Introduction
Supplemental oxygen is a common lifesaving strategy
used in neonatal intensive care units (1). Oxygen therapy utilizing
supraphysiological concentrations of oxygen (hyperoxia) is
routinely administered during mechanical ventilation for the
management of severe respiratory distress (2–4).
However, oxygen therapy can also cause oxygen toxicity, including
acute lung injury (ALI) (5,6).
Additionally, exposure to high concentrations of oxygen may induce
diffuse pulmonary injuries, vascular leakage, excessive
inflammation and pulmonary edema (7,8).
Hyperoxia-induced damage to lung tissues is attributed to the
generation of reactive oxygen species (ROS) and the subsequent
formation of more potent oxidants (9–11).
Therefore, excessive levels of ROS and the resultant oxidative
damage have an important role during the process of
hyperoxia-induced ALI (12,13).
As the understanding of the mechanism of hyperoxia-induced ALI is
incomplete, effective therapies have not yet been developed.
Sirtuins are protein deacetylases that hydrolyze one
oxidized nicotinamide adenine dinucleotide (NAD+) for each lysine
residue that they deacetylate. Thus, their activity is associated
with cellular energy levels (14,15).
Sirtuins were initially investigated as mediators of the increased
lifespan that is associated with calorie restriction in yeast
(16,17); however, recent studies indicate
that they are involved in a variety of functions, including genomic
stability, tumorigenesis, inflammation and metabolic diseases
(18). In mammals, sirtuins are
comprised of seven proteins (SIRT1-7), which have different
subcellular localizations. SIRT1 and 2 are present in the nucleus
and cytoplasm, SIRT3-5 are located in the mitochondria, and SIRT6
and 7 are located in the nucleus (19). The majority of studies have focused
on SIRT1 and 2, and the investigation of other SIRTs has been less
extensive.
SIRT3 is an important mitochondrial protein. It
controls various aspects of mitochondrial function by deacetylating
various mitochondrial matrix proteins, including antioxidant
effectors and proteins involved in the electron transport chain,
therefore acting as a tumor suppressor by limiting the production
of ROS. SIRT3 is important for mitochondrial function, by limiting
oxidative stress and reducing ROS production, which results in a
decrease in mitochondrial membrane potential (20). A previous study demonstrated that
SIRT3 enhanced the expression of certain antioxidant proteins,
including mitochondrial superoxide dismutase (SOD) (21).
SODs are enzymes that alternately catalyze the
dismutation, or partitioning, of the toxic superoxide radical into
either ordinary molecular oxygen or hydrogen peroxide. Superoxide
is produced as a by-product of oxygen metabolism and causes various
types of cell or tissue damage. Thus, SOD is a major antioxidant
defense in almost all living cells that are exposed to oxygen. The
following four isoforms of SOD are present in mammalian cells:
Manganese superoxide dismutase (MnSOD), copper-zinc superoxide
dismutase (CuZnSOD), extracellular SOD (ecSOD) and glutathione
peroxidase 1 (GPx1). Studies using exogenous MnSOD or genetically
altered mice overexpressing MnSOD demonstrated that MnSOD
inactivates mitochondrial ROS and ameliorates hyperoxia-induced ALI
(22). Additionally, CuZnSOD,
which is evenly distributed intracellularly, is present in the
nucleus and lysosomes. A previous study demonstrated that CuZnSOD
was expressed in the alveolar epithelium, mesenchymal cells,
fibroblasts and vascular endothelial cells of rat lungs (23). By contrast, ecSOD is primarily
located in the extracellular matrix and expressed in the bronchial
epithelium, alveolar epithelium and alveolar macrophages (24).
Based on the evidence discussed, we hypothesized
that SIRT3 may have pharmacological effects on hyperoxia-induced
ALI and that the potential antioxidative mechanism may be caused by
regulating the expression of SOD in mice. Therefore, the aim of the
current study was to investigate whether SIRT3 was able to inhibit
the oxidative damage observed during hyperoxia-induced ALI by
increasing the expression of SOD.
Materials and methods
Animals
Eighty adult pathogen-free female C57BL/6 mice
(6–8-weeks-old, weight 20±5 g) were provided by the SLRC Laboratory
(Shanghai, China). Animals were raised under standard conditions
and were provided with water and food ad ibitium, with a 12
h day/night cycle, and were acclimatized to their environment for
at least one week prior to the initiation of the experiments. The
study was approved the Ethics Committee of Shengli Oilfield Central
Hosptal. Animal care and handling were performed in accordance with
the National Institutes of Health guidelines.
Retrovirus preparation and
infection
Retroviral vectors containing either the SIRT3 gene
or SIRT3 small interfering RNA (siRNA) were constructed according
to the sequence information from NCBI (NCBI reference sequence:
NM_022433.2). RNAi design software (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was utilized for the sequence
design. The sequence for the SIRT3 siRNA was:
5′-GCAAUAGAUUUAAUGACAG-3′. Retrovirus vectors (SIRT3 overexpression
and SIRT3 siRNA) were transfected into 293 cells using calcium
phosphate precipitation (25).
After 24 h, the medium was changed to DMEM containing 10% FBS. The
cells were cultured for another 24 h, and then the supernatant
containing the lentivirus was harvested.
Animal model of hyperoxia-induced
ALI
Mice were randomly divided into the following four
groups: Control (n=20; mice subjected to normal air containing 21%
O2; Ctr group); hyperoxia-induced ALI model group (n=20;
mice subjected to 90% O2; Hyper group); vector-carrying
retrovirus (vector-RV)-treated ALI group (n=20; mice subjected to
90% O2 and received lentivirus containing only vector
via tail vein injection; Hyper + vector group) and
SIRT3-overexpressing retrovirus (SIRT3-RV)-treated ALI group (n=20;
mice subjected to 90% O2 and received lentivirus
containing SIRT3 via tail vein injection; Hyper + SIRT3 group). The
mice were treated with vector or SIRT3-RV 3 days after exposure to
90% O2.
To induce hyperoxia-induced ALI, mice were allowed
to roam free under normobaric pressures in chambers under 90%
O2 or normal air containing 21% O2.
O2 mixtures or normal air were delivered through the
chamber at 3 l/min and allowed to vent through a distal ventilation
port to maintain normobaric pressures. After 6 days of exposure,
mice were terminally anesthetized with ketamine intraperitoneally
(80 mg/kg body weight). Subsequently, under sterile conditions,
thoraxes were opened, and blood was sampled by cardiac puncture.
Simultaneously, three bronchoalveolar lavage (BAL) procedures were
performed, each with 1 ml of normal saline. Fluid and blood were
centrifuged (2,000 × g, for 10 min at 4°C) and the supernatant and
plasma were stored for further processing. Survival of mice
following ALI induction and group-specific treatments were
assessed, and the cumulative survival curve was produced using the
Kaplan-Meier method.
Bronchoalveolar lavage fluid (BALF)
collection and determination of cytokine and protein
concentration
At the end of the procedure, the right lungs were
ligated at the right main bronchus and the BALF was collected from
the left lung through a tracheal cannula with 5 ml of sterile PBS.
The collected BALF was centrifuged at 300 × g for 10 min at 4°C,
and the supernatants were stored at −70°C. The protein
concentration in the BALF supernatants was determined by the
Bradford method (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
with bovine serum albumin (Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) used as a reference standard.
BAL cell counts
The trachea was exposed via a midline incision and
the lungs were gently lavaged via a tracheal mannula with three
aliquots of 1 ml PBS (0.5 M, pH 7.4). The volume of the recovered
lavage fluid was recorded, and cell counts were determined using a
hemocytometer. Differential counts were performed on cells stained
with Wright-Giemsa stain as previously described (26).
Histopathological grading of
hyperoxia-induced ALI
Histopathological evaluation was performed on
paraffin-embedded tissues as described previously (27). Prior to removal, the lungs were
rinsed with PBS and then instilled with 1 ml of buffered formalin
through an angiocatheter inserted in the trachea. The lungs were
then paraffin embedded, and these paraffin blocks were sliced into
5 µm sections. Five random 5 µm thick paraffin-embedded tissue
sections from five different mice lungs taken at day 6 of ALI
treatment were stained with hematoxylin and eosin (H&E). The
histopathology analysis was performed using a conventional light
microscope (Olympus BX51; Olympus Corporation, Tokyo, Japan) and
images were captured using a Nikon DXM1200C digital camera (Nikon
Corporation, Tokyo, Japan).
To assess the severity of the lung injury, a
semi-quantitative histological index was used. Five sections were
randomly selected from each group of mice, and five fields were
examined per section. The lung histopathological changes were
scored on a scale of 0–5 according to the degree of congestion,
lung edema, infiltration of inflammatory cells and hemorrhage in
lung tissues (28). A score of 0
indicated no injury in lung tissues, 1 indicated modest injury, 2
indicated intermediate injury, 3 indicated widespread injury and 4
indicated severe injury. The overall score of hyperoxia-induced ALI
was generated based on the summation of all scores, and the mean +
standard error of the mean (SEM) of the scores were calculated for
the lungs of the normal air controls.
Cell culture
A549 cells, a tumor cell line derived from a human
lung carcinoma with properties of type II alveolar epithelial
cells, and 293 cells were purchased from Cell Resource Center of
Chinese Academy of Sciences (Shanghai, China). The cells were
cultured in Dulbecco's modified Eagle's medium (Invitrogen; Thermo
Fisher Scientific, Inc.) containing 10% heat-inactivated fetal calf
serum (Gibco/Invitrogen; Thermo Fisher Scientific, Inc.), and
cultured in a 5% CO2-95% air chamber at 37°C. For the
delivery of Ritrovirus into A549 cells, the cells were resuspended
with virus solution, and then the plates were centrifuged at 1,500
× g for 120 min at 4°C.
Western blotting
The levels of SIRT3 and MnSOD protein in lung tissue
were measured using western blot analysis. The lung tissues of
treated and control mice, and differentially treated cells, were
homogenized, washed with PBS, incubated in lysis buffer for 30 min
at 4°C, and a mixture of protease inhibitors was added
(Sigma-Aldrich; Merck Millipore) to obtain extracts of tissue or
cell proteins. The protein concentration in the supernatant was
determined using the Bradford assay. Briefly, total protein (50 µg)
was loaded into each lane. The proteins were transferred onto
polyvinylidene fluoride membranes following 10% SDS-PAGE, and the
membranes were blocked with 5% non-fat milk for 1 h at room
temperature. Subsequently, membranes were incubated with primary
antibodies overnight at 4°C, and the membranes were washed,
incubated with secondary antibodies for 1 h at room temperature,
and visualized by enhanced chemiluminescence reagent (Thermo Fisher
Scientific, Inc.). The following antibodies were used: Rabbit
anti-mouse SIRT3 polyclonal antibody (cat. no. ab86671; Abcam,
Cambridge, UK); rabbit anti-mouse MnSOD polyclonal antibody (cat.
no. PA1-125; Thermo Fisher Scientific, Inc.) and rabbit anti-mouse
β-actin antibody (cat. no. ab189073; Abcam). Horseradish
peroxidase-conjugated goat anti-rabbit IgG (cat. no. ab6721; Abcam)
was used as secondary antibody. The dilutions for all antibodies
were 1:1,000. ImageJ version 1.46r software (National Institutes of
Health, Bethesda, MD, USA) was used for densitometric analysis. The
experiment was repeated three times.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA from treated lung tissues and cells were
isolated using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). Total RNA (0.4 µg) was then treated with
RNase-free Dnase (1 U/sample, Sigma-Aldrich; Merck Millipore), and
cDNA was generated using random hexamer primers provided in the
RevertAid First-Strand cDNA synthesis kit (Applied Biosystems;
Thermo Fisher Scientific, Inc., Cat no. K1621). The products of RT
reaction were diluted and used as templates in subsequent qPCR or
stored at −20°C. qPCR analysis was performed using a sequence
detection system (7900HT Fast Real-Time PCR system; Applied
Biosystems; Thermo Fisher Scientific, Inc.). Specifically, diluted
cDNA sample was amplified using the SYBR Green PCR kit (Invitrogen;
Thermo Fisher Scientific, Inc.). Thermal cycling was initiated with
an initial denaturation step of 5 min at 95°C, followed by 40
cycles of 95°C for 10 sec and 60°C for 30 sec. The number of
replicates per sample was 40 and the 2−ΔΔCq method was
used to analyze the results (29).
The following mouse-specific primer pairs were used: β-actin,
5′-GGCCAGGTCATCACTATTG-3′ (forward) and 5′-GAGGTCTTTACGGATGTCAAC-3′
(reverse); CuZnSOD, 5′-GACAAACCTGAGCCCTAAG-3′ (forward) and
5′-CGACCTTGCTCCTTATTG-3′ (reverse); MnSOD, 5′-ATGTCTGTGGGAGTCCAA-3′
(forward) and 5′-TGAAGGTAGTAAGCGTGCTC-3′ (reverse); ecSOD,
5′-ATTTCAGTCTGGAGGGCT-3′ (forward), 5′-CACGAAGTTGCCAAAGTC-3′
(reverse); GPx1, 5′-GACTACACCGAGATGAACGAT-3′ (forward) and
5′-CACTTCGCACTTCTCAAACA-3′ (reverse); SIRT3,
5′-CATCGACGGGCTTGAGAGA-3′ (forward) and 5′-GGTCCCGTGGGCTTCAAC-3′
(reverse). The Primer Express 3.0 software (https://www.thermofisher.com/order/catalog/product/4363991)
was used to design the qPCR primers.
Lung wet/dry (W/D) ratio
The mice (40 in total) were anesthetized using
sodium pentobarbital (intraperitoneally, 40 mg/kg) and sacrificed
via exsanguination 6 days after ALI induction. Right lungs were
removed and the wet weights were obtained. Subsequently, the lungs
were dried at 80°C and weighed again 3 days after drying. The W/D
ratio was calculated to assess tissue edema. The W/D ratio was
calculated as follows: (wet weight-dry weight)/dry weight (30).
Measurement of oxidized/reduced
glutathione (GSH) ratio
The ratio of reduced GSH and oxidized GSH (GSSG) was
determined in lung tissue homogenates from treated ALI mice by
reaction with 5,5′-dithiobis-2-nitrobenzoic acid, using the
Glutathione Assay kit (Merck Millipore) according to the
manufacturer's instructions.
Lipid peroxidation
The lung tissues were immediately flash frozen in
liquid nitrogen at time of harvest and stored at −80°C to prevent
auto-oxidation. Lipid peroxidation, a well-defined mechanism of
cellular damage, was assessed by measuring the level of
8-isoprostane, an indicator of oxidative stress; 8-isoprostane
levels were determined using an 8-isoprostane ELISA kit (Cayman
Chemical Company, Ann Arbor, MI, USA) according to the
manufacturer's instructions.
Measurement of tissue SOD
activity
The BIOXYTECH® SOD-525 assay kit (OXIS
Health Products, Inc., Portland, OR, USA) was used according to the
manufacturer's instructions to measure SOD activity. Tissue SOD
activity was determined by spectrophotometric detection of formazan
production at 550 nm, as a result of inhibition of nitroblue
tetrazolium reduction, with xanthine-xanthine oxidase used as a
superoxide generator, as described previously (31).
Survival study in mice with
hyperoxia-induced ALI
Mice were randomly divided into four groups (n=10
per group) as mentioned above. The survival rates were recorded at
the indicated time points (day 1, 3, 5, 7, 9, 11, 13 and 15 after
treatment).
Statistical analysis
Data were analyzed using SPSS 13.0 software (SPSS,
Inc., Chicago, IL, USA) and expressed as the mean ± SEM.
Significant differences were assessed by one-way analysis of
variance followed by Fisher's least significant difference test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
SIRT3 expression is reduced in
hyperoxia-induced ALI
Previous studies have demonstrated that sirtuins are
highly conserved class III NAD+-dependent deacetylases
that target histone proteins (23,32–35).
As a member of the sirtuin family, SIRT3 is reported to target
mitochondrial proteins for lysine deacetylation and to regulate
cellular functions. Evidence indicates that SIRT3 may regulate the
mitochondrial adaptive antioxidant response (36–38).
Therefore, the present study induced ALI in mice by exposure to
high concentrations of O2 in order to investigate the
function of SIRT3 in hyperoxia-induced ALI. The expression of SIRT3
mRNA and protein in the lung tissues of hyperoxia-induced ALI mice
and control mice were then measured by RT-qPCR and western blot
analysis. Representative histological sections of lung tissues from
control mice (Fig. 1A) and
hyperoxia-induced ALI mice (Fig.
1B) are presented. It was observed that the lungs from
hyperoxia-induced ALI mice exhibited inflammatory infiltrations,
edema and thickening of the alveolar walls, which was not observed
in the control mice (Fig. 1A and
B). The lung injury index that represents the severity of lung
injury was significantly increased in the ALI group compared with
the control mice (P<0.05; Fig.
1C). To assess the expression of SIRT3, the mRNA and protein
levels of SIRT3 in different mice lung tissues were measured.
Compared with the controls, SIRT3 mRNA (Fig. 1D) and protein (Fig. 1E and F) expression in the lungs of
hyperoxia-induced ALI mice were significantly decreased
(P<0.05). These results indicated that SIRT3 may have an
important role in hyperoxia-induced ALI.
Retrovirus treatment enhances SIRT3
expression in lung tissue
To determine the effect of a SIRT3-containing
retrovirus on the expression of SIRT3 in the lung tissues,
hyperoxia-induced ALI mice were treated with SIRT3-RV and vector-RV
(retrovirus containing a blank vector as a control) via tail vein
injection 3 days after ALI induction and the expression of SIRT3
protein in the differentially treated lung tissues was measured 3
days after injection by western blot analysis. As presented in
Fig. 2, SIRT3 was overexpressed in
the lung of SIRT3-RV treated ALI mice (Hyper + SIRT3), compared
with the control ALI group (Hyper; P<0.05) and the
vector-RV-treated ALI group (Hyper + vector; P<0.05; Fig. 2).
Enhanced expression of SIRT3 protects
against hyperoxia-induced ALI in mice
To further investigate the function of SIRT3 in
hyperoxia-induced ALI, mice were treated with SIRT3-RV via tail
vein injection. The effect of SIRT3-RV on lung histopathology is
presented in Fig. 3. Treatment
with SIRT3-RV reduced visible lung damage caused by exposure to a
high concentration of O2, compared with untreated ALI
mice and vector-treated ALI mice (Fig.
3A-D). The comparison of lung injury scores between groups was
consistent with these findings; the Hyper + SIRT3 group
demonstrated a lower score than Hyper and Hyper + vector groups
(Fig. 3E). Additionally, compared
with Hyper and Hyper + vector groups, treatment with SIRT3-RV
significantly reduced the concentration of protein in BALF
(P<0.05; Fig. 3F), the wet/dry
ratio (P<0.05; Fig. 3G), and
the number of infiltrated neutrophils (P<0.05; Fig. 3H) in the lung tissue.
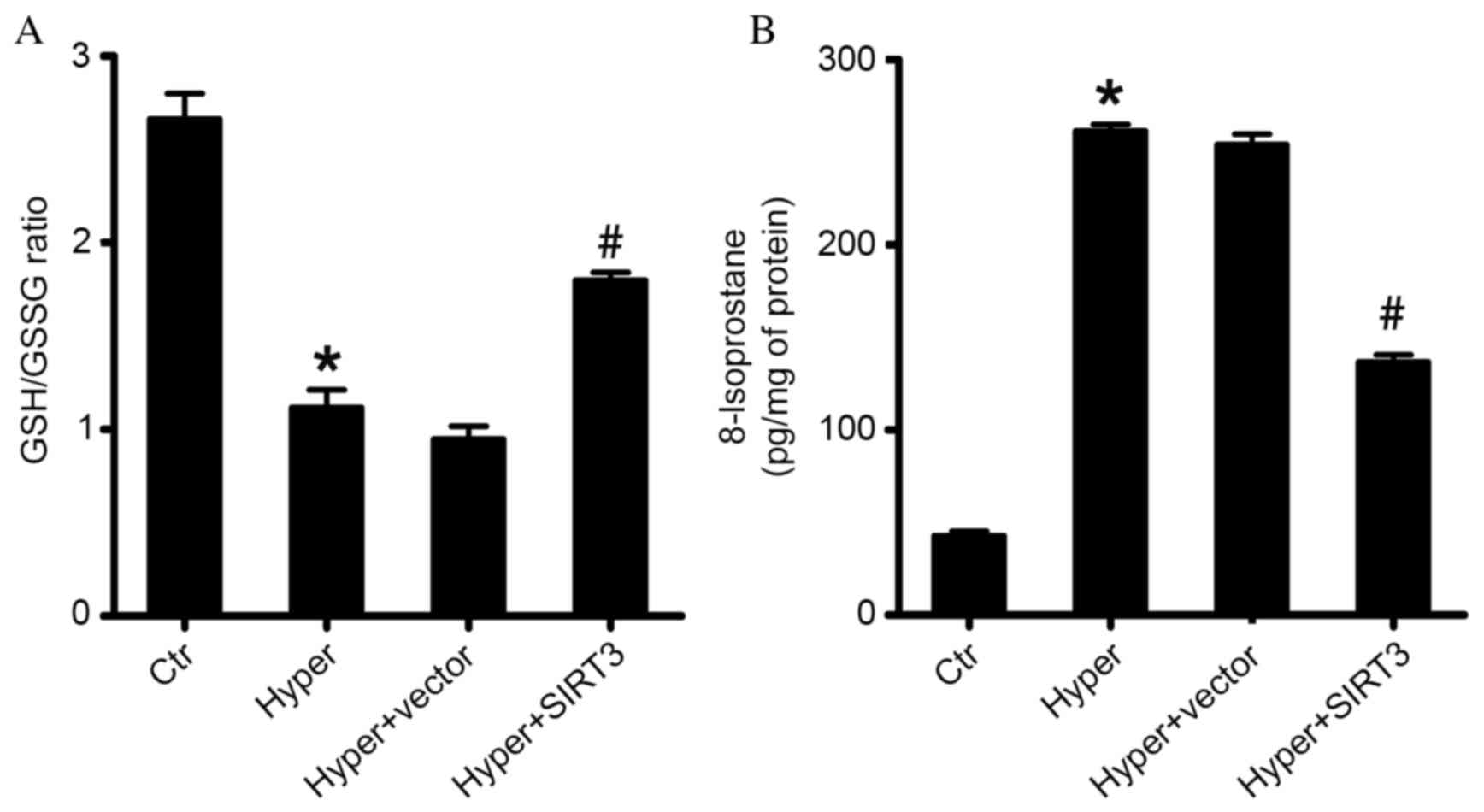
SIRT3 overexpression inhibits
oxidation and the level of 8-isoprostane in lung tissues
It is known that oxidative stress and lipid
peroxidation are involved in hyperoxia-induced ALI. The ratio of
reduced/oxidized GSH (GSH/GSSG) reflects the oxidative status of
tissues. To further determine the effect of SIRT3 on the
hyperoxia-induced oxidative damage of lung tissues, the GSH/GSSG
ratio in treated ALI mice was measured in the current study. It was
observed that, compared with the control group, there was
significant reduction in the ratio of GSH/GSSG in lung tissues of
hyperoxia-induced ALI mice (Hyper; P<0.05; Fig. 4A). Additionally, the current study
demonstrated that the overexpression of SIRT3 (Hyper + SIRT3)
significantly inhibited the reduction of the GSH/GSSG ratio caused
by high O2 exposure, compared with the untreated ALI
group (Hyper) and vector-treated ALI group (Hyper + vector;
P<0.05; Fig. 4A). 8-Isoprostane
is one of the most reliable biomarkers of lipid peroxidation and
oxidative stress. Therefore, the present study also measured the
level of 8-isoprostane in lungs and demonstrated that treatment
with SIRT3-RV significantly reduced the level of 8-isoprostane 3
days after treatment, compared with the untreated ALI group (Hyper)
and vector-treated group (Hyper + vector; P<0.05; Fig. 4B). These results indicated that
SIRT3 may have a potential antioxidative effect in
hyperoxia-induced ALI.
Overexpression of SIRT3 enhances the
total enzyme activity of SOD in ALI mice
Animal and human studies have indicated that acute
and chronic lung injury due to hyperoxia may be ameliorated by the
administration of antioxidants, such as SOD (39–43).
Therefore, the enzyme activity of SOD in the lung tissues of
differentially treated ALI mice was determined using a photometric
assay that measured the autoxidation of a tetracyclic catechol. The
current study demonstrated that SOD enzyme activity was
significantly decreased in hyperoxia-induced ALI mice (Hyper) after
6 days of O2 inhalation, compared with the control group
(P<0.05; Fig. 5).
Overexpression of SIRT3 in the lungs of hyperoxia-induced ALI mice
(Hyper + SIRT3) increased the level of SOD enzymatic activity to a
level comparable to the healthy controls (Fig. 5).
SIRT3 enhances the expression of MnSOD
but has no effect on the expression of other SODs
Increased production of ROS, including superoxide,
hydroxyl radicals and hydrogen peroxide is generally considered
essential for enhancing O2 toxicity (44–47).
Hyperoxia-induced injury increases the intracellular production of
ROS, which occurs via the mitochondria. Additionally, an increasing
number of studies suggest that SIRT3 may regulate the expression of
SOD (48–51). Therefore, we hypothesized that
SIRT3 may reduce hyperoxia-induced ALI by increasing the expression
of SODs in vivo, as these enzymes scavenge ROS. The
expression of MnSOD, CuZnSOD, ecSOD and GPx1 was measured by
RT-qPCR in the current study. As presented in Fig. 6, compared with the untreated ALI
group (Hyper) and vector-treated ALI group (Hyper + vector), SIRT3
overexpression (Hyper + SIRT3) significantly increased the
expression of MnSOD (P<0.05; Fig.
6A), while the expression of the other SODs investigated,
CuZnSOD (Fig. 6B), ecSOD (Fig. 6C) and GPx1 (Fig. 6D), was unchanged. These results
indicated that SIRT3 overexpression may increase the expression of
MnSOD in the lung tissue of hyperoxia-induced ALI mice and inhibit
hyperoxia-induced ALI through the antioxidative effect of MnSOD
in vivo.
 | Figure 6.mRNA expression of SOD enzymes in the
lung tissues of differentially treated mice. mRNA levels of (A)
MnSOD, (B) CuZnSOD, (C) ecSOD and (D) GPx1. #P<0.05
vs. hyperoxia-induced acute lung injury group (Hyper) or
vector-treated group (Hyper + vector). Data are presented as the
mean ± standard error of the mean. SOD, superoxide dismutase;
MnSOD, manganese SOD; Ctr, control; Hyper, hyperoxia; SIRT3,
sirtuin 3; CuZnSOD, copper-zinc SOD; ecSOD, extracellular SOD;
GPx1, glutathione peroxidase 1. |
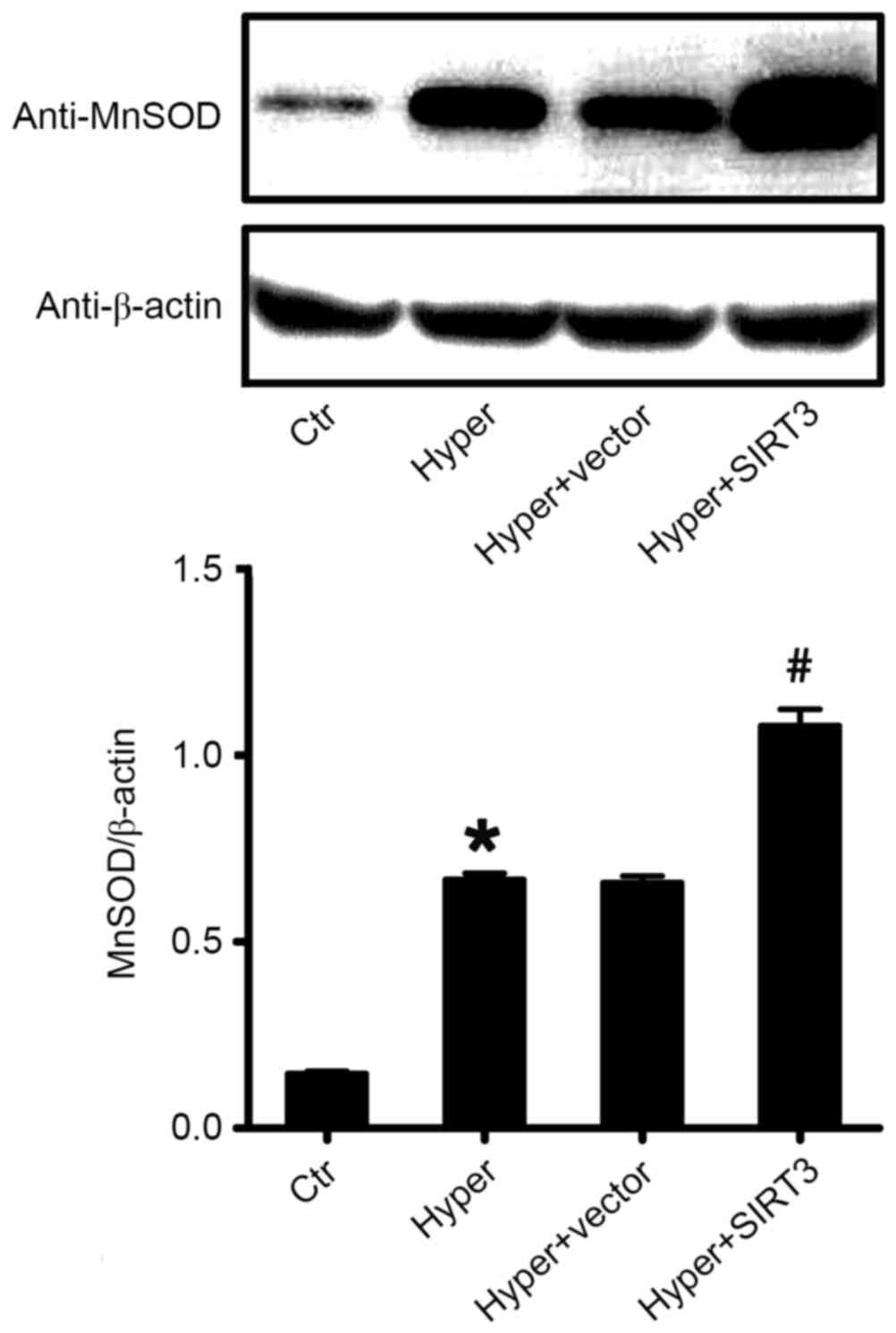
Western blot analysis of MnSOD in
mouse lung tissue
To further confirm the effect of SIRT3 on the
expression of the MnSOD protein, the current study detected the
protein levels of MnSOD in lung tissues using the western blot
analysis. As presented in Fig. 7,
MnSOD protein levels in the lungs of hyperoxia-induced ALI mice
(Hyper) was significantly increased compared with control mice.
Treatment with SIRT3-RV increased MnSOD protein levels induced by
O2 exposure compared with the untreated ALI mice (Hyper)
and the vector-treated ALI mice (Hyper + vector; P<0.05;
Fig. 7).
SIRT3 enhances the expression of MnSOD
in vitro
To further investigate the role SIRT3 may have in
hyperoxia-induced ALI injury, the current study increased or
decreased the expression of SIRT3 in human A549 cells using a
retrovirus overexpressing SIRT3 or SIRT3 siRNA, respectively,
followed by western blot analysis performed 24 h after cell
transfection. As presented in Fig.
8A, compared with the control, overexpression of SIRT3
significantly increased the expression of MnSOD in vitro
(P<0.05). The present study also demonstrated that the activity
of SODs was significantly increased by overexpression of SIRT3,
compared with the control (P<0.05; Fig. 8B). These results are consistent
with the observations from the in vivo studies.
SIRT3 overexpression improves survival
following hyperoxia-induced ALI
To evaluate the long-term beneficial effect of SIRT3
on hyperoxia-induced ALI, the survival rate was compared between
treated (Hyper + SIRT3) and control ALI mice (Hyper). The current
study demonstrated that the survival rate was significantly
improved in SIRT3-RV treated ALI mice compared with the untreated
ALI mice (P<0.05) and vector-treated ALI mice (P<0.05;
Fig. 9).
Discussion
Hyperoxia is a state of excess oxygen in tissues and
organs. Oxygen toxicity occurs when the partial pressure of
alveolar oxygen exceeds that which occurs when breathing normal
air. With continuous exposure to supraphysiological concentrations
of oxygen, a state of hyperoxia develops. Several studies have
demonstrated that exposure of lung tissue to high concentration of
oxygen causes oxidative stress and lung damage (5,52–54).
Lung oxidative stress results in an oxidant-antioxidant imbalance
that can lead to various lung diseases, including adult respiratory
distress syndrome. It is well established that oxidant production
within the lung can lead to ALI (55–58).
The present study demonstrated that SIRT3 overexpression reduced
hyperoxia-induced ALI by increasing the expression of MnSOD and
inhibiting the oxidative damage caused by hyperoxia induction.
Sirtuins are a family of highly conserved
NAD+-dependent deacetylases that act as cellular sensors
to detect energy availability and modulate metabolic processes. One
sirtuin, SIRT3, is central to the control of metabolic processes,
and is localized to the mitochondria, where the most damaging
oxidants are generated (33,59).
Therefore, we hypothesized that SIRT3 may have an important role in
hyperoxia-induced ALI. The results of this study demonstrated that
lung injury was induced by exposure to a high concentration of
O2, and reduced SIRT3 levels was dependent upon
hyperoxic exposure (Fig. 1). The
data indicated that SIRT3 may have an important role in the process
of hyperoxia-induced ALI.
To further investigate the effect of SIRT3 on
hyperoxia-induced ALI, the current study treated ALI mice with a
retroviral vector containing a SIRT3 gene (SIRT3-RV). The present
study demonstrated that SIRT3 overexpression had a beneficial
effect on hyperoxia-induced ALI (Fig.
3), including effects on the severity of lung damage (Fig. 3C), protein concentration in BALF
(Fig. 3D), the lung wet/dry ratio
(Fig. 3E) and the number of
infiltrating neutrophils (Fig.
3F). Furthermore, the current study also demonstrated that
SIRT3 overexpression increased the total enzyme activity of SODs
(Fig. 5). These observations
support the model that SIRT3 has a therapeutic effect on
hyperoxia-induced ALI due to its antioxidative effect. Previous
studies have reported that SIRT3 deacetylates MnSOD, thereby
increasing MnSOD activity (49,60).
To explore the mechanism of the antioxidative effect of SIRT3, the
mRNA expression of certain antioxidant enzymes that scavenge ROS,
including MnSOD, CuZnSOD, ecSOD and GPx1 in the lung tissues of
differentially treated mice were measured in the present study. The
results demonstrated that SIRT3 overexpression increased the
expression of MnSOD (Fig. 6A);
however, it did not alter the mRNA levels of the other antioxidant
enzymes, CuZnSOD (Fig. 6B), ecSOD
(Fig. 6C) and GPx1 (Fig. 6D). These results were consistent
with a previous report (61). To
confirm the effect of SIRT3 on MnSOD, this effect was investigated
in vitro. The current study inhibited or overexpressed SIRT3
in human A549 cells, and then detected the expression of SIRT3 and
MnSOD proteins by western blot analysis. As presented in Fig. 8, the results demonstrated that
SIRT3 significantly increased the expression of the MnSOD protein
in vitro, compared with the control group. These results
support the hypothesis that SIRT3 inhibits hyperoxia-induced ALI by
increasing the expression of MnSOD, and thus inhibiting the
oxidative damage caused by high concentration O2
exposure.
In conclusion, the results of the current study
demonstrated that SIRT3 inhibited hyperoxia-induced ALI. As a
mitochondrial protein, SIRT3 enhanced the expression of MnSOD and
reduced the oxidative injury caused by hyperoxic exposure. SIRT3
may be useful as a target for the treatment of hyperoxia-induced
ALI due to its potentially antioxidative effect.
References
|
1
|
Bhandari V: Hyperoxia-derived lung damage
in preterm infants. Semin Fetal Neonatal Med. 15:223–229. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cordingley JJ and Keogh BF: The pulmonary
physician in critical care. 8: Ventilatory management of ALI/ARDS.
Thorax. 57:729–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esteban A, Anzueto A, Alia I, Gordo F,
Apezteguía C, Pálizas F, Cide D, Goldwaser R, Soto L, Bugedo G, et
al: How is mechanical ventilation employed in the intensive care
unit? An international utilization review. Am J Respir Crit Care
Med. 161:1450–1458. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang H, Liao H, Ochani M, Justiniani M,
Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, et al:
Cholinergic agonists inhibit HMGB1 release and improve survival in
experimental sepsis. Nat Med. 10:1216–1221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kallet RH and Matthay MA: Hyperoxic acute
lung injury. Respir Care. 58:123–141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sinclair SE, Altemeier WA, Matute-Bello G
and Chi EY: Augmented lung injury due to interaction between
hyperoxia and mechanical ventilation. Crit Care Med. 32:2496–2501.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhandari V and Elias JA: Cytokines in
tolerance to hyperoxia-induced injury in the developing and adult
lung. Free Radic Biol Med. 41:4–18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McGrath-Morrow SA and Stahl J: Apoptosis
in neonatal murine lung exposed to hyperoxia. Am J Respir Cell Mol
Biol. 25:150–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Radomski A, Sawicki G, Olson DM and
Radomski MW: The role of nitric oxide and metalloproteinases in the
pathogenesis of hyperoxia-induced lung injury in newborn rats. Br J
Pharmacol. 125:1455–1462. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klings ES, Lowry MH, Li G, Jean JC,
Fernandez BO, Garcia-Saura MF, Feelisch M and Joyce-Brady M:
Hyperoxia-induced lung injury in gamma-glutamyl transferase
deficiency is associated with alterations in nitrosative and
nitrative stress. Am J Pathol. 175:2309–2318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Allen BW, Demchenko IT and Piantadosi CA:
Two faces of nitric oxide: Implications for cellular mechanisms of
oxygen toxicity. J Appl Physiol (1985). 106:662–667. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhandari V: Molecular mechanisms of
hyperoxia-induced acute lung injury. Front Biosci. 13:6653–6661.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Zoelen MA, Ishizaka A, Wolthuls EK,
Choi G, van der Poll T and Schultz MJ: Pulmonary levels of
high-mobility group box 1 during mechanical ventilation and
ventilator-associated pneumonia. Shock. 29:441–445. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sauve AA, Wolberger C, Schramm VL and
Boeke JD: The biochemistry of sirtuins. Annu Rev Biochem.
75:435–465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guarente L and Picard F: Calorie
restriction-the SIR2 connection. Cell. 120:473–482. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin SJ, Defossez PA and Guarente L:
Requirement of NAD and SIR2 for life-span extension by calorie
restriction in Saccharomyces cerevisiae. Science. 289:2126–2128.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin SJ, Kaeberlein M, Andalis AA, Sturtz
LA, Defossez PA, Culotta VC, Fink GR and Guarente L: Calorie
restriction extends Saccharomyces cerevisiae lifespan by increasing
respiration. Nature. 418:344–348. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michan S and Sinclair D: Sirtuins in
mammals: Insights into their biological function. Biochem J.
404:1–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Taylor DM, Maxwell MM, Luthi-Carter R and
Kazantsev AG: Biological and potential therapeutic roles of sirtuin
deacetylases. Cell Mol Life Sci. 65:4000–4018. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi T, Wang F, Stieren E and Tong Q:
SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial
function and thermogenesis in brown adipocytes. J Biol Chem.
280:13560–13567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park SH, Ozden O, Jiang H, Cha YI,
Pennington JD, Aykin-Burns N, Spitz DR, Gius D and Kim HS: Sirt3,
mitochondrial ROS, ageing, and carcinogenesis. Int J Mol Sci.
12:6226–6239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wispé JR, Warner BB, Clark JC, Dey CR,
Neuman J, Glasser SW, Crapo JD, Chang LY and Whitsett JA: Human
Mn-superoxide dismutase in pulmonary epithelial cells of transgenic
mice confers protection from oxygen injury. J Biol Chem.
267:23937–23941. 1992.PubMed/NCBI
|
|
23
|
Slot JW, Geuze HJ, Freeman BA and Crapo
JD: Intracellular localization of the copper-zinc and manganese
superoxide dismutases in rat liver parenchymal cells. Lab Invest.
55:363–371. 1986.PubMed/NCBI
|
|
24
|
Oury TD, Chang LY, Marklund SL, Day BJ and
Crapo JD: Immunocytochemical localization of extracellular
superoxide dismutase in human lung. Lab Invest. 70:889–898.
1994.PubMed/NCBI
|
|
25
|
Kingston RE, Chen CA and Rose JK: Calcium
phosphate transfection. Curr Protoc Mol Biol. 9:9 12003.PubMed/NCBI
|
|
26
|
Zhang Y, Lin X, Koga K, Takahashi K, Linge
HM, Mello A, Laragione T, Gulko PS and Miller EJ: Strain
differences in alveolar neutrophil infiltration and macrophage
phenotypes in an acute lung inflammation model. Mol Med.
17:780–789. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mantell LL, Shaffer TH, Horowitz S, Foust
R III, Wolfson MR, Cox C, Khullar P, Zakeri Z, Lin L, Kazzaz JA, et
al: Distinct patterns of apoptosis in the lung during liquid
ventilation compared with gas ventilation. Am J Physiol Lung Cell
Mol Physiol. 283:L31–L41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nishina K, Mikawa K, Takao Y, Maekawa N,
Shiga M and Obara H: ONO-5046, an elastase inhibitor, attenuates
endotoxin-induced acute lung injury in rabbits. Anesth Analg.
84:1097–1103. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Numata M, Suzuki S, Miyazawa N, Miyashita
A, Nagashima Y, Inoue S, Kaneko T and Okubo T: Inhibition of
inducible nitric oxide synthase prevents LPS-induced acute lung
injury in dogs. J Immunol. 160:3031–3037. 1998.PubMed/NCBI
|
|
31
|
Sun Y, Oberley LW and Li Y: A simple
method for clinical assay of superoxide dismutase. Clin Chem.
34:497–500. 1988.PubMed/NCBI
|
|
32
|
North BJ and Verdin E: Sirtuins:
Sir2-related NAD-dependent protein deacetylases. Genome Biol.
5:2242004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vassilopoulos A, Fritz KS, Petersen DR and
Gius D: The human sirtuin family: Evolutionary divergences and
functions. Hum Genomics. 5:485–496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feldman JL, Dittenhafer-Reed KE and Denu
JM: Sirtuin catalysis and regulation. J Biol Chem. 287:42419–42427.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sauve AA and Youn DY: Sirtuins:
NAD(+)-dependent deacetylase mechanism and regulation. Curr Opin
Chem Biol. 16:535–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hallows WC, Albaugh BN and Denu JM: Where
in the cell is SIRT3?-functional localization of an NAD+-dependent
protein deacetylase. Biochem J. 411:e11–e13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Buler M, Aatsinki SM, Izzi V and Hakkola
J: Metformin reduces hepatic expression of SIRT3, the mitochondrial
deacetylase controlling energy metabolism. PLoS One. 7:e498632012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hallows WC, Lee S and Denu JM: Sirtuins
deacetylate and activate mammalian acetyl-CoA synthetases. Proc
Natl Acad Sci USA. 103:pp. 10230–10235. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Davis JM, Rosenfeld WN, Richter SE, Parad
MR, Gewolb IH, Spitzer AR, Carlo WA, Couser RJ, Price A, Flaster E,
et al: Safety and pharmacokinetics of multiple doses of recombinant
human CuZn superoxide dismutase administered intratracheally to
premature neonates with respiratory distress syndrome. Pediatrics.
100:24–30. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakamura T and Ogawa Y: Prophylactic
effects of recombinant human superoxide dismutase in neonatal lung
injury induced by the intratracheal instillation of endotoxin in
piglets. Biol Neonate. 80:163–168. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jacobson JM, Michael JR, Jafri MH Jr and
Gurtner GH: Antioxidants and antioxidant enzymes protect against
pulmonary oxygen toxicity in the rabbit. J Appl Physiol (1985).
68:1252–1259. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tanswell AK, Olson DM and Freeman BA:
Liposome-mediated augmentation of antioxidant defenses in fetal rat
pneumocytes. Am J Physiol. 258:L165–L172. 1990.PubMed/NCBI
|
|
43
|
Walther FJ, Gidding CE, Kuipers IM,
Willebrand D, Bevers EM, Abuchowski A and Viau AT: Prevention of
oxygen toxicity with superoxide dismutase and catalase in premature
lambs. J Free Radic Biol Med. 2:289–293. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Freeman BA, Topolosky MK and Crapo JD:
Hyperoxia increases oxygen radical production in rat lung
homogenates. Arch Biochem Biophys. 216:477–484. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fox RB, Hoidal JR, Brown DM and Repine JE:
Pulmonary inflammation due to oxygen toxicity: Involvement of
chemotactic factors and polymorphonuclear leukocytes. Am Rev Respir
Dis. 123:521–523. 1981.PubMed/NCBI
|
|
46
|
Mantell LL and Lee PJ: Signal transduction
pathways in hyperoxia-induced lung cell death. Mol Genet Metab.
71:359–370. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jamieson D: Oxygen toxicity and reactive
oxygen metabolites in mammals. Free Radic Biol Med. 7:87–108. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ansari A, Rahman MS, Saha SK, Saikot FK,
Deep A and Kim KH: Function of the SIRT3 mitochondrial deacetylase
in cellular physiology, cancer, and neurodegenerative disease.
Aging Cell. 16:4–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tao R, Coleman MC, Pennington JD, Ozden O,
Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et
al: Sirt3-mediated deacetylation of evolutionarily conserved lysine
122 regulates MnSOD activity in response to stress. Mol Cell.
40:893–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Candas D and Li JJ: MnSOD in oxidative
stress response-potential regulation via mitochondrial protein
influx. Antioxid Redox Signal. 20:1599–1617. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bause AS, Matsui MS and Haigis MC: The
protein deacetylase SIRT3 prevents oxidative stress-induced
keratinocyte differentiation. J Biol Chem. 288:36484–36491. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rahman I and MacNee W: Lung glutathione
and oxidative stress: Implications in cigarette smoke-induced
airway disease. Am J Physiol. 277:L1067–L1088. 1999.PubMed/NCBI
|
|
53
|
Bargagli E, Olivieri C, Bennett D, Prasse
A, Muller-Quernheim J and Rottoli P: Oxidative stress in the
pathogenesis of diffuse lung diseases: A review. Respir Med.
103:1245–1256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mach WJ, Thimmesch AR, Pierce JT and
Pierce JD: Consequences of hyperoxia and the toxicity of oxygen in
the lung. Nurs Res Pract. 2011:2604822011.PubMed/NCBI
|
|
55
|
Borok Z, Buhl R, Grimes GJ, Bokser AD,
Hubbard RC, Holroyd KJ, Roum JH, Czerski DB, Cantin AM and Crystal
RG: Effect of glutathione aerosol on oxidant-antioxidant imbalance
in idiopathic pulmonary fibrosis. Lancet. 338:215–216. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kinnula VL, Fattman CL, Tan RJ and Oury
TD: Oxidative stress in pulmonary fibrosis: A possible role for
redox modulatory therapy. Am J Respir Crit Care Med. 172:417–422.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Johnson LN and Koval M: Cross-talk between
pulmonary injury, oxidant stress, and gap junctional communication.
Antioxid Redox Signal. 11:355–367. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vivekananda J, Lin A, Coalson JJ and King
RJ: Acute inflammatory injury in the lung precipitated by oxidant
stress induces fibroblasts to synthesize and release transforming
growth factor-alpha. J Biol Chem. 269:25057–25061. 1994.PubMed/NCBI
|
|
59
|
Guo X, Kesimer M, Tolun G, Zheng X, Xu Q,
Lu J, Sheehan JK, Griffith JD and Li X: The NAD(+)-dependent
protein deacetylase activity of SIRT1 is regulated by its
oligomeric status. Sci Rep. 2:6402012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sundaresan NR, Gupta M, Kim G, Rajamohan
SB, Isbatan A and Gupta MP: Sirt3 blocks the cardiac hypertrophic
response by augmenting Foxo3a-dependent antioxidant defense
mechanisms in mice. J Clin Invest. 119:2758–2771. 2009.PubMed/NCBI
|
|
61
|
Zhang B, Cui S, Bai X, Zhuo L, Sun X, Hong
Q, Fu B, Wang J, Chen X and Cai G: SIRT3 overexpression antagonizes
high glucose accelerated cellular senescence in human diploid
fibroblasts via the SIRT3-FOXO1 signaling pathway. Age (Dordr).
35:2237–2253. 2013. View Article : Google Scholar : PubMed/NCBI
|