Introduction
Hepatic fibrosis (HF) is a leading cause of varices,
ascites, and liver failure, and results in death in millions of
patients (1). HF is usually
associated with chronic liver diseases caused by infection, drugs,
metabolic disorders, or autoimmune ailments (2). Liver fibrosis is characterized by the
activation and proliferation of hepatic stellate cells (HSCs)
(3). Tremendous progress has been
achieved regarding the roles of inflammatory cells, growth factors,
cytokines, and chemokines in the control of HSC activation during
liver fibrogenesis (4,5); however, the underlying mechanism
remains unclear and needs further investigation.
It is well-established that interleukin (IL)-22 has
hepatoprotective and antifibrotic functions in acute liver injury
models. IL-22 is a member of the IL-10 cytokine family, and is
produced primarily by Th22, Th17, and Th1 cells (6,7).
IL-22 effects epithelial cells, hepatocytes, and pancreatic cells,
and induces innate immune responses as well as tissue protection
and repair (8). In the liver,
IL-22 protects the tissues from damage, and mediates tissue repair
through multiple mechanisms in hepatocytes (9,10).
Accumulating evidence suggests that IL-22 induces HSC senescence
through crosstalk with other signaling pathways such as JAK/STAT3,
SOCS3 and p53 pathways, thereby inhibiting liver fibrosis (11).
The Notch pathway represents a highly conserved
signaling network with essential roles in the regulation of key
cellular processes and functions, many of which are critical for
development (12–14). Furthermore, emerging evidence
indicates that it is also essential for fibrosis, thus affecting
the pathogenesis of chronic fibro proliferative diseases in diverse
organs and tissues (15,16). Bansal et al (16) demonstrated the functional effects
of Notch signaling on HSC activation and M1/M2 polarization of
macrophages in liver fibrosis, although the molecular mechanism
remains elusive. Although the Notch signaling pathway is involved
in human fibrotic diseases affecting the lung, kidney, and
peritoneum (12,13), the interaction between IL-22 and
Notch signaling in HSCs remains elusive. The evidence currently
available, suggests that IL-22 may downregulate Notch3 (17–19);
therefore, we concentrated on this member of the Notch family. In
this study, we confirmed that IL-22 inhibited HSC activation
through TGF-β-mediated downregulation of the Notch pathway.
Therefore, selective inhibition of Notch signaling may be a novel
anti-fibrotic strategy for liver fibrosis treatment.
Materials and methods
Cell lines and reagents
HSC-T6 cells were purchased from the Chinese Academy
of Sciences (Beijing, China), and cultured in Dulbecco's modified
Eagle's medium (DMEM) (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gemini, Australia). Cells were maintained at 37°C in a humidified
atmosphere containing 5% CO2. IL-22 was purchased from
R&D Systems (Minneapolis, MN, USA) and TGF-β1 from Cusabio
Biotech Co., Ltd. (Wuhan, China).
Cell viability assay
The effects of IL-22 on HSC-T6 cell viability were
evaluated with the Cell Counting Kit-8 (CCK-8) (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) cell proliferation assay.
Cells were seeded in 96-well plates at a density of
1×104 cells/well. After overnight growth, the cells were
incubated with various concentrations of IL-22 (0–1,000 pg/ml)
(R&D Systems). After cell treatment for 24 h, 10 µl CCK-8
solution was added into each well. Absorbance was measured at 450
nm with background at 655 nm, using a micro plate reader (Bio-Rad
Laboratories, Hercules, CA, USA). All experiments were repeated
three times.
Cell apoptosis assay
Apoptosis was assessed, after 24 h of treatment with
IL-22, with the APC Annexin V Apoptosis Detection kit and PE Active
Caspase-3 Apoptosis kit (both from BD Pharmingen, San Diego, CA,
USA) according to the manufacturers' instructions. Briefly, HSC-T6
cells were treated with IL-22, followed by staining in fluorescence
activated cell sorter (FACS) buffer (PBS, 2% bovine serum albumin,
0.1% sodium azide) for 10 min at 4°C. Finally, cells were washed
and assessed on BD FACSCalibur Flow Cytometer (BD Pharmingen). Data
were analyzed with the FlowJo software (Tree Star, Ashland, OR,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was extracted from cells using TRIzol
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer's instructions. Reverse transcription was performed
with a cDNA Reverse Transcription kit (Takara Bio, Inc., Otsu,
Japan). RT-qPCR was performed with a SYBR-Green Master Mix kit
(Takara Bio, Inc.). Relative mRNA levels were normalized to GAPDH
mRNA expression, and calculated by the 2−∆∆Cq method.
The primer sequences are summarized in Table I.
 | Table I.Primers used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers used for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Primer (5′-3′) | Base pairs (bp) |
|---|
| GAPDH | Forward:
CAGCTTTTGAAGGGGAACGC | 182 |
|
| Reverse:
TCATGCTCAGAAGTGGCTGG |
|
| Hes-1 | Forward:
TCAACACGACACCGGACAA | 120 |
|
| Reverse:
GTGCTTCACTGTCATTTCCAGA |
|
| Hes-5 | Forward:
AGCCGGTGGTGGAGAAGAT | 100 |
|
| Reverse:
AGTTTGGAGTTGGGCTGGTG |
|
| Hey-1 | Forward:
AGCTGAGATCTTGCAGATGACTGTG | 109 |
|
| Reverse:
AGCCAGGCATTCCCGAAAC |
|
| TGF-β | Forward:
ATTCCTGGCGTTACCTTGG | 120 |
|
| Reverse:
AGCCCTGTATTCCGTCTCCT |
|
| TNF-α | Forward:
CAGGTTCCGTCCCTCTCATA | 100 |
|
| Reverse:
TGCCAGTTCCACATCTCG |
|
| ICAM-1 | Forward:
ATGGACGCTCACCTTTAGCA | 109 |
|
| Reverse:
TCTCCCAGGCATTCTCTTTG |
|
Immunoblot
Samples were homogenized in lysis buffer containing
Tris-HCl, NP-40, NaCl, ethylene diamine-tetra acetic acid,
NaN3, phenylmethylsulfonyl fluoride, aprotinin, and
leupeptin (pH 7.5), and centrifuged at 16,000 rpm at 4°C for 10
min. Protein concentration was determined by BCA (Pierce
Biotechnology, Inc., Rockford, IL, USA). Equal amounts of total
protein (30 µg) were separated by 10–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto PVDF membranes. Antibodies against SMA were
obtained from Abcam (Cambridge, MA, USA). Anti-GAPDH was from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-Notch3 was
manufactured by Abcam (Cambridge, UK). Then membranes were
incubated with various primary antibodies overnight at 4°C,
followed by incubation with secondary antibodies and detection by
enhanced chemiluminescence (ECL) (Odyssey; LI-COR Biosciences,
Lincoln, NE, USA).
Statistical analysis
Data are mean ± standard deviation (SD) from three
independent experiments. Statistical analysis was performed by
unpaired Student's t-test, with two tailed P<0.05 considered
statistically significant. Multiple groups were assessed by one-way
analysis of variance and Dunnett's post hoc test. The SPSS 16.0
software (SPSS Inc., Chicago, IL, USA) was used for statistical
analysis.
Results
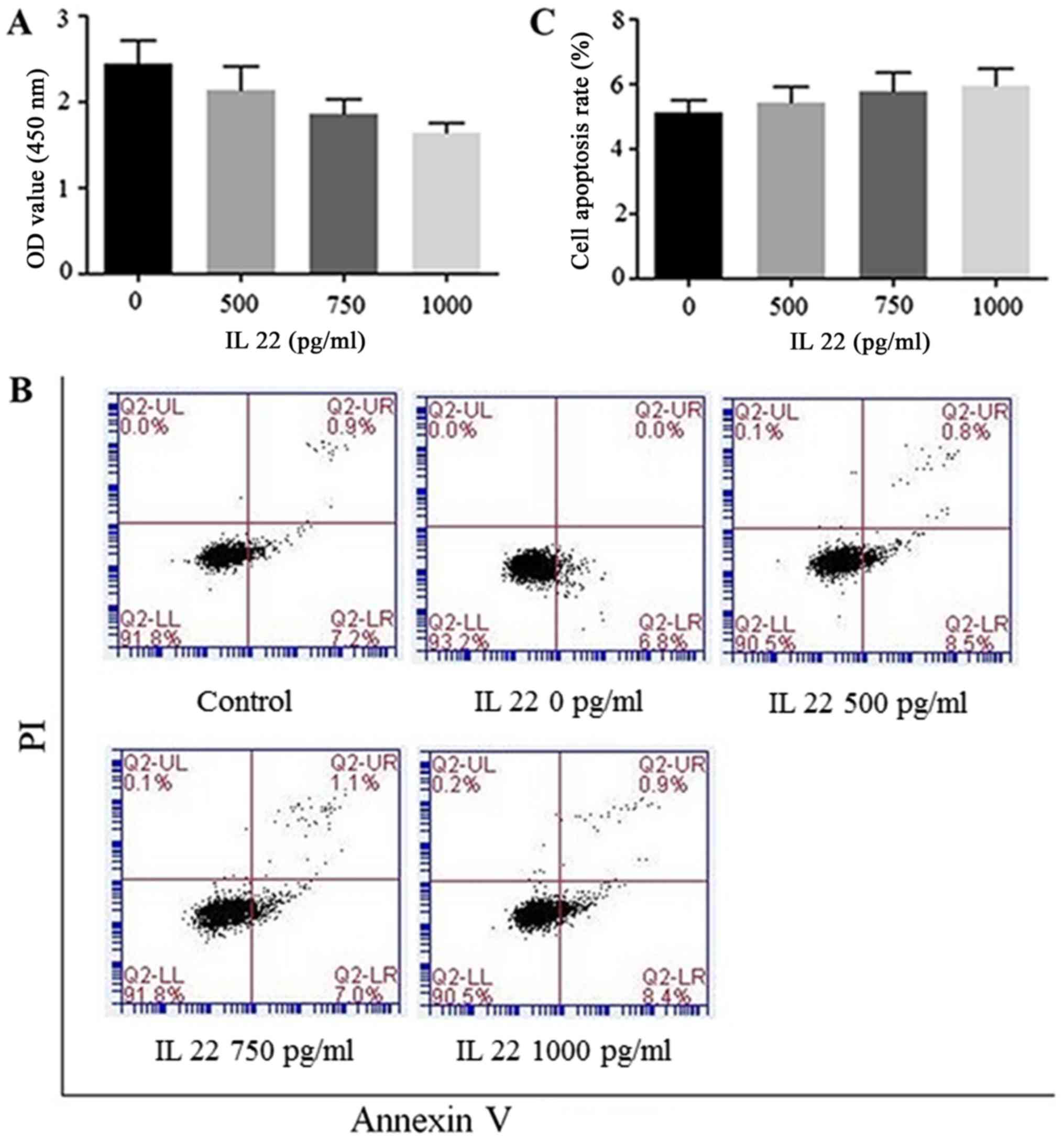
IL-22 treatment inhibits HSC-T6 cells
proliferation without inducing apoptosis
We first assessed the anti-proliferative effects of
IL-22 on hepatic cells. Increasing concentrations of IL-22 were
respectively applied to HSC-T6 cells, and cell proliferation was
determined by CCK-8 assay. Cell viability was significantly
decreased at high IL-22 amounts (Fig.
1A). However, IL-22 had no effect on apoptosis inHSC-T6 cells.
As shown in Fig. 1B and C, there
was no significant difference between IL-22 treatment and control
groups. Taken together, these results indicated that IL-22 may
exert anti-proliferative effects, independent of apoptosis.
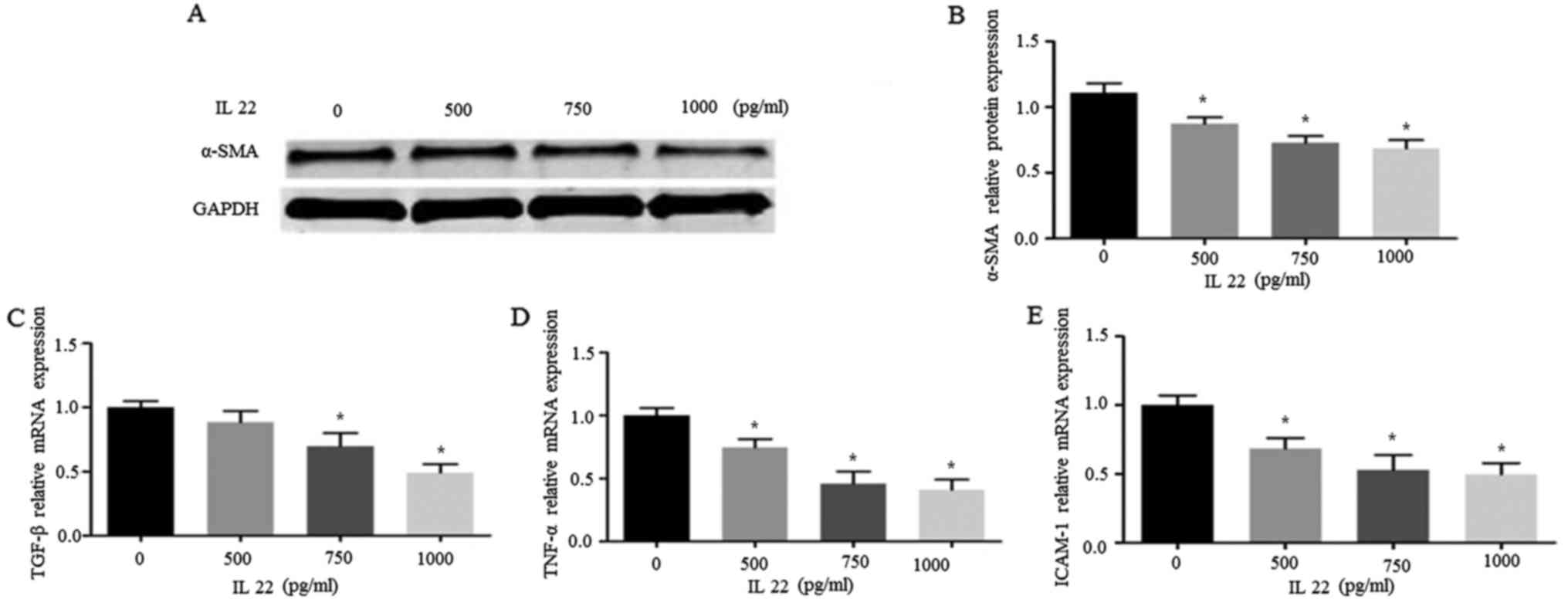
IL-22 downregulates α-SMA and
proinflammatory cytokines in HSC-T6 cells
Increased expression levels of α-SMA and collagen
are commonly recognized as biomarkers of HSC activation (3). Next, we assessed the expression
levels of α-SMA and multiple hepatocyte-associated proinflammatory
cytokines to verify the inhibitory effects of IL-22 on HSC-T6
proliferation. Interestingly, application of IL-22 inhibited α-SMA
expression in a dose-dependent manner (Fig. 2A). Furthermore, we assessed whether
common pro-inflammatory cascades were inhibited by IL-22. TGF-β and
tumor necrosis factor-α (TNF-α) are the most important mediators of
inflammatory responses in HSCs, while ICAM-1 is an essential cell
adhesion molecule (8,9). As shown in Fig. 2B-E, the mRNA levels of TGF-β,
TNF-α, and ICAM-1 were simultaneously decreased by IL-22 treatment
in HSC-T6 cells, suggesting that these effectors are downstream of
IL-22. Collectively, HSC inactivation by IL-22 was confirmed by the
downregulation of α-SMA and related-inflammatory cytokines.
TGF-β1 activates the Notch pathway in
HSC-T6 cells
We further explored the possible downstream
effectors upon TGF-β stimulation. Several studies previously
described a functional interaction between the TGF-β superfamily of
proteins and Notch signaling in multiple biological processes
(15,16). The results demonstrated that Notch3
levels were remarkably increased in HSC-T6 cells after TGF-β1
treatment, in a dose-dependent manner (Fig. 3A), in line with densitometry
analysis (Fig. 3B). Furthermore,
not only was the expression of Notch3 receptor increased by TGF-β1,
but Notch pathway activation was confirmed, as indicated by
substantial upregulation of the downstream effectors Hes family
basic helix-loop-helix transcription factor-1 (Hes-1), Hes-5 and
Hey-1 (Fig. 3C-E). Taken together,
these results indicated that TGF-β is responsible for the
activation of Notch signaling in HSC-T6 cells.
IL-22 treatment inhibits the Notch
pathway in HSC-T6 cells
As TGF-β1-induced Notch3 upregulation, we wondered
if such regulation can be altered by IL-22. Intriguingly, Notch3
expression was reduced after incubation with IL-22 (Fig. 4A and B). Meanwhile, the effect of
IL-22 on the TGF-β1-induced Notch3 increase was evaluated. The
results showed that IL-22 decreased Notch3 expression in a dose
dependent way in HSC-T6 cells (Fig. 4A
and B), suggesting Notch3 to be a downstream signal transducer
of IL-22-related TGF-β1 inhibition. These results further confirmed
that both Notch3 expression and pathway activation were impaired by
IL-22. Compared with the TGF-β1 group, combined treatment with
IL-22 significantly decreased Hes-1, Hes-5 and Hey-1 mRNA levels
(Fig. 4C-E). Taken together, these
findings suggested that IL-22 plays a role in regulation of the
Notch signaling pathway.
Discussion
The present study demonstrated that IL-22 reduces
HSC-T6 activation through Notch3 inhibition, suggesting the
protective role of IL-22 in liver fibrosis in vitro and
revealing a potential target for liver fibrosis.
Liver fibrosis is a chronic, but reversible wound
healing response characterized by a tight interplay between
inflammatory and matrix-producing cellular pathways (5). During this process, various signaling
pathways are involved in the activation of HSCs. Therefore,
targeting a specific signaling pathway may be helpful in
identifying effective treatment tools for liver fibrosis. To date,
several signaling pathways have been found to be closely related to
liver fibro-genesis. Notch signaling is considered a major
signaling mechanism in liver biology as well as multiple
pathological conditions, from liver damage to carcinogenesis
(20). In addition, Notch3 and
Jagged1 (mainly related to HSC activation) are upregulated in
diseased human livers as well as mouse models (21,22).
The activated Notch pathway is involved in some fibrotic diseases.
Zhu et al (23) reported
that Notch signaling is highly activated in rats with fibrotic
peritoneum induced by peritoneal dialysis fluid; indeed, blocking
Notch signaling significantly attenuates peritoneal fibrosis.
Studies also showed that Notch signaling is involved in the
activation of HSCs in vivo, with transient knockdown of
Notch3 antagonizing TGF-β1-induced expression of α-SMA and collagen
I in HSC-T6 cells (19). In the
present study, TGF-β1 was used to activate HSCs, and Notch3, TGF-β,
TNF-α, and ICAM-1 expression levels were significantly increased,
indicating that the Notch pathway is involved in TGF-β1 induced
activation of HSCs.
IL-22 inhibits HSCs through the signal transducer
and activator of transcription 3 (STAT3) signaling pathway
(10,24). Meanwhile, studies reported that
Notch is regulated by TGF-β1 (15,16);
however, whether TGF-β1 is involved in IL-22 induced inhibition of
HSCs remains unclear. In the present study, TGF-β1 significantly
increased the expression levels of Notch3, Hes-1, Hes-5 and Hey-1,
which were significantly rescued by IL-22. Taken together, these
findings suggested that IL-22 inhibits HSC activation may through
the regulation of TGF-β1/Notch signaling. However, how IL-22
modulates TGF-β1/Notch signaling, and whether this is directly
related to the expression of α-SMA, needs to be further
investigated.
In conclusion, this study firstly revealed a
biological interaction between the pro-inflammatory cytokine IL-22
and Notch signaling in preventing liver fibrosis, with TGF-β1
required for the regulatory process.
Glossary
Abbreviations
Abbreviations:
|
HSC
|
hepatic stellate cell
|
|
α-SMA
|
α-smooth muscle actin
|
|
TGF-β1
|
transforming growth factor-β1
|
|
TNF-α
|
tumor necrosis factor-α
|
|
ICAM-1
|
intercellular adhesion molecule-1
|
|
HF
|
hepatic fibrosis
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
ECL
|
electrochemiluminescence
|
|
SD
|
standard deviation
|
|
ANOVA
|
analysis of variance
|
References
|
1
|
Ginès P, Càrdenas A, Arroyo V and Rodès J:
Management of cirrhosis and ascites. N Engl J Med. 350:1646–1654.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pan CX, Tang J, Wang XY, Wu FR, Ge JF and
Chen FH: Role of interleukin-22 in liver diseases. Inflamm Res.
63:519–525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Puche JE, Saiman Y and Friedman SL:
Hepatic stellate cells and liver fibrosis. Compr Physiol.
3:1473–1492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sonnenberg GF, Fouser LA and Artis D:
Border patrol: Regulation of immunity, inflammation and tissue
homeostasis at barrier surfaces by IL-22. Nat Immunol. 12:383–390.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zenewicz LA and Flavell RA: Recent
advances in IL-22 biology. Int Immunol. 23:159–163. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sertorio M, Hou X, Carmo RF, Dessein H,
Cabantous S, Abdelwahed M, Romano A, Albuquerque F, Vasconcelos L,
Carmo T, et al: IL-22 and IL-22 binding protein (IL-22BP) regulate
fibrosis and cirrhosis in hepatitis C virus and schistosome
infections. Hepatology. 61:1321–1331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carmo RF, Cavalcanti MS and Moura P: Role
of interleukin-22 in chronic liver injury. Cytokine. 98:107–144.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang YM, Liu ZR, Cui ZL, Yang C, Yang L,
Li Y and Shen ZY: Interleukin-22 contributes to liver regeneration
in mice with concanavalin A-induced hepatitis after hepatectomy.
World J Gastroenterol. 22:2081–2091. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kong X, Feng D, Wang H, Hong F, Bertola A,
Wang FS and Gao B: Interleukin-22 induces hepatic stellate cell
senescence and restricts liver fibrosis in mice. Hepatology.
56:1150–1159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bolòs V, Grego-Bessa J and de la Pompa JL:
Notch signaling in development and cancer. Endocr Rev. 28:339–363.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nichols AM, Pan Y, Herreman A, Hadland BK,
De Strooper B, Kopan R and Huppert SS: Notch pathway is dispensable
for adipocyte specification. Genesis. 40:40–44. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu T, Hu B, Choi YY, Chung M, Ullenbruch
M, Yu H, Lowe JB and Phan SH: Notch1 signaling in FIZZ1 induction
of myofibroblast differentiation. Am J Pathol. 174:1745–1755. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bielesz B, Sirin Y, Si H, Niranjan T,
Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH and
Susztak K: Epithelial Notch signaling regulates interstitial
fibrosis development in the kidneys of mice and humans. J Clin
Invest. 120:4040–4054. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bansal R, van Baarlen J, Storm G and
Prakash J: The interplay of the Notch signaling in hepatic stellate
cells and macrophages determines the fate of liver fibrogenesis.
Sci Rep. 5:182722015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trehanpati N, Shrivastav S, Shivakumar B,
Khosla R, Bhardwaj S, Chaturvedi J, Sukriti, Kumar B, Bose S, Mani
Tripathi D, et al: Analysis of Notch and TGF-β signaling expression
in different stages of disease progression during hepatitis B virus
infection. Clin Transl Gastroenterol. 3:e232012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu DH, Guo XY, Qin SY, Luo W, Huang XL,
Chen M, Wang JX, Ma SJ, Yang XW and Jiang HX: Interleukin-22
ameliorates liver fibrogenesis by attenuating hepatic stellate cell
activation and downregulating the levels of inflammatory cytokines.
World J Gastroenterol. 21:1531–1545. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen YX, Weng ZH and Zhang SL: Notch3
regulates the activation of hepatic stellate cells. World J
Gastroenterol. 18:1397–1403. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Geisler F and Strazzabosco M: Emerging
roles of Notch signaling in liver disease. Hepatology. 61:382–392.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Y, Zheng S, Qi D, Zheng S, Guo J,
Zhang S and Weng Z: Inhibition of Notch signaling by a γ-secretase
inhibitor attenuates hepatic fibrosis in rats. PLoS One.
7:e465122012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei X, Wang JP, Hao CQ, Yang XF, Wang LX,
Huang CX, Bai XF, Lian JQ and Zhang Y: Notch signaling contributes
to liver inflammation by regulation of interleukin-22-producing
cells in hepatitis B virus infection. Front Cell Infect Microbiol.
6:1322016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu F, Li T, Qiu F, Fan J, Zhou Q, Ding X,
Nie J and Yu X: Preventive effect of Notch signaling inhibition by
a gamma-secretase inhibitor on peritoneal dialysis fluid-induced
peritoneal fibrosis in rats. Am J Pathol. 176:650–659. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mühl H: STAT3, a key parameter of
cytokine-driven tissue protection during sterile inflammation-the
case of experimental acetaminophen (Paracetamol)-induced liver
damage. Front Immunol. 7:1632016. View Article : Google Scholar : PubMed/NCBI
|