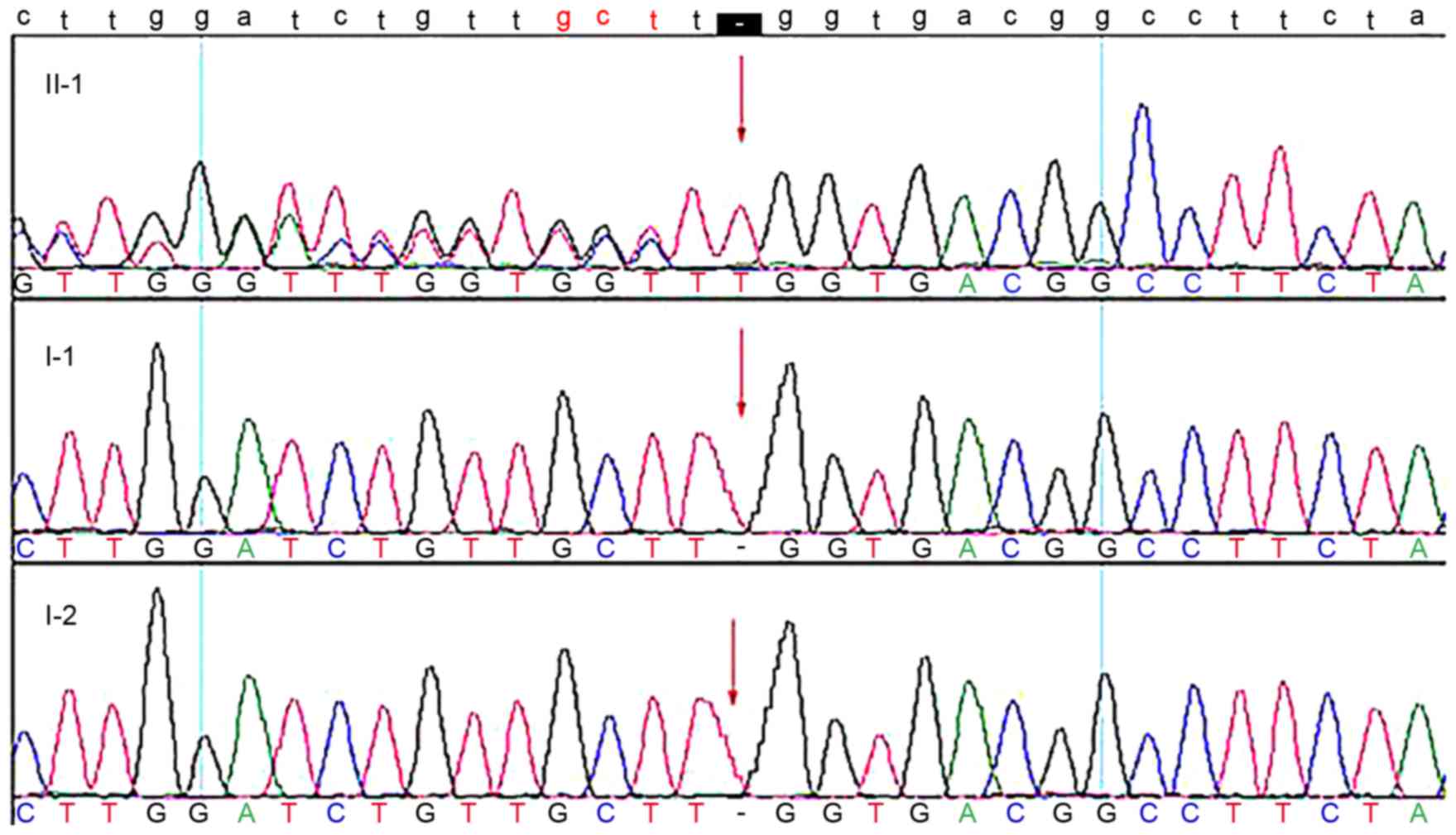
Sanger sequencing
The mutation was subsequently sequenced in three
members of the family via Sanger sequencing. The results of Sanger
sequencing demonstrated that the mutation was present in the
proband but absent in unaffected members (Fig. 3). The mutation results in a
premature stop codon and a truncated protein, which affects the
Notch signaling pathway, influencing multi-system development in
the embryonic period.
Discussion
Originally, the proband was diagnosed with chronic
intrahepatic cholestasis, which is a syndrome characterized by
jaundice, pruritus and hepatomegaly. His serum levels of bilirubin,
alkaline phosphatase, cholesterol and other indicators were greater
compared with normal values (31).
The primary function of the liver is the formation and secretion of
bile, which is necessary for lipid digestion. However, cholestasis
is a condition where bile does not form or does not flow from the
liver to duodenum, and may be caused by numerous factors. In the
process of bile formation, environmental factors, including viral
hepatitis and intrahepatic cholestasis of pregnancy, may lead to
hepatocellular cholestasis. However, the present study primarily
focused on genetic factors.
To investigate the underlying mechanisms of chronic
intrahepatic cholestasis, chip capture high-throughput sequencing
was performed on the DNA of the proband and Sanger sequencing was
conducted to validate the candidate mutation in this family. A
novel frame-shift variant was identified in the JAG1 gene,
which is the primary pathogenic gene of AGS. AGS, linked with the
JAG1 and NOTCH2 genes, is associated with dysfunction
of the liver, heart, skeleton and eyes, and a characteristic facial
appearance. The underlying pathogenic mechanisms remain unclear;
however, these may be attributed to haploinsufficiency (32–37).
A dominant-negative effect of putative truncated proteins is an
additional potential pathogenic factor (34). The majority of mutations in the
JAG1 gene lead to a truncated protein, impairing its ability
to attach to the cell membrane, and disrupting the Notch signaling
pathway, which is involved in embryonic development. JAG1 protein
is the ligand of the Notch receptor, and binding triggers a cascade
of proteolytic cleavage that releases the Notch intracellular
domain (NICD) from the plasma membrane. The NICD translocates to
the nucleus, where it forms a complex with the DNA binding protein
C-promoter binding factor 1, suppressor of hairless, lin-12 and
glp-1 (CSL). Components of the activated complex are recruited to
NICD-CSL, which may lead to the transcriptional activation of Notch
target genes (15,16). The Notch signaling pathway has been
reported to be linked to AGS, and is important in cell fate
determination in Drosophila melanogaster and
Caenorbabditis elegans (13,38,39).
It is highly conserved and essential for proper embryonic
development in all metazoan organisms. Furthermore, the JAG1
protein is a key signaling molecule on the cell surface. The
present study demonstrated that an insert mutation causes a
premature termination codon that encodes a truncated protein. The
truncated protein has a dominant negative effect in in vitro
cell culture systems and in vivo transgenic Drosophila
melanogaster (35,40,41).
The truncated protein lacks the transmembrane region necessary for
the protein product to embed in the cell membrane and contribute to
signaling. The conserved region of the JAG1 protein includes a
signal peptide (aa:1-21; cDNA:414-477), a delta-serrate-lin12-like
region (aa:186-230; cDNA:972-1104), EGF-like repeats (aa:235-863;
cDNA:1119-3003), a cysteine rich region (aa:864-1003;
cDNA:3006-3423) and a transmembrane (TM) domain (aa:1069-1094;
cDNA:3621-3696) (8,42). The mutation identified in the
present study, c.3254_3255insT (p.Leu1085PhefsX24), is located in
the TM region.
In conclusion, the present study identified a de
novo heterozygous mutation c.3254_3255insT (p.Leu1085PhefsX24)
in exon 26 of the JAG1 gene, which may be responsible for
AGS in the proband. In addition, these data expanded the genotypic
spectrum of JAG1 mutations associated with AGS, and provided
evidence that gene detection may be used for clinical diagnosis.
Additionally, these results suggested that targeted region capture
sequencing is a reliable, cost-effective and accurate clinical
molecular diagnosis method and may improve the accuracy of clinical
diagnosis for AGS and associated disorders.
Acknowledgements
The authors would like to thank the family for
participating in the present study, and the BGI-Shenzhen and The
Third People's Hospital of Shenzhen.
References
|
1
|
Alagille D, Odièvre M, Gautier M and
Dommergues JP: Hepatic ductular hypoplasia with characteristic
facies, vertebral malformations, retarded physical, mental, and
sexual development, and cardiac murmur. J Pediatr. 86:63–71. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alagille D, Estrada A, Hadchouel M,
Gautier M, Odièvre M and Dommergues JP: Syndromic paucity of
interlobular bile ducts (Alagille syndrome or arteriohepatic
dysplasia): Review of 80 cases. J Pediatr. 110:195–200. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Giannakudis J, Röpke A, Kujat A,
Krajewska-Walasek M, Hughes H, Fryns JP, Bankier A, Amor D,
Schlicker M and Hansmann I: Parental mosaicism of JAG1 mutations in
families with Alagille syndrome. Eur J Hum Genet. 9:209–216. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yuan ZR, Kohsaka T, Ikegaya T, Suzuki T,
Okano S, Abe J, Kobayashi N and Yamada M: Mutational analysis of
the Jagged 1 gene in Alagille syndrome families. Hum Mol Genet.
7:1363–1369. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim BJ and Fulton AB: The genetics and
ocular findings of Alagille syndrome. Semin Ophthalmol. 22:205–210.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Danks DM, Campbell PE, Jack I, Rogers J
and Smith AL: Studies of the aetiology of neonatal hepatitis and
biliary atresia. Arch Dis Child. 52:360–367. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Emerick KM, Rand EB, Goldmuntz E, Krantz
ID, Spinner NB and Piccoli DA: Features of Alagille syndrome in 92
patients: Frequency and relation to prognosis. Hepatology.
29:822–829. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krantz ID, Colliton RP, Genin A, Rand EB,
Li L, Piccoli DA and Spinner NB: Spectrum and frequency of jagged1
(JAG1) mutations in Alagille syndrome patients and their families.
Am J Hum Genet. 62:1361–1369. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crosnier C, Driancourt C, Raynaud N,
Dhorne-Pollet S, Pollet N, Bernard O, Hadchouel M and
Meunier-Rotival M: Mutations in JAGGED1 gene are predominantly
sporadic in Alagille syndrome. Gastroenterology. 116:1141–1148.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Spinner NB, Colliton RP, Crosnier C,
Krantz ID, Hadchouel M and Meunier-Rotival M: Jagged1 mutations in
Alagille syndrome. Hum Mutat. 17:18–33. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Krantz ID, Deng Y, Genin A, Banta
AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, et al: Alagille
syndrome is caused by mutations in human Jagged1, which encodes a
ligand for Notch1. Nat Genet. 16:243–251. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oda T, Elkahloun AG, Pike BL, Okajima K,
Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS
and Chandrasekharappa SC: Mutations in the human Jagged1 gene are
responsible for Alagille syndrome. Nat Genet. 16:235–242. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Artavanis-Tsakonas S, Matsuno K and
Fortini ME: Notch signaling. Science. 268:225–232. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Callahan R and Egan SE: Notch signaling in
mammary development and oncogenesis. J Mammary Gland Biol
Neoplasia. 9:145–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hansson EM, Lendahl U and Chapman G: Notch
signaling in development and disease. Semin Cancer Biol.
14:320–328. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Harper JA, Yuan JS, Tan JB, Visan I and
Guidos CJ: Notch signaling in development and disease. Clin Genet.
64:461–472. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Turnpenny PD and Ellard S: Alagille
syndrome: Pathogenesis, diagnosis and management. Eur J Hum Genet.
20:251–257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan ZR, Kohsaka T, Ikegaya T, Suzuki T,
Okano S, Abe J, Kobayashi N and Yamada M: Mutational analysis of
the Jagged 1 gene in Alagille syndrome families. Hum Mole Genet.
7:1363–1369. 1998. View Article : Google Scholar
|
|
19
|
Krantz ID, Smith R, Colliton RP, Tinkel H,
Zackai EH, Piccoli DA, Goldmuntz E and Spinner NB: Jagged1
mutations in patients ascertained with isolated congenital heart
defects. Am J Med Genet. 84:56–60. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Onouchi Y, Kurahashi H, Tajiri H, Ida S,
Okada S and Nakamura Y: Genetic alterations in the JAG1 gene in
Japanese patients with Alagille syndrome. J Hum Genet. 44:235–239.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pilia G, Uda M, Macis D, Frau F, Crisponi
L, Balli F, Barbera C, Colombo C, Frediani T, Gatti R, et al:
Jagged-1 mutation analysis in Italian Alagille syndrome patients.
Hum Mutat. 14:394–400. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Crosnier C, Attié-Bitach T, Encha-Razavi
F, Audollent S, Soudy F, Hadchouel M, Meunier-Rotival M and
Vekemans M: JAGGED1 gene expression during human embryogenesis
elucidates the wide phenotypic spectrum of Alagille syndrome.
Hepatology. 32:574–581. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Heritage ML, MacMillan JC, Colliton RP,
Genin A, Spinner NB and Anderson GJ: Jagged1 (JAG1) mutation
detection in an Australian Alagille syndrome population. Hum Mutat.
16:408–416. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Colliton RP, Bason L, Lu FM, Piccoli DA,
Krantz ID and Spinner NB: Mutation analysis of Jagged1 (JAG 1) in
Alagille syndrome patients. Hum Mutat. 17:151–152. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Giannakudis J, Röpke A, Kujat A,
Krajewska-Walasek M, Hughes H, Fryns JP, Bankier A, Amor D,
Schlicker M and Hansmann I: Parental mosaicism of JAG1 mutations in
families with Alagille syndrome. Eur J Hum Genet. 9:209–216. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Röpke A, Kujat A, Gräber M, Giannakudis J
and Hansmann I: Identification of 36 novel Jagged1 (JAG1) mutations
in patients with Alagille syndrome. Hum Mutat. 21:1002003.
View Article : Google Scholar
|
|
27
|
McDaniell R, Warthen DM, Sanchez-Lara PA,
Pai A, Krantz ID, Piccoli DA and Spinner NB: NOTCH2 mutations cause
Alagille syndrome, a heterogeneous disorder of the notch signaling
pathway. Am J Hum Genet. 79:169–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kamath BM, Bauer RC, Loomes KM, Chao G,
Gerfen J, Hutchinson A, Hardikar W, Hirschfield G, Jara P, Krantz
ID, et al: NOTCH2 mutations in Alagille syndrome. J Med Genet.
49:138–144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Penton AL, Leonard LD and Spinner NB:
Notch signaling in human development and disease. Semin Cell Dev
Biol. 23:450–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu J and Ng PC: SIFT Indel: Predictions
for the functional effects of amino acid insertions/deletions in
proteins. PLoS One. 8:e779402013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rudzki C, Ishak KG and Zimmerman HJ:
Chronic intrahepatic cholestasis of sarcoidosis. Am J Med.
59:373–387. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Morrissette JD, Colliton RP and Spinner
NB: Defective intracellular transport and processing of JAG1
missense mutations in Alagille syndrome. Hum Mol Genet. 10:405–413.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu F, Morrissette JJ and Spinner NB:
Conditional JAG1 mutation shows the developing heart is more
sensitive than developing liver to JAG1 dosage. Am J Hum Genet.
72:1065–1070. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Boyer J, Crosnier C, Driancourt C, Raynaud
N, Gonzales M, Hadchouel M and Meunier-Rotival M: Expression of
mutant JAGGED1 alleles in patients with Alagille syndrome. Hum
Genet. 116:445–453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun X and Artavanis-Tsakonas S: Secreted
forms of DELTA and SERRATE define antagonists of Notch signaling in
Drosophila. Development. 124:3439–3448. 1997.PubMed/NCBI
|
|
36
|
Wong MK, Prudovsky I, Vary C, Booth C,
Liaw L, Mousa S, Small D and Maciag T: A non-transmembrane form of
Jagged-1 regulates the formation of matrix-dependent chord-like
structures. Biochem Biophys Res Commun. 268:853–859. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vas V, Szilágyi L, Pálóczi K and Uher F:
Soluble Jagged-1 is able to inhibit the function of its multivalent
form to induce hematopoietic stem cell self-renewal in a surrogate
in vitro assay. J Leukoc Biol. 75:714–720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kopan R and Weintraub H: Mouse notch:
Expression in hair follicles correlates with cell fate
determination. J Cell Biol. 121:631–641. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Robey E, Chang D, Itano A, Cado D,
Alexander H, Lans D, Weinmaster G and Salmon P: An activated form
of Notch influences the choice between CD4 and CD8 T cell lineages.
Cell. 87:483–492. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wong MK, Prudovsky I, Vary C, Booth C,
Liaw L, Mousa S, Small D and Maciag T: A non-transmembrane form of
Jagged-1 regulates the formation of matrix-dependent chord-like
structures. Biochem Biophys Res Commun. 268:853–859. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Small D, Kovalenko D, Kacer D, Liaw L,
Landriscina M, Di Serio C, Prudovsky I and Maciag T: Soluble Jagged
1 represses the function of its transmembrane form to induce the
formation of the Src-dependent chord-like phenotype. J Biol Chem.
276:32022–32030. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lindsell CE, Shawber CJ, Boulter J and
Weinmaster G: Jagged: A mammalian ligand that activates Notch1.
Cell. 80:909–917. 1995. View Article : Google Scholar : PubMed/NCBI
|