Introduction
Myocardial infarction is a serious disease, which
contributes to high mortality rates in modern society. Myocardial
reperfusion therapies, including percutaneous coronary
intervention, thrombolytic therapy and coronary artery bypass
grafts, have been regarded as the most effective approaches for
rescuing ischemic myocardial tissue. However, regardless of the
type of effective reperfusion therapy used for myocardial
infarction, reperfusion itself can cause further myocardial injury,
which can even attenuate the therapeutic benefit (1). Myocardial I/R can induce local
myocardial inflammation, including promoting the release of various
cytokines, including interleukin (IL)-17A, tumor necrosis factor-α
(TNF-α) and IL-6, and promoting the activation of inflammatory
cells, including neutrophils, which is one of the crucial
pathophysiological processes in myocardial I/R injury (2–4).
Apoptosis is another pathological result of myocardial I/R, which
can result in reversible or irreversible damage of myocardial
tissue, accompanied with the inflammatory processes mentioned above
(5).
IL-23, as a newly-identified member of the IL-12
family, has attracted increased attention (6). IL-23, predominantly produced by
activated macrophages and dendritic cells, is a heterodimeric
cytokine composed of IL-23p19 and IL-12/IL-23p40 subunits (7). Previous studies have confirmed that
IL-23, as a pro-inflammatory cytokine, is critical in infections,
autoimmune diseases, tumors and inflammatory diseases, including
myocardial I/R injury, by promoting the expression of inflammatory
cytokines and inflammatory responses (8–11).
IL-17A, expressed at high levels in myocardial I/R injury, has been
described as a downstream pro-inflammatory cytokine. The inhibition
of IL-17A release can reduce myocardial I/R injury and improve
cardiac function (12). Studies
have also indicated that IL-23 can regulate the expression of
IL-17A (8,11). The present study attempted to test
the hypothesis that IL-23 aggravates myocardial I/R injury by
promoting inflammatory responses and myocardial apoptosis, which
may be associated with the high expression of IL-17A and
upregulation of the Janus kinase 2/signal transducers and
activators of transcription 3 (JAK2-STAT3) signaling pathway.
Materials and methods
Construction of adenovirus (Ad)
vectors
IL-23 and empty plasmid Ads (termed Ad-IL-23 and
Ad-GFP) were constructed by cloning the IL-23 cDNA or empty
plasmid. The recombinant viruses were then amplified in HEK 293
cells (American Type Culture Collection, Manassas, VA, USA) by
transfection and finally purified using an Adeno-X™ purification
kit (Microbix Biosystems, Toronto, ON, Canada) in order to reach
the titer of 1011 pfu/ml.
Animal preparation and experimental
design
All experimental protocols conformed to the
Guideline for the Care and Use of Laboratory Animals published by
the US National Institutes of Health (NIH Publication, revised
1996) (13) and were approved by
the Renmin Hospital of Wuhan University Animal Care and Use
Committee (Wuhan, China). Male Sprague-Dawley rats (200–250 g) were
supplied by the Experimental Animal Center of Vital River
Laboratories (Beijing, China) and randomly assigned into the
following six treatment groups: Group 1, sham-operated control (SO;
n=10) rats subjected to surgery without myocardial ischemia; group
2, I/R (n=10) rats subjected to occlusion of the left anterior
descending coronary artery (LAD) for 30 min, followed by
reperfusion for 4 h; group 3, Ad-GFP+I/R (n=6) rats treated with
Ad-GFP (1011 pfu/ml, 100 µl per rat, intramyocardially)
for 72 h prior to LAD occlusion; group 4, Ad-IL-23+I/R (n=6) rats
treated with Ad-IL-23 (1011 pfu/ml, 100 µl per rat,
intramyocardially) for 72 h prior to LAD occlusion; group 5,
anti-IL-23+I/R (n=6) rats treated with anti-IL-23 neutralized
monoclonal antibodies (200 µg per rat, i.v. into the tail vein)
from Biosynthesis Biotechnology Co., Ltd. (Beijing, China) 30 min
prior to LAD occlusion; group 6, Ad-IL-23+AG490+I/R (n=6) rats
treated with Ad-IL-23 (1011 pfu/ml, 100 µl per rat,
intramyocardially) for 72 h and injection of AG490 (an inhibitor of
JAK2-STAT3) from MedChemExpress USA (Monmouth Junction, NJ, USA; 3
mg/kg, i.v. into tail vein), 30 min prior to LAD occlusion.
Sodium pentobarbital (2.5%; 45 mg/kg, i.p.) was used
as the anesthetic in surgery. The I/R used in the rats model was
performed, as previously described (14). Changes in ST segment elevation in
Leads-II and regional cyanosis of the myocardial surface were
considered to be signs of successful establishment of the
myocardial I/R model. Subsequently, a half dose (22.5 mg/kg, i.p.)
of 2.5% sodium pentobarbital was injected into the postoperative
rats. Samples, including 2 ml of blood from the jugular vein and
heart tissue in the infarct area (white) and risk area (5 mm around
the infarct area) were obtained immediately and frozen at −80°C for
subsequent assays.
Assessment of myocardial injury
A previously described double-staining technique was
used to determine the infarct size (14). In brief, following 4 h reperfusion,
the LAD was occluded again and 2 ml Evans Blue dye (1%;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) was injected
via the jugular vein. The heart was excised and frozen at −80°C for
15 min, and was then cut into 1.5 mm transverse sections from the
apex to the base, which were subsequently incubated in 1.0%
2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich; Merck
Millipore) for 15 min at 37°C. The sizes of the infarct area
(white) and the area at risk (red and white) were measured using an
image analyzer (Image-Pro Plus 3.0; Media Cybernetics, Inc., Silver
Spring, MD, USA). The percentage of the risk area volume (% infarct
area/risk + infarct area) was used to determine the infarct
size.
The serum levels of lactate dehydrogenase (LDH) and
creatine kinase (CK) were measured in the blood samples, which were
collected, centrifuged (2,050 × g, 5 min, 4°C) and stored at −20°C
until analyses. Standard techniques using commercialized assay kits
were used according to the manufacturer's protocol (Nanjing
Jiancheng Bioengineering Institute, Najing, China) for the
analyses. Values are expressed in international units (IU) per
liter.
Assessment of myocardial inflammatory
parameters
The levels of TNF-α and IL-6 in myocardial tissues
were measured using commercial enzyme-linked immunosorbent assay
kits (TNF-α and IL-6; Nanjing Jiancheng Bioengineering Institute)
according to the manufacturer's protocol. The sensitivity of the
assay was 1 pg/ml.
Cardiac myocyte protein concentrations were measured
using western blot assays as described previously (15). In brief, total protein content was
determined using a bicinchoninic acid protein assay kit (Beyotime
Institute of Biotechnology, Haimen, China) according the
manufacturer's protocol. Protein extracts (40 µg) of the cardiac
tissues in radioimmuno-precipitation assay lysis buffer (Beyotime
Institute of Biotechnology) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, transferred onto
polyvinylidene difuoride membranes (EMD Millipore, Billerica, MA,
USA) and probed with primary antibodies at 4°C overnight, including
anti-IL-23 (cat no. bs-1193R; diluted 1:300; Beijing Bioss
Biological Co., Ltd., Beijing, China), anti-IL-17A (cat no.
bs-1183R; diluted 1:300; Beijing Bioss Biological Co., Ltd,
Beijing, China) and β-actin (cat no. BM0627; diluted 1:500; Wuhan
Boster Bioengineering Co, Ltd., Wuhan, China). Following incubation
with the appropriate secondary antibodies (The horseradish
peroxidase-conjugated mouse IgG (cat no. BA1051); The horseradish
peroxidase-conjugated rabbit IgG (cat no. BA1054); diluted
1:50,000; Wuhan Boster Bioengineering Co, Ltd.) for 2 h at room
temperature, the specific bands were visualized using an ECL
detection system according to the manufacturer's protocol.
Assessment of myocardial
apoptosis
Myocardial apoptosis was examined using the
TdTmediated dUTP nick-end labeling (TUNEL) assay, as described
previously (16). In brief,
following being fixed in 4% paraformaldehyde and embedded in
paraffin, the myocardial tissue samples were cut into 5 µm blocks.
The percentages of apoptotic cells were evaluated in the five
slides of each block using the TUNEL assay. Subsequently, five
fields (magnification, ×200) were randomly selected on each section
under a light microscope (Leica Microsystems, Inc., Buffalo Grove,
IL, USA). Cells (positive brown cells and normal blue cells) were
counted using the MIAS 4.0 medical image analysis system (Beijing
Bingyang Keji Corporation, Beijing, China). The apoptosis index
(positive cells/total cells × 100%) was used to express myocardial
apoptosis.
The levels of B-cell lymphoma 2 (Bcl-2),
Bcl-2-associated X protein (Bax) and caspase-3 in myocardial
tissues were assessed using western blot assays, as mentioned
above. Anti-Bcl-2 ((cat no. sc-7382; diluted 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), anti-Bax (cat no. BS6420;
diluted 1:1,000; Bioworld Technology, Minneapolis, MN, USA) and
anti-caspase-3 (cat no. 19677-1-AP; diluted 1:1,000; Wuhan Mitaka
Biotechnology Co, Ltd., Wuhan, China) were used as primary
antibodies (4°C, overnight), and the secondary antibodies were as
described above (cat no. BA1051; cat no. BA1054).
Assessment of JAK2-STAT3 signaling
pathway activation
The levels of JAK2, phosphorylated (P-)JAK2, STAT3
and P-STAT3 were assessed using western blot assays, as mentioned
above. Anti-JAK2 (cat no. sc-21870; diluted 1:1,000; Santa Cruz
Biotechnology, Inc.), anti-P-JAK2 (cat no. bs-3206R; diluted 1:200;
Beijing Bioss Biological Co., Ltd.), anti-STAT3 (cat no. sc-483;
diluted 1:500; Santa Cruz Biotechnology, Inc.) and anti-P-STAT3
(cat no. BS4181; diluted 1:800; Bioworld Technology) were used as
primary antibodies (4°C, overnight). And the secondary antibodies
were the horseradish peroxidase-conjugated mouse IgG (cat no.
BA1051), the horseradish peroxidase-conjugated rabbit IgG (cat no.
BA1054) and the horseradish peroxidase-conjugated goat IgG (cat no.
BA1060); diluted 1:50,000; Wuhan Boster Bioengineering Co,
Ltd..
Statistical analysis
Statistical analysis was performed using SPSS 17.0
(SPSS, Inc., Chicago, IL, USA). All values are expressed as the
mean ± standard deviation. Student's t-test was used for
between-group comparisons. One-way analysis of variance or a Welch
test was used for comparisons among groups, and Tukey's post hoc
test was used for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
Myocardial expression of IL-23 during
myocardial I/R
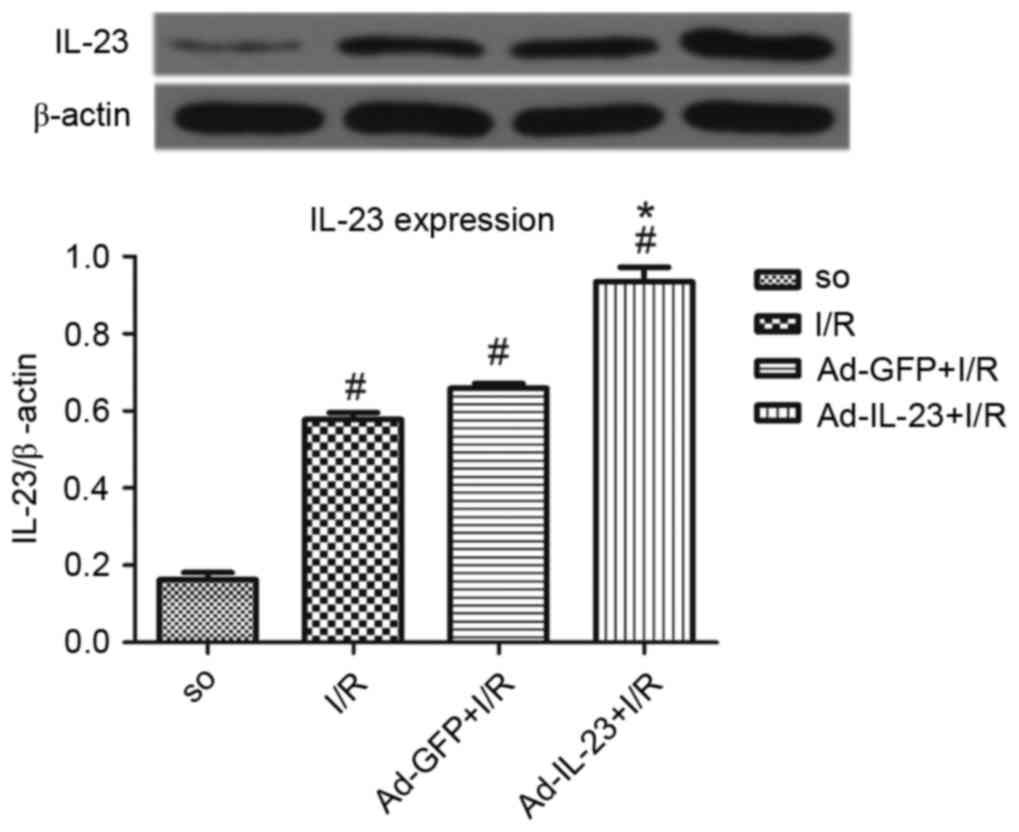
Compared with the SO group, the I/R group had a
significantly higher expression level of IL-23 (0.578±0.018, vs.
0.163±0.018; P<0.05). The level of IL-23 in the Ad-IL-23 group
was significantly increased, compared with that in the I/R group
(0.935±0.038; P<0.05). However, no significant difference was
found between the Ad-GFP group and I/R group (0.659±0.013;
P>0.05; Fig. 1).
IL-23 promotes myocardial injury
following I/R
Compared with the I/R group, the infarct size in the
Ad-IL-23 group was significantly increased (52.4±0.7, vs.
42.4±1.5%; P<0.05). However, no significant difference was found
between the Ad-GFP group and I/R group (42.5±1.9%; P>0.05). The
addition of AG490 significantly inhibited the effect of Ad-IL-23
with regard to decreasing infarct size, compared with that in the
Ad-IL-23 group (47.2±1.3%; P<0.05; Fig. 2A).
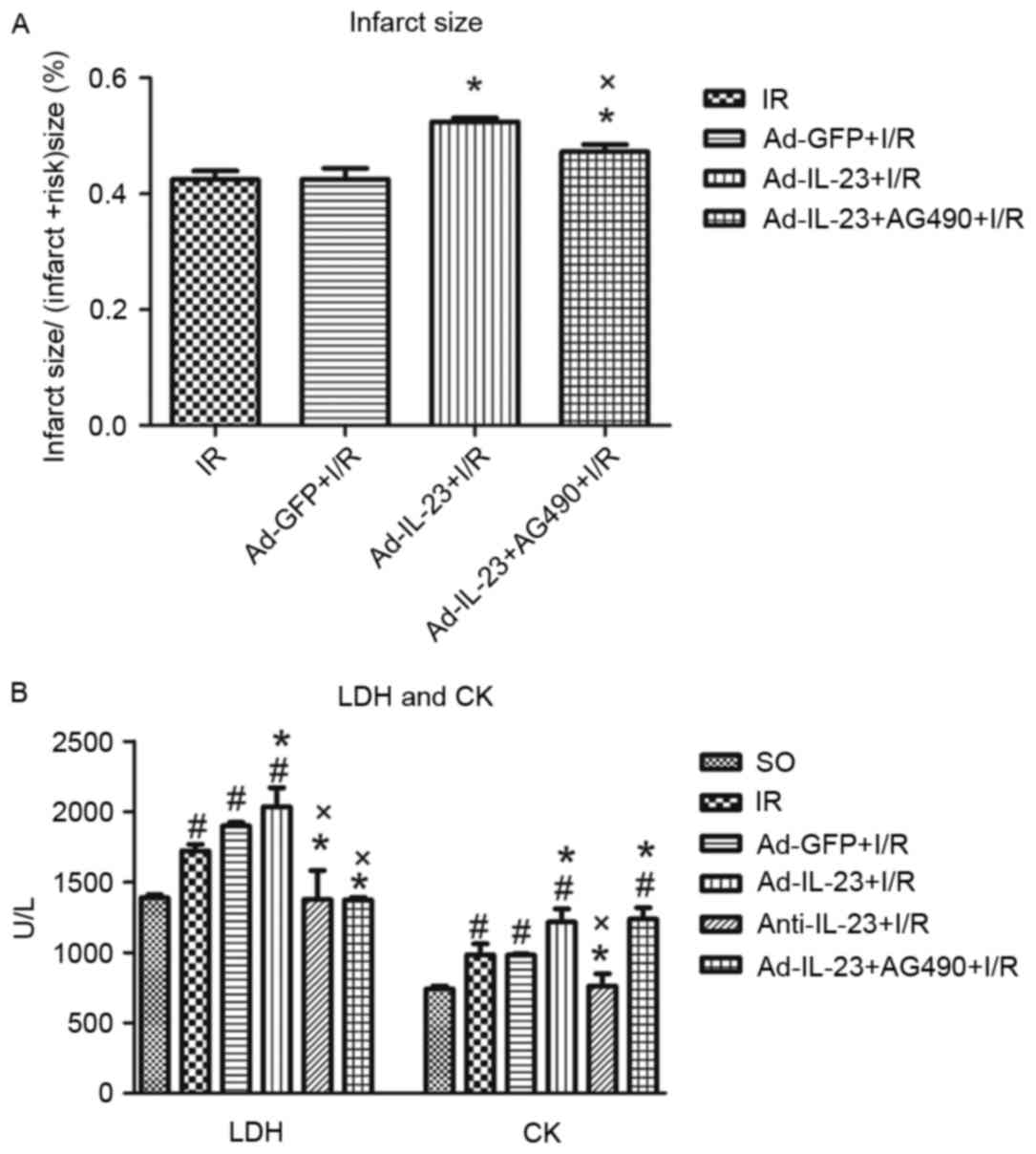 | Figure 2.Ad-IL-23 increases I/R-induced
myocardial injury, which is partially attenuated by AG490. (A)
Myocardial infarct size (n=5). (B) Serum levels of LDH and CK
(n=6). #P<0.05, vs. SO group; *P<0.05, vs. I/R
group; xP<0.05, vs. Ad-IL-23+I/R group. SO, sham
operated; I/R, ischemia/reperfusion; IL-23, interleukin 23; Ad,
adenovirus; LDH, lactate dehydrogenase; CK, creatine kinase. |
Following 4 h reperfusion, the levels of LDH
(1,721.2±106.1 U/l) and CK (985.2±172.3 U/l) in the I/R group were
significantly increased (P<0.05), compared with that in the SO
group. However, no significant difference was found between the
Ad-GFP group and I/R group in LDH (1,899.0±44.0 U/l) or CK
(981.5±17.1 U/l; P>0.05). Ad-IL-23 increased the high expression
levels of LDH (2,035.4±270.4 U/l) and CK (1215.7±187.5 U/l) induced
by myocardial I/R (P<0.05) compared with the I/R group. However,
the levels of LDH (1,376.6±411.1 U/l) and CK (762.2±175.5 U/l) in
the anti-IL-23 group were decreased, compared with those in the I/R
group (P<0.05). Furthermore, the addition of AG490 partially
inhibited the effect of Ad-IL-23 with regard to decreasing the
expression level of LDH (1,374.1±27.3 U/l) compared with that in
the Ad-IL-23 group (P<0.05; Fig.
2B).
IL-23 intensifies inflammatory
responses
Compared with the SO group, a higher expression
level of IL-17A was observe in the I/R group (0.451±0.105, vs.
0.160±0.030; P<0.05). Ad-IL-23 enhanced the increased expression
of IL-17A induced by myocardial I/R (0.673±0.149; P<0.05).
However, anti-IL-23 significantly inhibited the I/R-induced
increase of IL-17A (0.250±0.055; P<0.05). Furthermore, the
addition of AG490 significantly inhibited the effect of Ad-IL-23
(0.572±0.112; P<0.05; Fig.
3A.
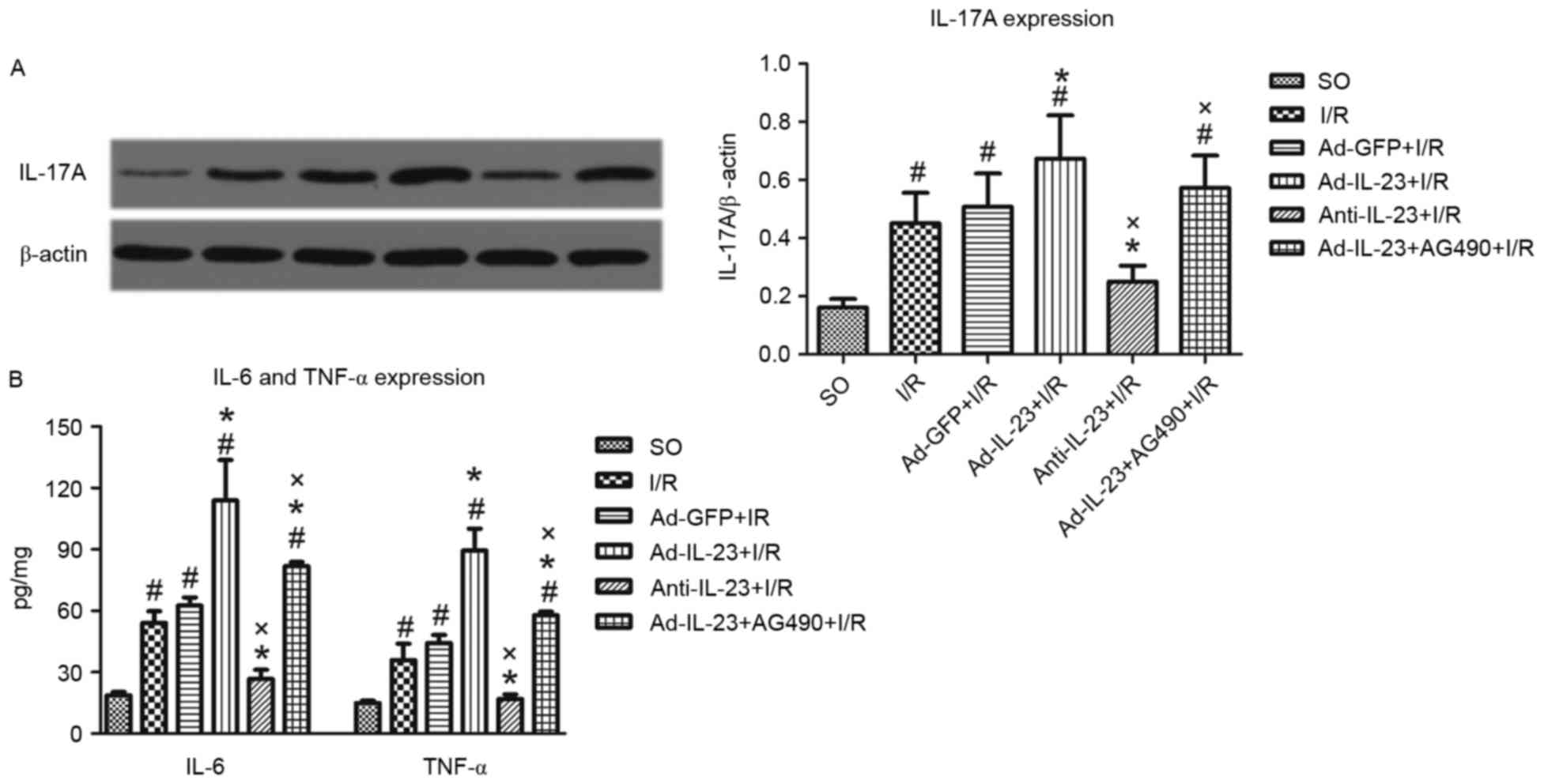 | Figure 3.Ad-IL-23 intensifies the high
expression levels of IL-17A, TNF-α and IL-6 induced by myocardial
I/R, which is inhibited by AG490. (A) Levels of IL-17A in
myocardial tissues from infarct and risk regions (n=6). (B)
Expression levels of TNF-α and IL-6 in myocardial tissues from
infarct and risk regions (n=6). #P<0.05, vs. SO
group; *P<0.05, vs. I/R group; xP<0.05, vs.
Ad-IL-23+I/R group. SO, sham operated; I/R, ischemia/reperfusion;
IL, interleukin; Ad, adenovirus; TNF-α, tumor necrosis
factor-α. |
Following 4 h reperfusion, the levels of TNF-α
(35.9±14.0 pg/mg) and IL-6 (54.1±9.8 pg/mg) were significantly
increased (P<0.05), compared with those in the SO group.
Ad-IL-23 increased the high expression levels of TNF-α (89.5±18.5
pg/mg) and IL-6 (113.9±34.3 pg/mg) induced by myocardial I/R
(P<0.05). However, anti-IL-23 decreased the expression levels of
TNF-α (17.0±3.8 pg/mg) and IL-6 (26.7±7.8 pg/mg), compared with
those in the I/R group (P<0.05). Furthermore, the addition of
AG490 significantly inhibited the effect of Ad-IL-23 with regard to
decreasing the expression levels of TNF-α (57.8±3.0 pg/mg) and IL-6
(81.9±3.4 pg/mg), compared with those in Ad-IL-23 group (P<0.05;
Fig. 3B.
IL-23 accelerates myocardial
apoptosis
Ad-IL-23 significantly increased the I/R-induced
myocardial apoptosis (26.6±1.0, vs. 18.7±0.5%; P<0.05), however,
its effect was inhibited by AG490 (21.9±1.1%; P<0.05; Fig. 4A.
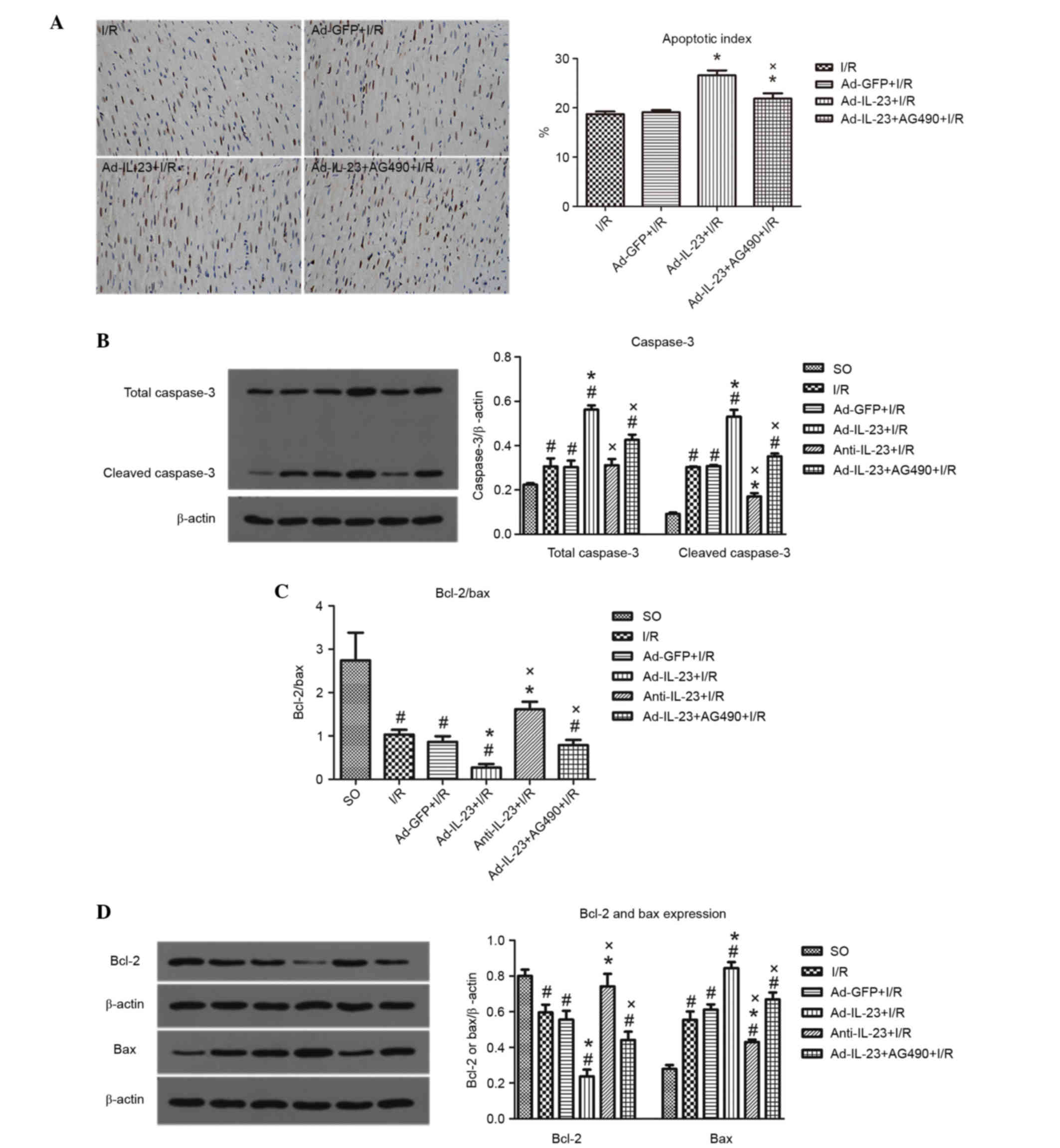 | Figure 4.Ad-IL-23 increases I/R-induced
myocardial apoptosis, which is partially inhibited by AG490. (A)
Images and apoptotic indices (magnification, ×200) of myocardial
tissues from infarct regions (n=5). (B) Expression levels of total
caspase-3 and cleaved caspase-3 in myocardial tissues from infarct
and risk regions (n=6). (C) Ratio of Bcl-2 to Bax in myocardial
tissues from infarct and risk regions (n 6). (D) Levels of Bcl-2
and Bax in myocardial tissues from infarct and risk regions (n=6).
#P<0.05, vs. SO group; *P<0.05, vs. I/R group;
xP<0.05, vs. Ad-IL-23+I/R group. SO, sham operated;
I/R, ischemia/reperfusion; IL-23, interleukin-23; Ad, adenovirus;
Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated X protein. |
Following 4 h reperfusion, the levels of total
caspase-3 (0.306±0.062) and cleaved caspase-3 (0.303±0.006) were
significantly increased (P<0.05), compared with those in the SO
group. Ad-IL-23 significantly increased the high expression levels
of total caspase-3 (0.562±0.033) and cleaved caspase-3
(0.530±0.055) induced by myocardial I/R (P<0.05). However,
anti-IL-23 decreased the I/R-induced high level of cleaved
caspase-3 (0.171±0.026, P<0.05) but not total caspase-3
(0.312±0.049; P>0.05). The effects of Ad-IL-23 on total
caspase-3 (0.426±0.041) and cleaved caspase-3 (0.352±0.022) were
inhibited by AG490 (P<0.05; Fig.
4B.
Compared with the SO group, the I/R group had a
lower ratio of Bcl-2 to Bax (1.03±0.11, vs. 2.74±0.64; P<0.05).
However, the ratio was even lower in the Ad-IL-23 group (0.27±0.08;
P<0.05), and the addition of AG490 significantly inhibited the
effect of Ad-IL-23 (0.79±0.01; P<0.05). The ratio of Bcl-2 to
Bax in the anti-IL-23 group was significantly higher, compared with
that in the I/R group (1.62±0.17; P<0.05; Fig. 4C. The expression levels of Bcl-2
showed the same trend, whereas the expression of Bax showed an
opposite trend to that of Bcl-2 (Fig.
4D).
IL-23 regulates the JAK2-STAT3
signaling pathway
Compared with the I/R group, the Ad-IL-23 group had
significantly higher (P<0.05) expression levels of P-JAK2
(1.033±0.092, vs. 0.695±0.036) and P-STAT3 (0.968±0.063, vs.
0.687±0.033), however, this not the case with total JAK2
(0.948±0.074, vs. 0.963±0.074) or STAT3 (0.823±0.086, vs.
0.800±0.061; P>0.05). The levels of P-JAK2 and P-STAT3 in the
anti-IL-23 group were significantly lower, compared with those in
the I/R group (0.426±0.023 and 0.383±0.066, respectively;
P<0.05; Fig. 5A and B).
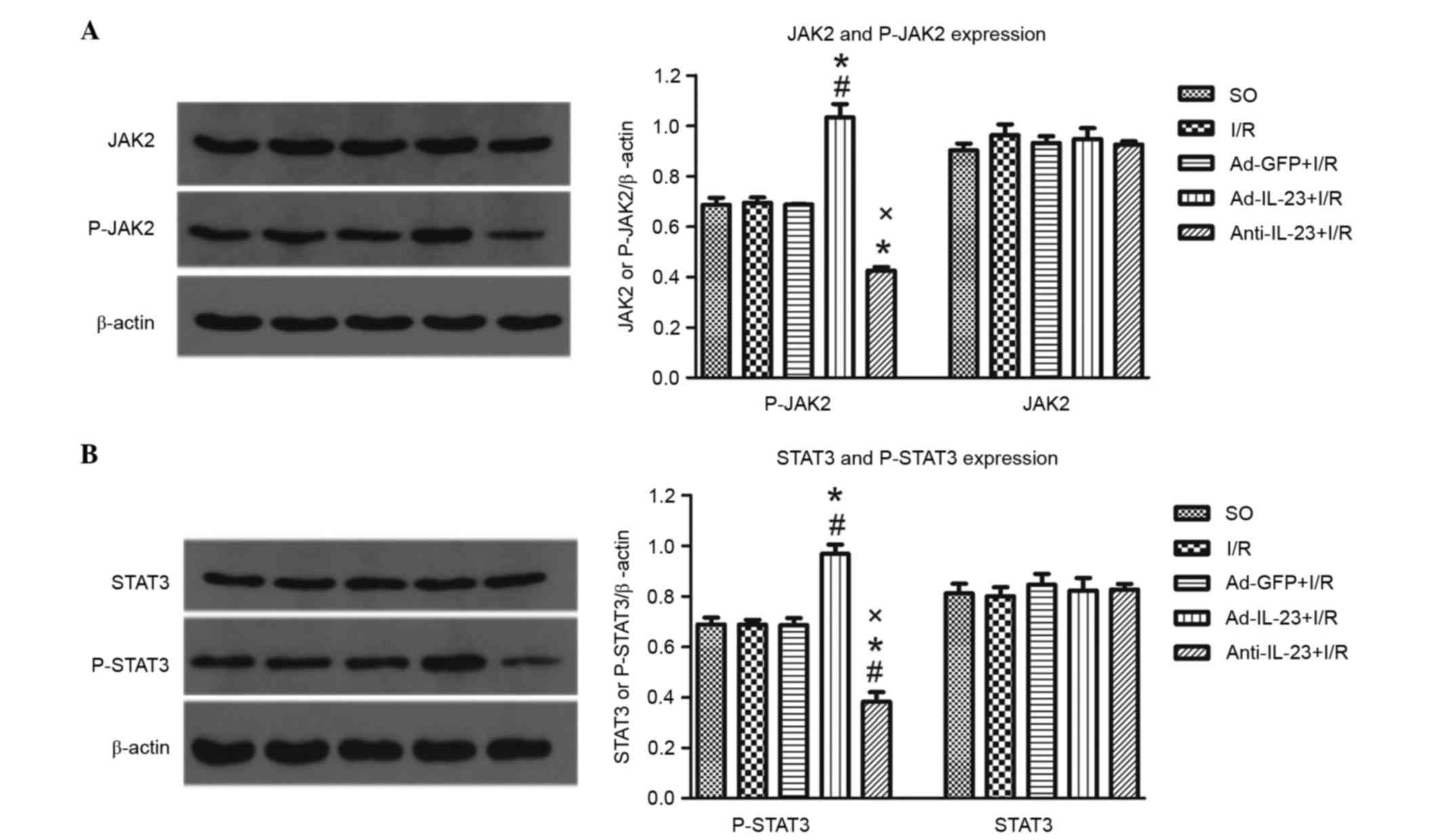 | Figure 5.Ad-IL-23 activates the JAK2-STAT3
signaling pathway. (A) Ad-IL-23 increased the expression level of
P-JAK2, but not total JAK2 in myocardial tissues in infarct and
risk regions (n=6). (B) Ad-IL-23 increased the expression level of
P-STAT3, but not total STAT3, in myocardial tissues from infarct
and risk regions (n=6). #P<0.05, vs. SO group;
*P<0.05, vs. I/R group; xP<0.05, vs. Ad-IL-23+I/R
group. SO, sham operated; I/R, ischemia/reperfusion; IL-23,
interleukin-23; Ad, adenovirus; JAK2, Janus kinase 2; P-JAK2,
phosphorylated JAK2; STAT3, signal transducer and activator of
transcription 3; P-STAT3, phosphorylated STAT3. |
Discussion
Myocardial inflammation is one of the crucial
pathophysiological processes in myocardial I/R injury, which can be
promoted by the release of various cytokines. The inflammatory
responses ultimately aggravate tissue injury (17). Previous studies have demonstrated
that the components of adaptive immunity and innate immunity are
involved in myocardial I/R injury (18). The heterodimeric cytokine IL-23,
primarily secreted by activated dendritic cells and macrophages,
functions as a link between innate and adaptive immunity by
promoting the proliferation of immune cells and secretion of
cytokines (19).
In the present study, it was found that I/R
significantly increased the expression of IL-23 in myocardial
tissues, which was consistent with the findings of previous
studies, indicating that macrophages can rapidly respond to
endogenous stimulating factors following tissue injury and have a
pathogenic role through their secretion of pro-inflammatory factors
(20,21).
In the present study, it was found that IL-23
promoted inflammatory responses via increasing the expression
levels of TNF-α and IL-6 in myocardial I/R. Previous studies have
demonstrated that IL-23 is important in several inflammatory
diseases, including myocardial I/R injury (8–11).
Once released from activated dendritic cells and macrophages, IL-23
functions as a pro-inflammatory stimulus, which induces the
differentiation of naive lymphocytes, including CD4+ T
cells, and the secretion of inflammatory cytokines, including
IL-1β, IFN-γ, TNF-α and IL-6 (8,22–24),
indicating that IL-23 may reinforce the inflammatory process and
intensify myocardial I/R injury through this mechanism.
The present study also found that the upregulation
of IL-23 significantly increased the expression level of IL-17A. As
an early pro-inflammatory cytokine, IL-17A is crucial in myocardial
I/R injury (12,25). Previous studies have suggested
that, in addition to directly aggravating myocardial I/R injury,
IL-17A stimulates endothelial cells and macrophages, and increases
the secretion of pro-inflammatory cytokines, including IL-1, IL-6,
TNF-α and c-reactive protein, which further reinforces the
inflammatory response and myocardial I/R injury (12). In addition, IL-17A can regulate the
expression of granulocyte colony stimulating factor and chemokines,
including CXC motif chemokine ligand (CXCL)l, CXCL5 and IL-18, to
amplify and collect neutrophils, which may be another mechanism
promoting myocardial inflammation and injury (12). Li et al (25) demonstrated that the expression of
IL-17A was increased in myocardial I/R, and that the inhibition of
IL-17A with anti-IL-17A significantly reduced levels of cardiac
troponin T and myocardial infarct size, ameliorateing myocardial
I/R injury. These results suggested that IL-23 may promote
inflammatory responses and myocardial I/R injury by accelerating
the secretion of IL-17A. Previous studies have demonstrated that
the generation of IL-17A induced by IL-23 is mediated by activation
of the JAK2, phosphoinositide 3-kinase/AKT, nuclear factor-κB and
STAT3 signaling pathways (26–29),
which may also be pathways through which IL-23 regulates the
expression of IL-17A in myocardial I/R, although the precise
mechanism remains to be elucidated.
Apoptosis is another important pathophysiological
process during myocardial I/R, and a previous study found that the
inhibition of apoptosis protected the heart from I/R injury
(30). The present study found
that IL-23 significantly reduced the ratio of Bcl-2 to Bax and
increased the apoptotic index, indicating that IL-23 accelerated
myocardial apoptosis during I/R injury. Liao et al (12) suggested that the upregulation
(exogenous IL-17A treatment) or downregulation (anti-IL-17A
monoclonal antibody treatment or IL-17A-knockout) of IL-17A
markedly affected the severity of I/R injury through mediating
cardiomyocyte apoptosis and neutrophil infiltration. Another study
(11) reported that IL-17A, as a
downstream pro-inflammatory cytokine, can regulate cardiomyocyte
apoptosis. Therefore, it was hypothesized that IL-23 aggravates
cardiomyocyte apoptosis by regulating the release of IL-17A. In the
present study, it was demonstrated that IL-23 had a promoting
effect in myocardial I/R injury via regulation of the expression of
IL-17A, which is an novel finding, although previous studies have
demonstrated that the IL-23/IL-17A axis is important in several
inflammatory diseases and I/R in other organs (25,31,32).
In the present study, it was found that IL-23
activated the JAK2-STAT3 signaling pathway, and inhibiting this
pathway suppressed the pro-inflammatory and pro-apoptotic effects
of IL-23. The JAK2-STAT3 signaling pathway is a primary downstream
signaling pathway of IL-23, which is an important signaling pathway
in various cardiovascular diseases, including myocardial I/R injury
(33–35). Previous studies have demonstrated
that, once activated by IL-23, the JAK2-STAT3 pathway induces the
activation and differentiation of memory T cells, including
CD4+ T cells, into IL-17A secretory cells (Th17/Thil-17)
and upregulates the secretion of IL-17A (36). In addition to IL-17A, activated
memory T cells can secrete a mass of pro-inflammatory factors,
including IL-6 and TNF-α, which are also crucial cytokines in the
inflammatory process of myocardial I/R injury (22–25).
The promoting effects of IL-17A in myocardial I/R injury suggested
that the JAK2-STAT3 signaling pathway may be involved in IL-17A
release and the promoting effects of IL-23 in myocardial I/R
injury.
In conclusion, the present study suggested that
IL-23 promoted myocardial I/R injury by increasing inflammatory
responses and myocardial apoptosis, which may be associated with
high expression levels of IL-17A and upregulation of the JAK2-STAT3
signaling pathway.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81500274 and
81370308) and the Natural Science Foundation of Hubei Province
(grant no. 2015CFB207).
References
|
1
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Andrassy M, Volz HC, Igwe JC, Funke B,
Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK,
et al: High-mobility group box-1 in ischemia-reperfusion injury of
the heart. Circulation. 117:3216–3226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xiong J, Xue FS, Yuan YJ, Wang Q, Liao X
and Wang WL: Cholinergic anti-inflammatory pathway: A possible
approach to protect against myocardial ischemia reperfusion injury.
Chin Med J (Engl). 123:2720–2726. 2010.PubMed/NCBI
|
|
4
|
Yang M, Chen J, Zhao J and Meng M:
Etanercept attenuates myocardial ischemia/reperfusion injury by
decreasing inflammation and oxidative stress. PLoS One.
9:e1080242014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Frangogiannis NG, Smith CW and Entman ML:
The inflammatory response in myocardial infarction. Cardiovasc Res.
53:31–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oppmann B, Lesley R, Blom B, Timans JC, Xu
Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al: Novel p19 protein
engages IL-12p40 to form a cytokine, IL-23, with biological
activities similar as well as distinct from IL-12. Immunity.
13:715–725. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yannam GR, Gutti T and Poluektova LY:
IL-23 in infections, inflammation, autoimmunity and cancer:
Possible role in HIV-1 and AIDS. J Neuroimmune Pharmacol. 7:95–112.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Langrish CL, Chen Y, Blumenschein WM,
Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA and
Cua DJ: IL-23 drives a pathogenic T cell population that induces
autoimmune inflammation. J Exp Med. 201:233–240. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morrison PJ, Ballantyne SJ and Kullberg
MC: Interleukin-23 and T helper 17-type responses in intestinal
inflammation: From cytokines to T-cell plasticity. Immunology.
133:397–408. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yao J, Liu L and Yang M: Interleukin-23
receptor genetic variants contribute to susceptibility of multiple
cancers. Gene. 533:21–25. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu H, Li J, Wang S, Liu K, Wang L and
Huang L: Hmgb1-TLR4-IL-23-IL-17A axis promote ischemia-reperfusion
injury in a cardiac transplantation model. Transplantation.
95:1448–1454. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao YH, Xia N, Zhou SF, Tang TT, Yan XX,
Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, et al: Interleukin-17A
contributes to myocardial ischemia/reperfusion injury by regulating
cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll
Cardiol. 59:420–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Research NRCU: Guide for the care and use
of laboratory animals. National academies press (US); Washington
(DC): 1996
|
|
14
|
Hu X, Cui B, Zhou X, Xu C, Lu Z and Jiang
H: Ethyl pyruvate reduces myocardial ischemia and reperfusion
injury by inhibiting high mobility group box 1 protein in rats. Mol
Biol Rep. 39:227–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu H, Su Z, Wu J, Yang M, Penninger JM,
Martin CM, Kvietys PR and Rui T: The alarmin cytokine, high
mobility group box 1, is produced by viable cardiomyocytes and
mediates the lipopolysaccharide-induced myocardial dysfunction via
a TLR4/phosphatidylinositol 3-kinase gamma pathway. J Immunol.
184:1492–1498. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ni J, Hu G, Xiong J, Shen J, Shen J, Yang
L, Tang M, Zhao Y, Ying G, Yu G, et al: Involvement of
interleukin-17A in pancreatic damage in rat experimental acute
necrotizing pancreatitis. Inflammation. 36:53–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Steffens S, Montecucco F and Mach F: The
inflammatory response as a target to reduce myocardial ischaemia
and reperfusion injury. Thromb Haemost. 102:240–247.
2009.PubMed/NCBI
|
|
18
|
Linfert D, Chowdhry T and Rabb H:
Lymphocytes and ischemia-reperfusion injury. Transplant Rev
(Orlando). 23:1–10. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun J, Walsh M, Villarino AV, Cervi L,
Hunter CA, Choi Y and Pearce EJ: TLR ligands can activate dendritic
cells to provide a MyD88-dependent negative signal for Th2 cell
development. J Immunol. 174:742–751. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Sun R, Wei H and Tian Z:
High-mobility group box 1 (HMGB1)-Toll-like receptor
(TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced
damage-associated lethal hepatitis: Interaction of γδ T cells with
macrophages. Hepatology. 57:373–384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McAllister F, Henry A, Kreindler JL, Dubin
PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman
SJ, et al: Role of IL-17A, IL-17F, and the IL-17 receptor in
regulating growth-related oncogene-alpha and granulocyte
colony-stimulating factor in bronchial epithelium: Implications for
airway inflammation in cystic fibrosis. J Immunol. 175:404–412.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stark MA, Huo Y, Burcin TL, Morris MA,
Olson TS and Ley K: Phagocytosis of apoptotic neutrophils regulates
granulopoiesis via IL-23 and IL-17. Immunity. 22:285–294. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vanden ES, Goriely S, De Wit D, Willems F
and Goldman M: IL-23 up-regulates IL-10 and induces IL-17 synthesis
by polyclonally activated naive T cells in human. Eur J Immunol.
35:469–475. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li L, Huang L, Vergis AL, Ye H, Bajwa A,
Narayan V, Strieter RM, Rosin DL and Okusa MD: IL-17 produced by
neutrophils regulates IFN-gamma-mediated neutrophil migration in
mouse kidney ischemia-reperfusion injury. J Clin Invest.
120:331–342. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY,
Heo SB, Jin HT, Min SY, Ju JH, Park KS, et al: STAT3 and NF-kappaB
signal pathway is required for IL-23-mediated IL-17 production in
spontaneous arthritis animal model IL-1 receptor
antagonist-deficient mice. J Immunol. 176:5652–5661. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Herndon TM, Pirone DM, Tsokos GC and Chen
CS: T cell-to-T cell clustering enhances NF-kappaB activity by a
PI3K signal mediated by Cbl-b and Rho. Biochem Biophys Res Commun.
332:1133–1139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hunter CA: New IL-12-family members: IL-23
and IL-27, cytokines with divergent functions. Nat Rev Immunol.
5:521–531. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim KW, Cho ML, Park MK, Yoon CH, Park SH,
Lee SH and Kim HY: Increased interleukin-17 production via a
phosphoinositide 3-kinase/Akt and nuclear factor kappaB-dependent
pathway in patients with rheumatoid arthritis. Arthritis Res Ther.
7:R139–R148. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xie L, Pi X, Wang Z, He J, Willis MS and
Patterson C: Depletion of PHD3 protects heart from
ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis.
J Mol Cell Cardiol. 80:156–165. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hillyer P, Larché MJ, Bowman EP,
McClanahan TK, de Waal Malefyt R, Schewitz LP, Giddins G, Feldmann
M, Kastelein RA and Brennan FM: Investigating the role of the
interleukin-23/-17A axis in rheumatoid arthritis. Rheumatology
(Oxford). 48:1581–1589. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shichita T, Sugiyama Y, Ooboshi H,
Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ,
et al: Pivotal role of cerebral interleukin-17-producing
gammadeltaT cells in the delayed phase of ischemic brain injury.
Nat Med. 15:946–950. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boengler K, Hilfiker-Kleiner D, Drexler H,
Heusch G and Schulz R: The myocardial JAK/STAT pathway: From
protection to failure. Pharmacol Ther. 120:172–185. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Knight RA, Scarabelli TM and Stephanou A:
STAT transcription in the ischemic heart. Jakstat. 1:111–117.
2012.PubMed/NCBI
|
|
35
|
Jacoby JJ, Kalinowski A, Liu MG, Zhang SS,
Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, et al:
Cardiomyocyte-restricted knockout of STAT3 results in higher
sensitivity to inflammation, cardiac fibrosis, and heart failure
with advanced age. Proc Natl Acad Sci USA. 100:pp. 12929–12934.
2003; View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mathur AN, Chang HC, Zisoulis DG,
Stritesky GL, Yu Q, O'Malley JT, Kapur R, Levy DE, Kansas GS and
Kaplan MH: Stat3 and Stat4 direct development of IL-17-secreting Th
cells. J Immunol. 178:4901–4907. 2007. View Article : Google Scholar : PubMed/NCBI
|