Introduction
Deafness is one of the most common human health
problems, affecting one in 700–1,000 newborns (1). It has been estimated that ≥50% of
cases of congenital deafness have genetic causes. At present, the
etiology of hearing loss remains largely unknown, although gene
alterations (2–15) and environmental factors, including
aminoglycoside antibiotics (AmAn), are now generally considered to
be involved in this condition (16). AmAn, including gentamicin and
kanamycin, are of great clinical importance for the treatment of
bacterial infections. However, the use of these drugs is frequently
associated with toxicity, involving the renal, auditory and
vestibular systems (17). In
familial cases of ototoxicity, AmAn hypersensitivity is frequently
maternally transmitted, suggesting that mutations in mitochondrial
(mt)DNA may be the molecular basis for this susceptibility
(18). Among the mtDNA genes, 12S
ribosomal (r)RNA and transfer (t)RNA are hot-spots for pathogenic
mutations associated with deafness (19,20).
Indeed, it has been reported that deafness-associated mtDNA primary
mutations impair mitochondrial translation, leading to deficient
respiration (21). However, only
mild mitochondrial dysfunctions are observed in cells carrying
these mtDNA mutations, suggesting that these mutations are
necessary but insufficient to produce a clinical phenotype
(22). Therefore, there may be
other modifying factors that modulate the phenotypic manifestation
of these mtDNA mutations (23).
To investigate the modifying factors that contribute
to the clinical expression of deafness-associated mtDNA mutations,
a mutational screening on deaf patients from Tianjin, China was
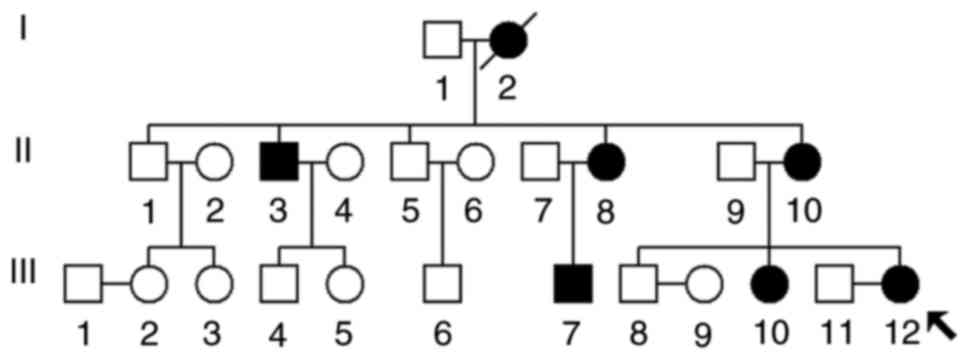
performed in the present study. The present study describes a Han
Chinese family with maternally-transmitted hearing loss, in which
analysis of the complete mtDNA sequence demonstrated the occurrence
of 12S rRNA A1555G and tRNAIle A4317G mutations. To
further understand the possible role of A4317G mutation, the mtDNA
copy number in the patients harboring this mutation was analyzed.
In addition, to determine the contributions of the gap junction
protein β2 (GJB2) and tRNA
5-methylaminomethyle-2-thiouridylate methyltransferase
(TRMU) genes to the phenotypic manifestation of the A1555G
mutation, a mutational analysis of these genes in individuals with
hearing impairment in this family was conducted.
Patients and methods
Subjects
A screening program was performed for mtDNA
mutations in deaf patients from Tianjin City, China. A Han Chinese
family, as presented in Fig. 1,
was enrolled in October, 2017 through the otology clinic of The PLA
254 Hospital (Tianjin, China). Informed consent was obtained from
the participants prior to their participation in the study, in
accordance with the requirements of the Ethics Committee of The PLA
254 Hospital. In addition, 300 healthy age- and sex-matched
controls (including 100 females and 200 males; aged 20–50 years
with an average of 30.5 years) were obtained from a panel of
unaffected subjects with Han Chinese ancestry. The control subjects
were healthy individuals, and did not have any family history of
mitochondrial disorders. The present study was approved by the
Ethics Committee of The PLA 254 Hospital.
Clinical examinations
A comprehensive history was taken and a physical
examination was performed on all available members of this Chinese
pedigree to identify any syndromic findings, history of exposure to
AmAn and genetic factors associated with hearing impairment. An
age-appropriate audiological examination was additionally
performed, which included pure-tone audiometry (PTA), auditory
brainstem response and distortion product otoacoustic emissions.
The PTA was calculated from the sum of the audiometric thresholds
at 500, 1,000, 2,000, 4,000 and 8,000 Hz. The severity of hearing
impairment was classified into five grades: Normal (<26 dB);
mild (26–40 dB); moderate (41–70) dB; severe (71–90) dB; and
profound (>90 dB).
Screening for variants in the
mitochondrial genome
The maternally-transmitted pattern in the studied
family suggested the involvement of the mitochondria and thus
analysis of variants in the mitochondrial genome was performed.
Genomic DNA was isolated from whole blood samples (3 ml) from the
participants using PAXgene Blood DNA Isolation kits (Qiagen, Inc.,
Valencia, CA, USA). The entire mitochondrial genomes of the
matrilineal relatives were amplified by polymerase chain reaction
(PCR) in 24 overlapping fragments using sets of the light-strand
(L) and heavy-strand (H) oligonucleotide primers, as described
elsewhere (24). The sequences of
the primers are the same as stated in a previous study (24). The PCR primers were supplied by BGI
(Shenzhen, China) and the PCR mixture included 200 µM dNTP, 10X
buffer, Taq DNA polymerase and 15 mmol/l Mg2+ (Takara
Biotechnology Co., Ltd., Dalian, China). The PCR was performed as
follows: 95°C for 5 min, 94°C for 10 sec, 60°C for 30 sec and 72°C
for 1 min (for 30 cycles), followed by 72°C extension for 5 min.
Finally, 2 µl PCR product was analyzed using 1.5% agarose gel,
which contained ethidium bromide, electrophoresis at 130 V for 30
min. Following electrophoresis, a BandPeeper (Maestrogen, Inc.,
Hsinchu, Taiwan) instrument was used to detect the results using
the Invitrogen™ E-Gel™ Imager software (version 2.2.4; Invitrogen;
Thermo Fisher Scientific Inc., Waltham, MA, USA). Each fragment was
purified and subsequently analyzed by direct sequencing in an ABI
3700 automated DNA sequencer using the Big Dye Terminator Cycle
sequencing reaction kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The resultant sequence data were compared with
the updated consensus Cambridge sequence (GenBank accession no.
NC_012920; www.ncbi.nlm.nih.gov/genbank) (25) and the Mitomap database (www.mitomap.org).
Phylogenetic analysis
A total of 17 vertebrate mtDNA sequences were used
in the inter-species analysis. The conservation index (CI) was
calculated by comparing the human nucleotide variants with 16 other
vertebrates. The CI was defined as the percentage of species from
the list of 16 different vertebrates that possessed the wild-type
nucleotide at that position. CI ≥75% was regarded as being
indicative of functional potential.
Mutational analysis of the GJB2
gene
DNA fragments spanning the entire coding region of
the GJB2 gene were amplified by PCR using appropriate
primers, in accordance with a previous investigation (26). The sequences of the primers are as
stated in a previous study (26).
PCR amplification and subsequent sequencing analysis were performed
as described previously (26). The
results were compared with the wild-type GJB2 sequence to
identify potential mutations (GenBank accession no. M86849).
Genotyping analysis of TRMU gene
A previous study demonstrated that the TRMU
exon 1 A10S mutation may modulate the phenotypic manifestation of
deafness-associated mitochondrial 12S rRNA mutations (27). To determine whether TRMU
serves an active role in the expression of deafness, mutational
screening on TRMU exon 1 was conducted in matrilineal
relatives in this pedigree and in healthy controls. The primer
information and PCR conditions have been described elsewhere
(27). The PCR segments were
analyzed and compared with the TRMU genomic sequence
(GenBank accession no. AF_448221).
mtDNA copy number analysis
The mtDNA copy number was measured using
quantitative (q)PCR and the 2−ΔΔCq method (28). mtDNA content was normalized using a
single copy nuclear β-globin gene. The following primers were used
for qPCR analysis: β-Globin gene forward, 5′-CTATGGGACGCTTGATGT-3′
and reverse, 5′-GCAATCATTCGTCTGTTT-3′. For mtDNA forward,
5′-CACCAGCCTAACCAGATTTC-3′ and reverse,
5′-GGGTTGTATTGATGAGATTAGT-3′. Standard curves were generated for
the two fragments and their respective amplification efficiencies
calculated to test whether the 2−ΔΔCq method was
appropriate. The 20 µl PCR reaction solution contained 2X Taqman
Universal PCR Master Mix (Takara Biotechnology Co., Ltd.), 500
nmol/l of each primer, 200 nmol/l Taqman Probe and 100 ng of total
DNA. PCR conditions were 2 min at 50°C and 10 min at 95°C, followed
by 40 cycles of denaturation for 15 sec at 95°C and 60 sec
annealing/extension at 60°C. All experiments were repeated three
times.
Statistical analysis
Statistical significance was evaluated by
independent Student's t-test using SPSS software version 18.0
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Clinical features of the Han Chinese
family with hearing loss
The proband (III-12) was a 28-year-old woman from
Tianjin, China. As presented in Fig.
1 and Table I, the proband
began to suffer bilateral hearing loss at 1 year of age (108 dB
right ear and 108 dB left ear). Subsequent to taking a
comprehensive history and performing a physical examination, it was
noted that the proband (III-12) had begun to use gentamycin (5
mg/kg/dose; 10 days) at the age of 15 years. Following 2 weeks of
use, the proband exhibited irreversible hearing impairment. In
addition, two relatives (II-3 and II-10) on the maternal side of
the family had used AmAn in childhood and also developed profound
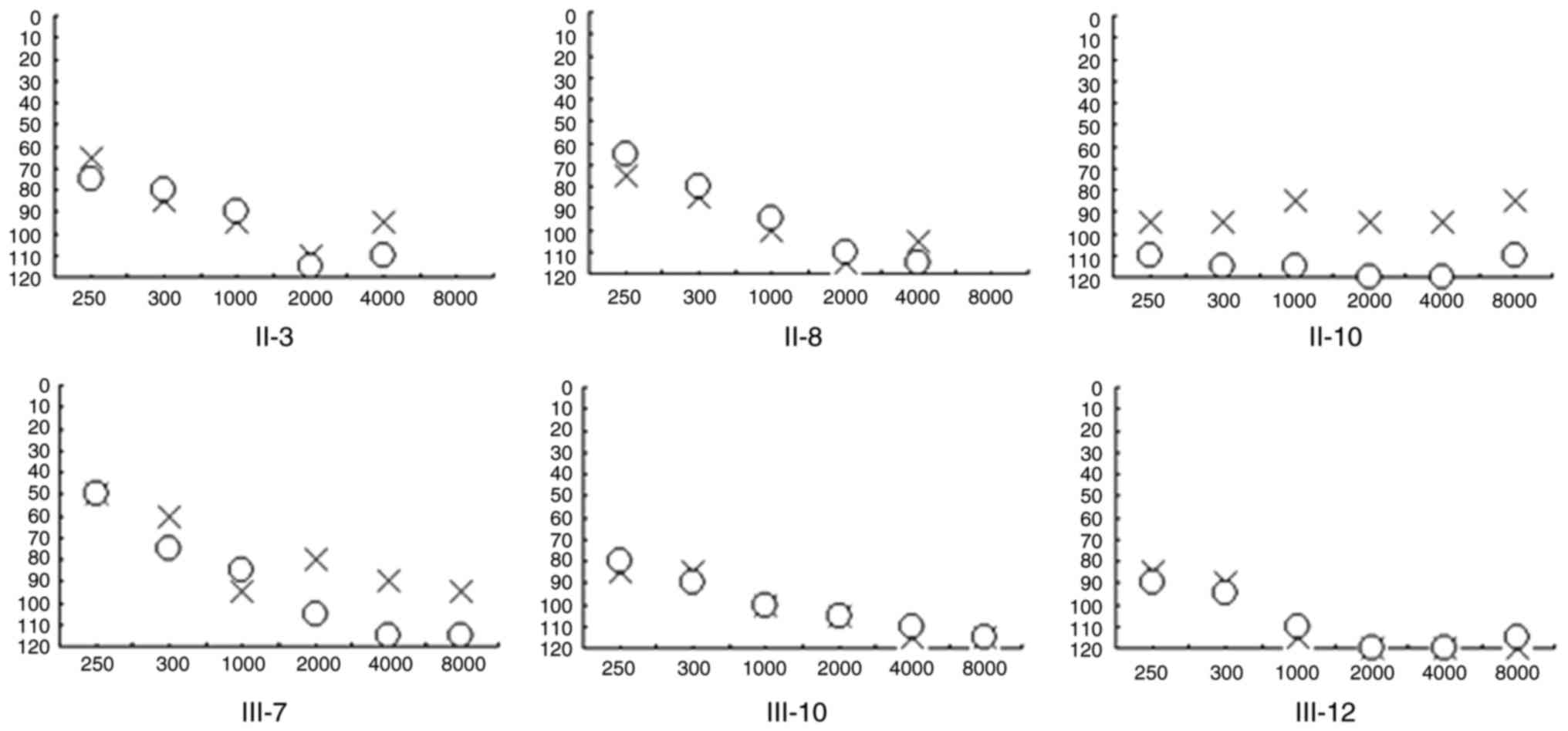
hearing impairment. As presented in Fig. 2, audiological evaluation of the
deaf patients in this family demonstrated that the majority of them
had profound hearing loss with a slope-shaped pattern. In addition,
this family exhibited a high penetrance of hearing loss, of 66.6%
or 33.3% when AmAn was included or excluded, respectively.
 | Table I.Clinical characterization of the
deafness patients in the family. |
Table I.
Clinical characterization of the
deafness patients in the family.
| Subject | Sex | Age at test,
years | Age at onset,
years | PTA (left ear) | PTA (right
ear) | Level of hearing
loss |
|---|
| II-3 | Male | 55 | 50 | 90 | 94 | Profound |
| II-8 | Female | 63 | 53 | 96 | 93 | Profound |
| II-10 | Female | 60 | 58 | 78 | 115 | Profound |
| III-7 | Male | 35 | 30 | 78 | 91 | Profound |
| III-10 | Female | 30 | 21 | 99 | 100 | Profound |
| III-12 | Female | 28 | 1 | 108 | 108 | Profound |
Genotype analysis of mitochondrial
genes
The fact that the deafness in this family was
maternally inherited indicated that mtDNA dysfunction may be
responsible for the clinical manifestation of hearing loss. The
mtDNA sequence variants in matrilineal relatives in this pedigree
were thus screened using PCR and direct sequencing. As presented in
Table II, Sanger sequencing led
to the identification of 27 genetic polymorphisms. These included
10 variants in the D-loop gene, three variants in the 12S rRNA
gene, two variants in the 16S rRNA and one variant in the tRNA
gene, in addition to a 9-bp common deletion in the junction between
tRNALys and CO2, while other variants were primarily
localized to protein-coding genes. In addition, six missense
mutations were noted, including ND2 C5178A (Leu to Met), A8 C8414T
(Leu to Phe), A6 A8701G (Thr to Ala) and A8860G (Thr to Ala), and
CytB C14766T (Thr to Ile) and A15326G (Thr to Ala). These variants
in rRNAs, tRNA and polypeptides were further evaluated by
phylogenetic analysis against other organisms including mouse
(29), bovine (30) and Xenopus laevis (31) sequences. However, none of these
variants exhibited evolutionary conservation, with the exception of
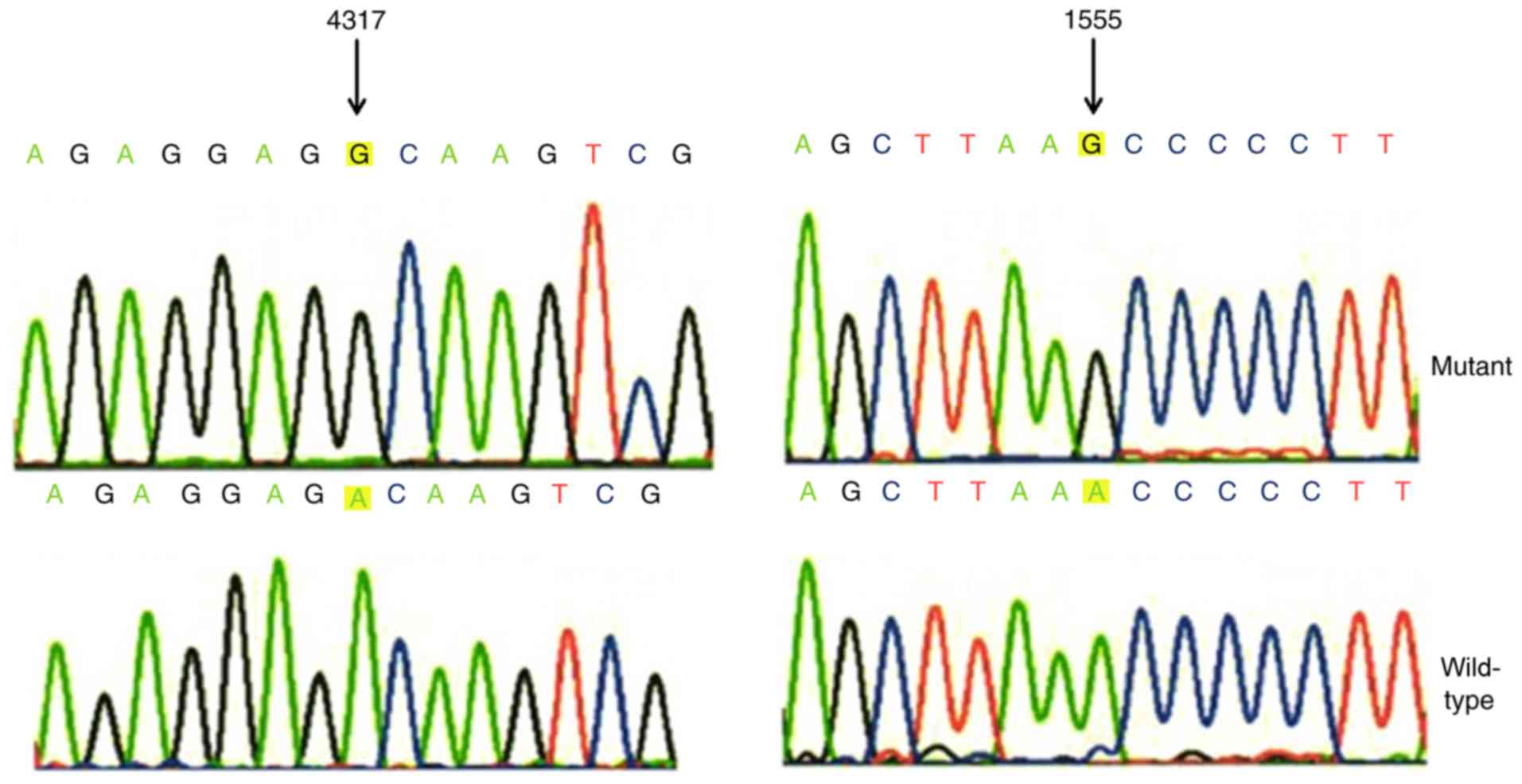
the A1555G and A4317G mutations (Figs.
3–5). In addition, A1555G and
A4317G mutations were not detected in healthy subjects, which
indicated that these mutations may be involved in the pathogenesis
of deafness in this family (P<0.05; data is not shown).
| Table II.Mitochondrial DNA sequence variants
in the family with hearing loss. |
Table II.
Mitochondrial DNA sequence variants
in the family with hearing loss.
| Gene |
Positionb | Alternations | Conservation
(H/B/M/X)a | Previously
reported |
|---|
| D-loop | 73 | A to G | – | Yes |
|
| 150 | A to G | – | Yes |
|
| 199 | T to C | – | Yes |
|
| 315 | C to CC | – | Yes |
|
| 489 | T to C | – | Yes |
|
| 16183 | A to C | – | Yes |
|
| 16189 | T to C | – | Yes |
|
| 16310 | A to G | – | Yes |
|
| 16355 | C to T | – | Yes |
|
| 16519 | T to C | – | Yes |
| 12S rRNA | 750 | A to G | A/A/A/− | Yes |
|
| 1438 | A to G | A/A/A/G | Yes |
|
| 1555 | A to G | A/A/A/A | Yes |
| 16S rRNA | 2706 | A to G | A/G/A/A | Yes |
|
| 3010 | G to A | G/G/A/A | Yes |
| ND1 | 3396 | T to C | – | Yes |
|
| 3970 | C to T | – | Yes |
|
| 4248 | T to C | – | Yes |
|
tRNAIle | 4317 | A to G | A/A/A/A | Yes |
| ND2 | 5178 | C to A (Leu to
Met) | T/T/T/T | Yes |
| CO1 | 7028 | C to T | – | Yes |
| NC7 | 8281–8289 | 9-bp del | – | Yes |
| A8 | 8414 | C to T (Leu to
Phe) | F/M/L/W | Yes |
| A6 | 8701 | A to G (Thr to
Ala) | M/M/M/F | Yes |
|
| 8860 | A to G (Thr to
Ala) | T/A/A/T | Yes |
| CytB | 14766 | C to T (Thr to
Ile) | T/S/T/S | Yes |
|
| 15326 | A to G (Thr to
Ala) | T/M/I/I | Yes |
Screening for mutations in the GJB2
gene
Mutations in the GJB2 gene have been reported
to be important causes of non-syndromic hearing loss (32). Therefore, to determine the
contribution of GJB2 to the expression of deafness
mutational analysis of the GJB2 coding region in the
matrilineal relatives and controls in this family was conducted.
For this, PCR was performed, the obtained products purified, and
subsequently analyzed by Sanger sequencing. However, no functional
variants in the GJB2 gene were detected (data not
shown).
Mutational analysis of the TRMU
gene
A previous study demonstrated that TRMU acts
as a nuclear modified gene and is responsible for the 2-thiolation
modification of mt-tRNAs, in addition to modulating the phenotypic
manifestation of deafness-associated 12S rRNA mutations (27). Therefore, the TRMU A10S
mutation was screened in matrilineal relatives and controls in the
family in the present study. However, no variants in TRMU
exon 1 were identified.
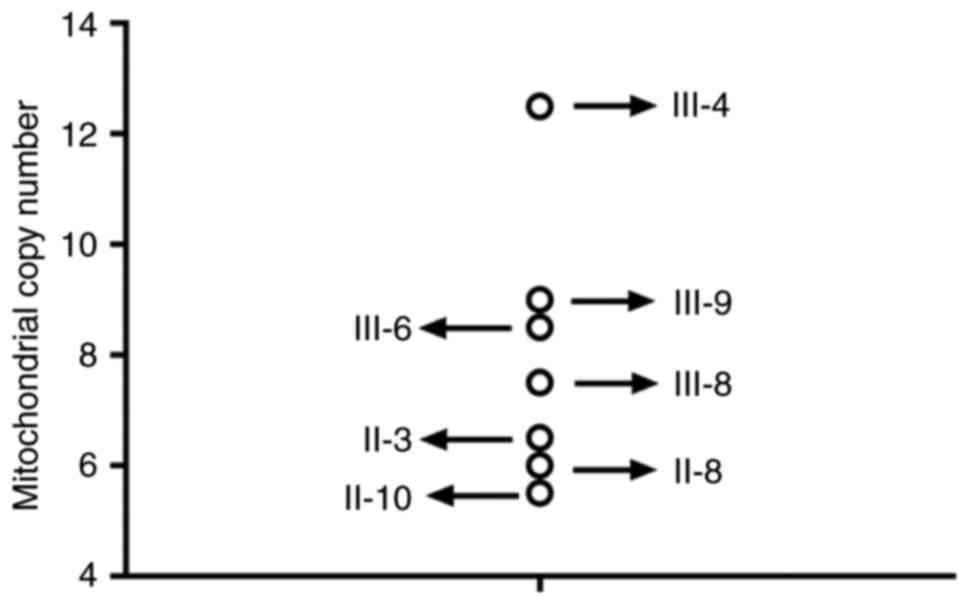
MtDNA content analysis
As presented in Fig.
6, it was observed that the patients carrying the mitochondrial
A4317G and A1555G mutations had a lower mtDNA copy number than the
healthy controls, suggesting that these mutations may decrease
mtDNA content and cause mitochondrial dysfunction.
Discussion
The present study reported the molecular and genetic
characterizations of a Han Chinese family with
maternally-transmitted and AmAn-induced hearing loss. Hearing
impairment as the sole clinical phenotype was presented only in the
maternal lineage of this pedigree. This family exhibited high
penetrance and expressivity of deafness; in particular, the
penetrance of hearing loss was 66.6% when the effect of AmAn was
included and 33.3% when it was excluded. In addition, the age at
onset of deafness ranged between 1 and 58 years, with an average of
35 years and the matrilineal relatives in this family had an
earlier age at onset of hearing loss, suggesting that mitochondrial
sequence variants may be risk factors that may be used for early
molecular diagnosis and prevention of deafness.
Mutational analysis of the complete mitochondrial
genome demonstrated the presence of the A1555G mutation, together
with a set of genetic polymorphisms belonging to the human
mitochondrial haplogroup B4c1b2 (33). The well-known A1555G mutation,
which was first described in a large Arab-Israel pedigree with
hearing loss (34), has been
associated with AmAn-induced and non-syndromic hearing loss in
numerous families worldwide (35–37).
Although biochemical data indicated that the A1555G mutation led to
sensitivity to AmAn (21,22), the incomplete penetrance and mild
mitochondrial dysfunction suggested that this mutation alone was
insufficient to produce the clinical phenotype. Therefore, other
modifying factors, including environmental factors, the use of
AmAn, mitochondrial variants/haplogroups and nuclear genes, may
contribute to the expression of deafness. Notably, the absence of
any functional variants in nuclear genes (GJB2 and
TRMU) suggested that these genes may not serve important
roles in the phenotypic expression of the A1555G mutation in this
family; therefore, other nuclear genes may contribute to the
phenotypic variability.
The mitochondrial haplogroup has been demonstrated
to modulate the phenotypic expression of the deafness-associated
A1555G mutation (23). For
example, the haplogroup Y2 specific tRNAGlu A14693G
variant may increase the penetrance and expressivity of the
deafness-associated A1555G mutation (23). In the present study, sequence
analysis of the complete mitochondrial genomes of the affected
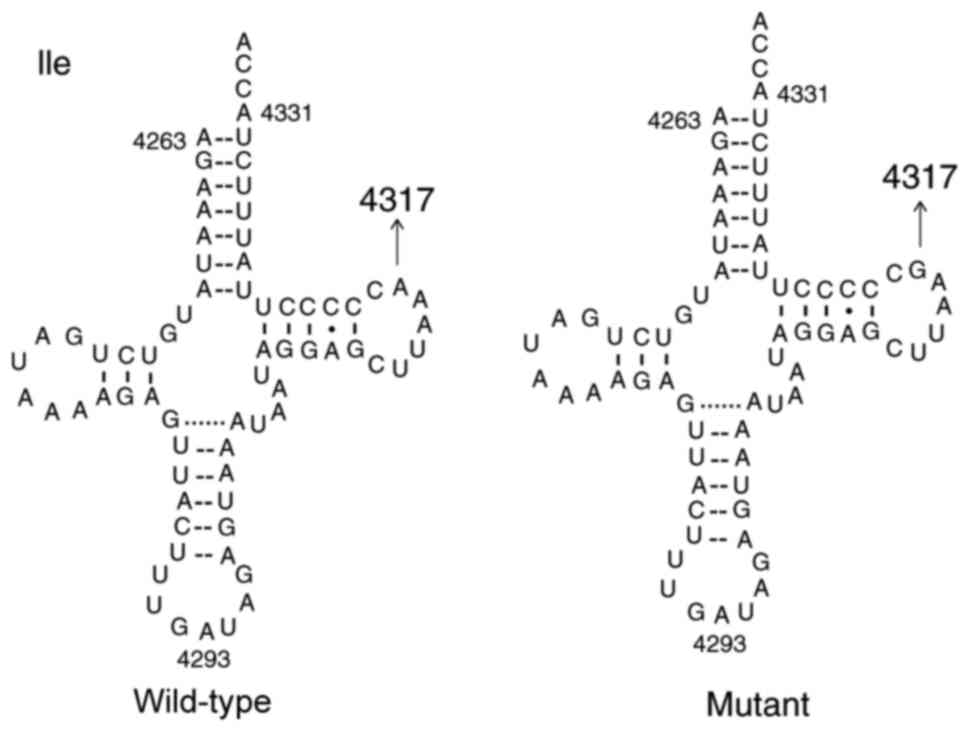
individuals in this family identified the occurrence of the A4317G
mutation. This mutation was localized at the TΨC arm of the
tRNAIle gene (position 59) using the Mitomap database,
which is highly conserved from bacteria to human mitochondria
(38,39). Notably, the A4317G mutation creates
a novel base-pairing and may exert a structural effect. In fact,
the presence of G59 facilitates the formation of an additional base
pair with C54. This may in turn lead to complete rearrangement of
the T stem and shortening of the T loop (40,41).
Furthermore, this mutation has been reported to be associated with
essential hypertension in the Han Chinese population (42). Biochemical analysis has also
demonstrated that the A4317G mutation decreases the aminoacylation
ability of tRNAIle (43). In addition, it was identified that
this mutation significantly decreased the mtDNA copy number when
compared with that in controls. Indeed, mtDNA content has been
shown to be a measure indicative of the cell number or mass of
mitochondria (44). A previous
experimental study suggested that alterations in mtDNA serve a
fundamental role in the increase in reactive oxygen species;
maintenance of mtDNA copy number was demonstrated to be essential
for the preservation of mitochondrial function and cell growth
(45). Therefore, it may be
speculated that A4317G mutation may lead to the failure of tRNA
metabolism, and subsequently impair mitochondrial protein
synthesis, thereby worsening mitochondrial dysfunction (represented
by a malfunction in biochemical processes, characterized by
mitochondrial membrane potential collapse and decreased ATP
production, which also has an essential role in the mediation of
apoptosis) induced by A1555G mutation (46). Therefore, the tRNAIle
A4317G mutation may contribute to the high penetrance and
expressivity of the hearing loss associated with the mtDNA A1555G
mutation in this Chinese pedigree. The principal limitation of the
present study study was the small sample size; further studies are
required, with a greater number of deaf patients and controls.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC and D-JH designed the study. YC performed the
experiments and analyzed the data. D-JH wrote the paper. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Informed consent was obtained from the participants
prior to their participation in the present study, in accordance
with the Ethics Committee of PLA 254 Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lin X, Teng Y, Lan J, He B, Sun H and Xu
F: GRHL2 genetic polymorphisms may confer a protective effect
against sudden sensorineural hearing loss. Mol Med Rep.
13:2857–2863. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nikopoulos K, Farinelli P, Giangreco B,
Tsika C, Royer-Bertrand B, Mbefo MK, Bedoni N, Kjellström U, El
Zaoui I, Di Gioia SA, et al: Mutations in CEP78 cause cone-rod
dystrophy and hearing loss associated with primary-cilia defects.
Am J Hum Genet. 99:770–776. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Namburi P, Ratnapriya R, Khateb S, Lazar
CH, Kinarty Y, Obolensky A, Erdinest I, Marks-Ohana D, Pras E,
Ben-Yosef T, et al: Bi-allelic truncating mutations in CEP78,
encoding centrosomal protein 78, cause cone-rod degeneration with
sensorineural hearing loss. Am J Hum Genet. 99:777–784. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Franzè A, Esposito G, Di Domenico C, Iossa
S, Sauchelli G, Fioretti T, Cavaliere M, Auletta G, Corvino V,
Laria C, et al: SLC26A4 genotypes associated with enlarged
vestibular aqueduct malformation in south Italian children with
sensorineural hearing loss. Clin Chem Lab Med. 54:e259–e263.
2016.PubMed/NCBI
|
|
5
|
Bakhchane A, Bousfiha A, Charoute H,
Salime S, Detsouli M, Snoussi K, Nadifi S, Kabine M, Rouba H, Dehbi
H, et al: Update of the spectrum of GJB2 gene mutations in 152
Moroccan families with autosomal recessive nonsyndromic hearing
loss. Eur J Med Genet. 59:325–329. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim SJ, Lee S, Park HJ, Kang TH, Sagong B,
Baek JI, Oh SK, Choi JY, Lee KY and Kim UK: Genetic association of
MYH genes with hereditary hearing loss in Korea. Gene. 591:177–182.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guan J, Wang D, Cao W, Zhao Y, Du R, Yuan
H, Liu Q, Lan L, Zong L, Yang J, et al: SIX2 haploinsufficiency
causes conductive hearing loss with ptosis in humans. J Hum Genet.
61:917–922. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu GJ, Shi LS, Zhou H, Yang Y, Chen J and
Gao X: A novel compound heterozygous mutation of SLC26A4 in two
Chinese families with nonsyndromic hearing loss and enlarged
vestibular aqueducts. Mol Med Rep. 16:9011–9016. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong SH, Wang WH, Chen PH, Li SY and Yang
JJ: Functional analysis of a nonsyndromic hearing loss-associated
mutation in the transmembrane II domain of the GJC3 gene. Int J Med
Sci. 14:246–256. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xia W, Hu J, Liu F, Ma J, Sun S, Zhang J,
Jin K, Huang J, Jiang N, Wang X, et al: New role of LRP5,
associated with nonsyndromic autosomal-recessive hereditary hearing
loss. Hum Mutat. 38:1421–1431. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee KY, Choi SY, Bae JW, Kim S, Chung KW,
Drayna D, Kim UK and Lee SH: Molecular analysis of the GJB2, GJB6
and SLC26A4 genes in Korean deafness patients. Int J Pediatr
Otorhinolaryngol. 72:1301–1309. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou XL, He LX, Yu LJ, Wang Y, Wang XJ,
Wang ED and Yang T: Mutations in KARS cause early-onset hearing
loss and leukoencephalopathy: Potential pathogenic mechanism. Hum
Mutat. 38:1740–1750. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cai XZ, Li Y, Xia L, Peng Y, He CF, Jiang
L, Feng Y, Xia K, Liu XZ, Mei LY and Hu ZM: Exome sequencing
identifies POU4F3 as the causative gene for a large chinese family
with non-syndromic hearing loss. J Hum Genet. 62:317–320. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Palombo F, Al-Wardy N, Ruscone GA, Oppo M,
Kindi MN, Angius A, Al Lamki K, Girotto G, Giangregorio T, Benelli
M, et al: A novel founder MYO15a frameshift duplication is the
major cause of genetic hearing loss in oman. J Hum Genet.
62:259–264. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ryu N, Lee S, Park HJ, Lee B, Kwon TJ, Bok
J, Park CI, Lee KY, Baek JI and Kim UK: Identification of a novel
splicing mutation within SLC17A8 in a Korean family with hearing
loss by whole-exome sequencing. Gene. 627:233–238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li D, Sun J, Zhao L, Guo W, Sun W and Yang
S: Aminoglycoside increases permeability of osseous spiral laminae
of cochlea by interrupting MMP-2 and MMP-9 balance. Neurotox Res.
31:348–357. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sagwa EL, Souverein PC, Ribeiro I,
Leufkens HG and Mantel-Teeuwisse AK: Differences in
VigiBase® reporting of aminoglycoside and
capreomycin-suspected ototoxicity during tuberculosis treatment.
Pharmacoepidemiol Drug Saf. 26:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding Y, Leng J, Fan F, Xia B and Xu P: The
role of mitochondrial DNA mutations in hearing loss. Biochem Genet.
51:588–602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Z, Li R, Chen J, Liao Z, Zhu Y, Qian Y,
Xiong S, Heman-Ackah S, Wu J, Choo DI and Guan MX: Mutational
analysis of the mitochondrial 12S rRNA gene in Chinese pediatric
subjects with aminoglycoside-induced and non-syndromic hearing
loss. Hum Genet. 117:9–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng J, Ji Y and Guan MX: Mitochondrial
tRNA mutations associated with deafness. Mitochondrion. 12:406–413.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guan MX, Fischel-Ghodsian N and Attardi G:
A biochemical basis for the inherited susceptibility to
aminoglycoside ototoxicity. Hum Mol Genet. 9:1787–1793. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guan MX, Fischel-Ghodsian N and Attardi G:
Biochemical evidence for nuclear gene involvement in phenotype of
non-syndromic deafness associated with mitochondrial 12S rRNA
mutation. Hum Mol Genet. 5:963–971. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu J, Qian Y, Li Z, Yang A, Zhu Y, Li R,
Yang L, Tang X, Chen B, Ding Y, et al: Mitochondrial haplotypes may
modulate the phenotypic manifestation of the deafness-associated
12S rRNA 1555A>G mutation. Mitochondrion. 10:69–81. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rieder MJ, Taylor SL, Tobe VO and
Nickerson DA: Automating the identification of DNA variations using
quality-based fluorescence re-sequencing: Analysis of the human
mitochondrial genome. Nucleic Acids Res. 26:967–973. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Andrews RM, Kubacka I, Chinnery PF,
Lightowlers RN, Turnbull DM and Howell N: Reanalysis and revision
of the cambridge reference sequence for human mitochondrial DNA.
Nat Genet. 23:1471999. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ding Y, Xia BH, Liu Q, Li MY, Huang SX and
Zhuo GC: Allele-specific PCR for detecting the deafness-associated
mitochondrial 12S rRNA mutations. Gene. 591:148–152. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guan MX, Yan Q, Li X, Bykhovskaya Y,
Gallo-Teran J, Hajek P, Umeda N, Zhao H, Garrido G, Mengesha, et
al: Mutation in TRMU related to transfer RNA modification modulates
the phenotypic expression of the deafness-associated mitochondrial
12S ribosomal RNA mutations. Am J Hum Genet. 79:291–302. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schmittgen TD, Zakrajsek BA, Mills AG,
Gorn V, Singer MJ and Reed MW: Quantitative reverse
transcription-polymerase chain reaction to study mRNA decay:
Comparison of endpoint and real-time methods. Anal Biochem.
285:194–204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bibb MJ, Van Etten RA, Wright CT, Walberg
MW and Clayton DA: Sequence and gene organization of mouse
mitochondrial DNA. Cell. 26:167–180. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gadaleta G, Pepe G, De Candia G,
Quagliariello C, Sbisà E and Saccone C: The complete nucleotide
sequence of the Rattus norvegicus mitochondrial genome: Cryptic
signals revealed by comparative analysis between vertebrates. J Mol
Evol. 28:497–516. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roe BA, Ma DP, Wilson R and Wong JF: The
complete nucleotide sequence of the xenopus laevis mitochondrial
genome. J Biol Chem. 260:9759–9774. 1985.PubMed/NCBI
|
|
32
|
Dai ZY, Sun BC, Huang SS, Yuan YY, Zhu YH,
Su Y and Dai P: Correlation analysis of phenotype and genotype of
GJB2 in patients with non-syndromic hearing loss in china. Gene.
570:272–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kong QP, Bandelt HJ, Sun C, Yao YG, Salas
A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, et al: Updating the
East Asian mtDNA phylogeny: A prerequisite for the identification
of pathogenic mutations. Hum Mol Genet. 15:2076–2086. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Prezant TR, Agapian JV, Bohlman MC, Bu X,
Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L and Rotter JI:
Mitochondrial ribosomal RNA mutation associated with both
antibiotic-induced and non-syndromic deafness. Nat Genet.
4:289–294. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
el-Schahawi M, de Munain López A, Sarrazin
AM, Shanske AL, Basirico M, Shanske S and DiMauro S: Two large
Spanish pedigrees with nonsyndromic sensorineural deafness and the
mtDNA mutation at nt 1555 in the 12s rRNA gene: Evidence of
heteroplasmy. Neurology. 48:453–456. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan EF, Xia W, Huang JT, Hu L, Liao X,
Dai X and Liu SM: A sensitive and convenient method for clinical
detection of non-syndromic hearing loss-associated common
mutations. Gene. 628:322–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu J, Li Z, Zhu Y, Yang A, Li R, Zheng J,
Cai Q, Peng G, Zheng W, Tang X, et al: Mitochondrial 12S rRNA
variants in 1642 han chinese pediatric subjects with
aminoglycoside-induced and nonsyndromic hearing loss.
Mitochondrion. 10:380–390. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Suzuki T and Nagao A: Human mitochondrial
tRNAs: Biogenesis, function, structural aspects, and diseases. Annu
Rev Genet. 45:299–329. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pütz J, Giegé R and Florentz C: Diversity
and similarity in the tRNA world: Overall view and case study on
malaria-related tRNAs. FEBS Lett. 584:350–358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tanaka M, Ino H, Ohno K, Hattori K, Sato
W, Ozawa T, Tanaka T and Itoyama S: Mitochondrial mutation in fatal
infantile cardiomyopathy. Lancet. 336:14521990. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lechowicz U, Pollak A, Frączak A,
Rydzanicz M, Stawiński P, Lorens A, Skarżyński PH, Skarżyński H,
Płoski R and Ołdak M: Application of next-generation sequencing to
identify mitochondrial mutations: Study on m.7511T>C in patients
with hearing loss. Mol Med Rep. 17:1782–1790. 2018.PubMed/NCBI
|
|
42
|
Zhu HY, Wang SW, Liu L, Chen R, Wang L,
Gong XL and Zhang ML: Genetic variants in mitochondrial tRNA genes
are associated with essential hypertension in a chinese Han
population. Clin Chim Acta. 410:64–69. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Degoul F, Brule H, Cepanec C, Helm M,
Marsac C, Leroux J, Giegé R and Florentz C: Isoleucylation
properties of native human mitochondrial tRNAIle and tRNAIle
transcripts. Implications for cardiomyopathy-related point
mutations (4269, 4317) in the tRNAIle gene. Hum Mol Genet.
7:347–354. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ding Y, Zhuo G and Zhang C: The
mitochondrial tRNALeu(UUR) A3302G mutation may be associated with
insulin resistance in woman with polycystic ovary syndrome. Reprod
Sci. 23:228–233. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jeng JY, Yeh TS, Lee JW, Lin SH, Fong TH
and Hsieh RH: Maintenance of mitochondrial DNA copy number and
expression are essential for preservation of mitochondrial function
and cell growth. J Cell Biochem. 103:347–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shibata SB, Ranum PT, Moteki H, Pan B,
Goodwin AT, Goodman SS, Abbas PJ, Holt JR and Smith RJH: RNA
interference prevents autosomal-dominant hearing loss. Am J Hum
Genet. 98:1101–1113. 2016. View Article : Google Scholar : PubMed/NCBI
|