Introduction
Head and neck squamous cell carcinoma (HNSCC) is a
leading cause of cancer mortality worldwide, with an estimated
600,000 new cases diagnosed per year (1,2). The
primary risk factors for HNSCC are tobacco and alcohol consumption
and infection of the oropharynx with human papilloma virus
(3–5). Standard-of-care treatment for HNSCC
includes surgery, radiation, and chemotherapy, often involving a
combination of these approaches. In addition, cetuximab, an
antibody targeting the epidermal growth factor receptor (EGFR), and
the checkpoint inhibitors nivolumab and pembrolizumab have been
approved for treatment of HNSCC (6–10).
Despite the availability of these agents and approaches, HNSCC
patients that receive therapy intended to be curative develop
second primary tumors (SPTs) at an alarmingly high rate of 3–6% per
year (11–15). The development of SPTs is a major
cause of death and is attributed to the ‘condemned’ nature of the
mucosa, or epithelial field cancerization, resulting from chronic
exposure to carcinogens (16).
Efforts to develop a chemoprevention strategy to
prevent the development of SPTs in HNSCC have focused on evaluation
of retinoids, EGFR inhibitors, and nonsteroidal anti-inflammatory
drugs (NSAIDs). In clinical testing, high-dose isotretinoin, a
vitamin A analogue, demonstrated chemopreventive activity against
HNSCC SPTs, but was poorly tolerated, while low-dose isotretinoin
proved ineffective at preventing SPTs (17–19).
Erlotinib, an EGFR inhibitor, has demonstrated chemopreventive
activity in a preclinical model of carcinogen-induced HNSCC, but
clinical application was hindered by issues of effectiveness and
tolerability (20).
Epidemiological evidence from the National Cancer Institute's
Prostate, Lung, Colorectal, and Ovarian randomized screening trial,
and other studies, has suggested a chemopreventive effect of NSAIDs
for HNSCC (21–27). More recently, a retrospective
analysis of 266 HNSCC patients found a dramatic survival benefit
associated with regular use of NSAIDs (28). This benefit was limited to patients
with genetic alterations in PIK3CA, the gene encoding
phosphatidylinositol (3)-kinase a,
as patients with wild-type PIK3CA did not display a survival
benefit with regular NSAID use (28).
An alternative strategy for chemoprevention in HNSCC
involves the use of naturally-occurring vegetable-derived
compounds. Compelling epidemiological evidence shows that diets
rich in cruciferous vegetables are linked to reduced risk for
developing HNSCC and, more specifically, SPTs (29–33).
Cruciferous vegetables contain high levels of glucoraphinin, which
is metabolized upon consumption to sulforaphane (34). Sulforaphane readily disables the
negative regulatory protein kelch-like ECH-associated protein 1,
resulting in liberation of the transcription factor nuclear factor
erythroid 2-related factor 2 (NRF2) from destruction by the
proteasome (34–36). This results in elevation of NRF2
protein levels and induction of a large number of NRF2 target
genes, many of which act to promote detoxication of cells from
environmental carcinogens (34).
Known NRF2 target genes include NAD(P)H quinone oxidoreductase 1
(NQO1), glutamate-cysteine ligase catalytic subunit (GCLC),
glutathione S-transferases, and aldo-keto reductases. In
preclinical models, treatment with sulforaphane has been shown to
prevent carcinogen-induced cancers of breast, skin, and stomach
(37–40). We previously reported that
sulforaphane prevented the development of HNSCC tumors in mice
exposed to the chemical carcinogen 4-nitroquinoline-1-oxide
(41). Importantly, consumption of
vegetable extracts rich in glucoraphinin or sulforaphane has been
shown to promote detoxication from common airborne pollutants in
healthy human volunteers (42–44).
Further development of sulforaphane as a chemopreventive strategy
against HNSCC SPTs in humans requires identification of robust
biomarkers of sulforaphane activity in normal and malignant
epithelium of the oral cavity and upper aerodigestive tract. RNA
and protein profiling following sulforaphane treatment has been
performed in a variety of murine and human cancer models and has
identified a broad number of pharmacodynamic markers of
sulforaphane activity, including genes involved in xenobiotic
metabolism and response to oxidative stress (45–50).
However, biomarkers of sulforaphane pharmacodynamic activity in
HNSCC cells, as well as normal epithelial cells derived from the
head and neck region, has not been investigated.
An alternative or parallel mechanism whereby
sulforaphane exerts chemopreventive activity may involve modulation
of anti-tumor immunity. Administration of sulforaphane has been
shown to enhance the activities of natural killer (NK) cells with
associated anti-tumor effects in murine models of melanoma,
prostate cancer, and leukemia (51–53).
Further, sulforaphane modestly induced expression of the NK cell
activating ligands MICA/MICB, members of the natural killer group
2D (NKG2D) ligand family, following treatment of A549 lung cancer
cells and MDA-MB-231 breast cancer cells (54). The impact of sulforaphane on
expression of NK cell activating ligands in HNSCC cells is
unknown.
In the present study we performed RNA and protein
profiling following sulforaphane treatment of HNSCC cell lines, as
well as a normal mucosal epithelial cell line, to identify robust
biomarkers of sulforaphane pharmacodynamic activity. We identified
the HMOX1 and HSPA1A genes as highly upregulated and reliable
biomarkers of sulforaphane activity. In addition, while
sulforaphane treatment led to modest NRF2-dependent upregulation of
MICA/MICB in HNSCC cells, enhanced sensitization to NK
cell-mediated killing following sulforaphane treatment was not
broadly observed in a panel of HNSCC cell line models.
Materials and methods
Cell lines and chemicals
Cal 27 (ATCC® CRL-2095), FaDu
(ATCC®, HTB-43), Het-1A (ATCC®, CRL-2692) and
NK-92 (ATCC® CRL-2407) cells were purchased from the
American Type Culture Collection (ATCC). PE/CA-PJ34 (clone C12)
(ECACC, 97062513) was purchased from Sigma-Aldrich; Merck KGaA.
HSC-2, HSC-3, and HSC-4 were obtained from the Health Science
Research Resources Bank (Osaka, Japan). All HNSCC cell lines were
cultured in DMEM, 10% FBS and 1% penicillin-streptomycin. NK-92
cells were cultured in Alpha Minimum Essential Medium without
ribonucleosides and deoxyribonucleosides, but containing 2 mM
L-glutamine, 1.5 g/l sodium bicarbonate, 0.2 mM inositol, 0.1 mM
2-mercaptoethanol, 0.02 mM folic acid, 12.5% horse serum, 12.5% FBS
and 100 UI/ml IL-2. Cell lines were authenticated every 6 months
during the course of experiments via short-tandem repeat testing
(UC Berkeley DNA Sequencing Facility). Mycoplasma testing was also
performed during the course of this study. NK-92 was free of
mycoplasma, but other HNSCC cell lines were all mycoplasma
positive. Cell lines were passaged for a period of 3 months (~24
passages) after thawing from liquid nitrogen.
R,S-sulforphane was purchased from LKT
Laboratiories, Inc. Recombinant human IL-2 was obtained from
PeproTech. IL-2 was reconstituted in 100 mM acetic acid and diluted
in PBS containing 0.1% BSA.
Treatment of cells
HNSCC cells or Het-1A cells were plated in 6 cm
dishes (2 million/dish) 24 h prior to treatment. The cells were
then treated with either vehicle (0.1% DMSO) or 10 mM sulforaphane
for 8 or 16 h for quantitative PCR or immunoblotting experiments,
respectively.
Reverse-transcription quantitative PCR
(RT-qPCR)
Total RNAs from cultured cells and murine tissues
were purified using miRNeasy® Mini Kit (Qiagen). cDNAs
were synthesized using Superscript III First-Strand cDNA Synthesis
System (Life Technologies; Thermo Fisher Scientific, Inc.)]
according to the manufacturer's instructions. PCR reactions were
performed using SYBR Green PCR Master Mix (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and a Bio-Rad CFX96 C1000 Touch™
Thermal Cycler. Quantification was performed using the
2−∆∆Cq method (55).
Gene-specific primers for NQO1, GCLC, and GAPDH were as previously
described (41). Primers for
AKR1C2, AKR1C18, AKR1C19, HMOX1 and HSPA1A were from Qiagen.
Relative mRNA levels were standardized to the mRNA levels of GAPDH
gene.
Probing the human oxidative stress
plus PCR array
RNAs from cultured cells were purified as described
above and cDNAs were synthesized by the RT2 First Strand
kit (Qiagen) and used to probe the Human Oxidative Stress Plus RT2
profiler PCR Array (Qiagen, cat. #330231), according to the
manufacturer's recommendations. Gene expression data were analyzed
using the Web-Based PCR Array Data Analysis from SABiociences.
Treatment of mice
C57BL/6 mice (5–6 weeks; 5 mice/group) were treated
by vehicle (PBS) or sulforaphane (6 µmol/mouse) via oral gavage for
6 or 18 h. Following treatment, mice were sacrificed and tissues
harvested.
Immunoblotting
Cells were washed with ice-cold PBS twice and then
lysed with RIPA lysis buffer (150 mM Tris, pH 7.4, 100 mM NaF, 120
mM NaCl, 100 µM sodium orthovannadate, and 1X protease inhibitor
cocktail and phosphatase inhibitor cocktail; Roche Diagnostics).
Lysates (20 µg) were resolved by SDS-PAGE, transferred to PVDF
Membranes (Bio-Rad, #1620177), and incubated with primary
antibodies at 4°C overnight, followed by incubation with horse
radish peroxidase-conjugated secondary antibodies (Bio-Rad,
#170-6516)] for 1 h at room temperature. Immunoreactive bands were
visualized by chemiluminescence (Santa Cruz Biotechnology, #SC2048
or Thermo Fisher Scientific, #1856194). Antibodies against NRF2
(#12721), β-tubulin (#2146), and GAPDH (#5174) were from Cell
Signaling Technology. Antibodies against HMOX1 (#A11919) and HSPA1A
(#A12948) were from ABclonal. Anti-MICA/B (#SC-2093) was from Santa
Cruz Biotechnology.
Crystal violet assays
Cells were seeded in 96-well plates and incubated
overnight. The following day, cells were treated with different
concentrations of sulforaphane for 48 h, then stained with crystal
violet for 30 min. Crystal violet solution was removed from the
wells and the plates were washed under tap water before being dried
for 24 h. Crystal violet-stained material was dissolved with 100 mM
sodium citrate solution and subsequently quantified using a
colorimetric plate reader at OD590.
Cytotoxicity assays
NK-92 cell-mediated cytotoxicity was assessed using
the CytoTox 96® Non-Radioactive Cytoxicity Assay
(Promega Corporation, #G1780), according to the manufacturer's
protocol. HNSCC cells were pre-treated with vehicle (0.1% DMSO) or
sulforaphane for 48 h, then washed with medium twice. NK-92 cells
and pre-treated HSNCC cells were counted and plated in round-bottom
96-well plates at ratios of 2.5:1, 5:1, and 10:1. Wells containing
NK-92 cells alone or HNSCC cells alone served as controls for
spontaneous LDH release of effector cells and target cells,
respectively. To assess the target cell maximum LDH release, lysis
buffer was added for one hour to wells containing HNSCC cells
alone, followed by harvesting of the supernatant. Prior to
supernatant harvest, 96-well plates were centrifuged at 250 × g for
5 min then kept in a 37°C incubator for 5 h. Plates were then
centrifuged again at 250 × g for 5 min and 50 µl of supernatant
from each well was transferred into a new 96-well plate. 50 µl/well
of reconstituted substrate mix was then added to the wells. Plates
were subsequently incubated at room temperature in the dark for 20
to 30 min, followed by addition of 50 µl stop solution to each well
and reading of absorbance at 490 nm. The cytotoxicity mediated by
NK-92 cells was calculated as follows: %
cytotoxicity=(ELR-ESR-TSR-MB)/(TMR-TSR-LBB), where ELR,
experimental LDH release; ESR, effector spontaneous release; TSR,
target spontaneous release; MB, medium background; TMR, target
spontaneous release; and LBB, lysis buffer background.
RNA interference
Cells in 6-cm dishes were transfected with 25 pmol
of siRNA oligonucleotides mixed with Lipofectamine RNAiMAX (Thermo
Fisher Scientific, Inc., #13778500). NRF2 siRNA oligonucleotides
and non-target siRNA (siNT) were obtained from Sigma-Aldrich; Merck
KGaA. The target sequence for NRF2 siRNAs was:
5′-UGACAGAAGUUGACAAUUA-3′.
Statistical analysis
Statistical differences between sulforaphane and
vehicle treatment groups were determined using ANOVA followed by
adjustment for multiple comparison by Bonferroni's method. Error
bars for all figures represent SD.
Results
Identification of oxidative
stress-related genes induced by sulforaphane in HNSCC cells
We first sought to identify potential biomarkers of
sulforaphane activity in HNSCC cells, as well as normal mucosal
epithelial cells. Two HNSCC cell lines, PE/CA-PJ34 and FaDu, and a
putative normal, non-tumorigenic mucosal epithelial cell line,
Het-1A (56), were treated for 8 h
with vehicle or 10 µM sulforaphane, followed by preparation of
cellular RNAs. The RNAs were converted to cDNAs, then used to probe
Human Oxidative Stress Plus PCR Arrays. This array enables
expression profiling of 84 different human genes related to
oxidative stress. Comparison of RNA expression levels in
sulforaphane-treated vs. vehicle-treated cells allowed
identification of genes whose expression was induced greater than
2-fold by sulforaphane treatment. In both HNSCC cell lines we
observed >2-fold induction of 11 genes (Fig. 1A and B). HMOX1, encoding heme
oxygenase 1, and HSPA1A, encoding a member of the heat shock
protein 70 family, were the most strongly induced genes in both
HNSCC cell lines, with induction levels ranging from roughly 10- to
20-fold. In the normal epithelial cell line, Het-1A, 9 genes were
found to be induced >2-fold by sulforaphane treatment, with
HMOX1 again being the most potently upregulated (~18-fold; Fig. 1C). HSPA1A was the third most
potently induced gene in Het-1A (~10-fold).
We next sought to validate the findings we obtained
with the Oxidative Stress Array by performing RT-qPCR analyses.
PE/CA-PJ34, FaDu, and Het-1A cells were again treated with vehicle
or 10 µM sulforaphane for 8 h and RNAs were prepared. RT-qPCR was
then performed for 4 genes (HMOX1, HSPA1A, AKR1C2, GCLC) that were
found to be upregulated in the Array studies. In addition, RT-qPCR
was used to assess expression of NQO1, a known downstream target of
sulforaphane (45). As shown in
Fig. 2A-C, HMOX1 and HSPA1A were
strongly upregulated by sulforaphane in all three cell lines, with
induction levels ranging from roughly 8-fold to 27-fold. Similar
upregulation of HMOX1 and HSPA1A was observed in three additional
HNSCC cell lines (Cal 27, HSC-2, HSC-3; Fig. S1). In the HNSCC cell lines, HMOX1
and HSPA1A were the most potently induced genes, whereas AKR1C2
(~29-fold induction) was the most upregulated in Het-1A.
Surprisingly, NQO1 was only weakly upregulated by sulforaphane.
Collectively, these experiments suggest that RNAs
for oxidative stress genes, particularly HMOX1 and HSPA1A, may
represent valuable biomarkers of sulforaphane activity in HNSCC and
normal epithelium of the upper aerodigestive tract.
Sulforaphane induction of oxidative
stress proteins in HNSCC cells
We next confirmed that sulforaphane induction of
mRNAs for oxidative stress genes was accompanied by upregulation of
the corresponding oxidative stress proteins. Five HNSCC cells lines
(PE/CA-PJ34, FaDu, Cal 27, HSC-2, HSC-3) and Het-1A cells were
treated with vehicle or sulforaphane (10 mM) for 16 h, followed by
immunoblot detection of HMOX1, HSPA1A, or the control protein GAPDH
(Fig. 3). As shown, sulforaphane
treatment led to upregulation of HMOX1 and HSPA1A in all cell lines
examined (Fig. 3), although the
fold induction was less than was observed with mRNA induction for
these proteins (Fig. 2). It should
be noted that while we observed sulforaphane induction of HMOX1 in
all cell lines, we did not detect nuclear translocation of the
protein (data not shown).
Sulforaphane induction of oxidative
stress genes in vivo
We next determined whether the oxidative stress
genes induced by sulforaphane in HNSCC cells and Het-1A cells are
induced in vivo in wild-type C57BL/6 mice. Mice were treated
by oral gavage (5 per group) with a single dose of vehicle (6 or 18
h) or a single dose (6 µmol) of sulforaphane (6 or 18 h). The dose
of 6 µmol/mouse was chosen, as we have previously shown that this
dose is well tolerated and prevents the development of
carcinogen-induced HNSCC tumors in mice (41). Following treatment, mice were
sacrificed and RNAs purified from liver or peripheral blood
mononuclear cells (PBMCs) for analysis by RT-qPCR (Fig. 4A and B). Since the AKR1C2 gene is
human-specific, we instead analyzed the related murine genes
AKR1C18 and AKR1C19. In liver, sulforphane treatment, primarily at
the 18-h time-point, led to >2-fold upregulation of all 5 genes
analyzed (HMOX1, HSPA1A, AKRC18, AKRC19, GCLC). HSPA1A was unique
in being induced only at the 6-h treatment time-point. In PBMCs,
only HMOX1, AKRC19, and GCLC were upregulated >2-fold, and only
at the 18-h time-point. Immunoblotting of protein lysates from
liver tissue revealed elevated expression HMOX1 protein in 3 of 5
mice treated with sulforaphane (18 h; Fig. 4C), validating upregulation at the
protein level.
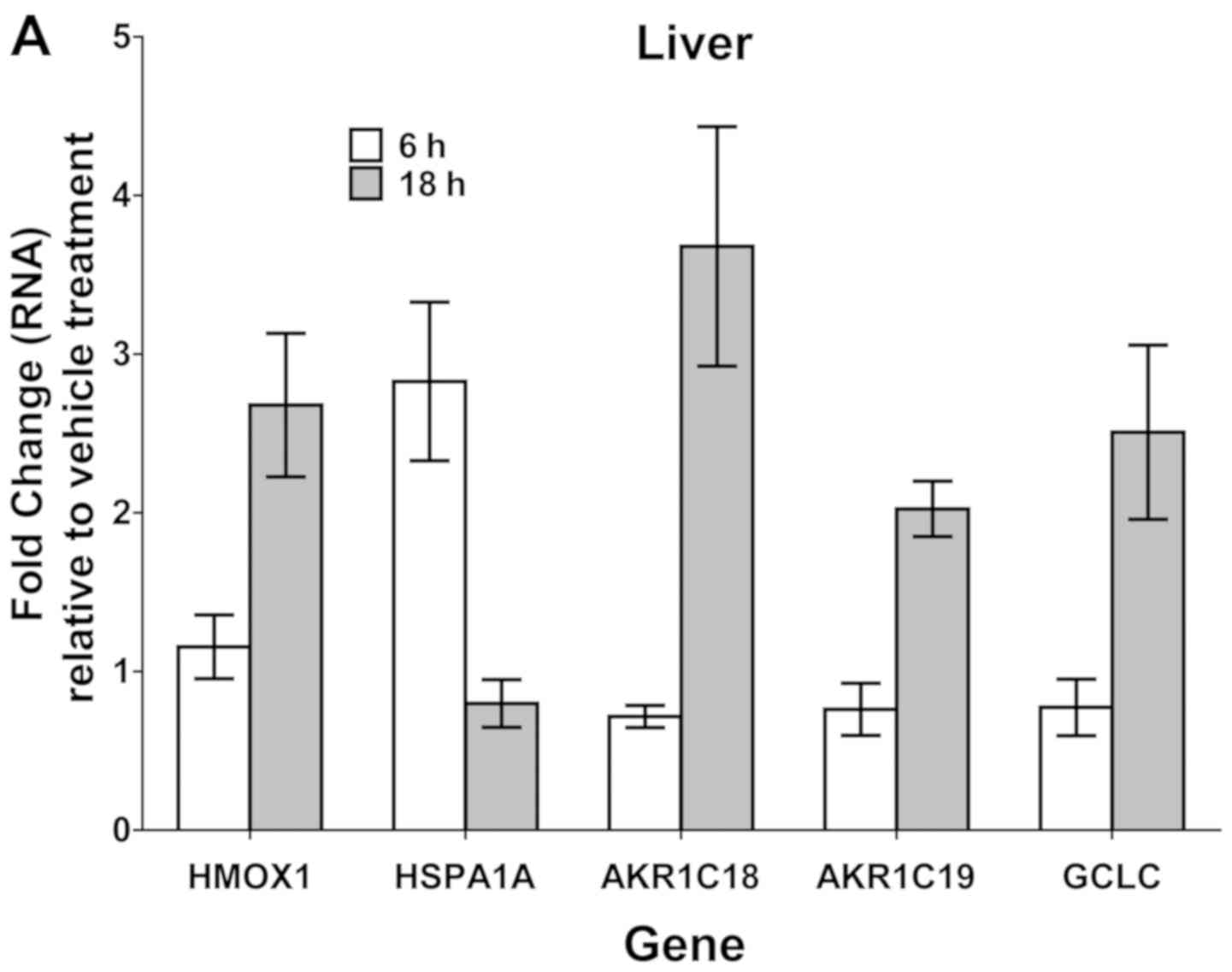 | Figure 4.SF induction of oxidative
stress-associated genes in vivo. Wild-type C57BL/6 mice (n=5
per treatment group) were treated via oral gavage with single doses
of vehicle (PBS) or SF (6 µmol/mouse). At 6 or 18 h after
treatment, mice were sacrificed and tissues harvested. (A) RT-qPCR
analysis of liver gene expression (data from five mice in each
treatment group were combined), demonstrating gene induction by SF
treatment relative to vehicle treatment. (B) RT-qPCR analysis of
RNA expression in PBMCs. Error bars in (A) and (B) represent the
standard deviation of the combined five specimens. The experiment
was performed twice with similar results. (C) Immunoblot analysis
of HMOX1 protein levels in liver from mice treated with vehicle (18
h) or SF (18 h). Numbers below the HMOX1 blot indicate the ratio of
HMOX1 to β-tubulin, as determined by densitometry. The experiment
was performed twice with similar results. AKR1C, aldo-keto
reductase family 1 member C; GCLC, glutamate-cysteine ligase
catalytic subunit; HMOX1, heme oxygenase 1; HSPA1A, heat shock
protein family A (Hsp70) member 1A; PBMC, peripheral blood
mononuclear cell; RT-qPCR, reverse transcription-quantitative PCR;
SF, sulforaphane. |
Impact of sulforaphane on NKG2D and
DNAM-1 ligands in HNSCC cells
We next examined the impact of sulforaphane on
expression of genes encoding members of the NKG2D ligand family
(MICA, MICB, ULBP1-6), as well as the DNAM-1 ligands CD112 and
CD155. Both NKG2D ligands and DNAM-1 ligands stimulate NK cell
cytotoxicity. Treatment of HNSCC cells with sulforaphane resulted
in a modest upregulation of RNA for MICA, with little apparent
effect on other members of the NKG2D ligand family, as assessed by
RT-qPCR (Fig. 5A). Immunoblotting
with an antibody that cross-reacts with both MICA and MICB was also
performed (Fig. 5B). Consistent
with findings at the RNA level, sulforaphane treatment of the HNSCC
cell lines PE/CA-PJ34 and Cal 27 also induced upregulation of
MICA/B protein. Modest induction of RNAs for CD112 and CD155 was
seen after 12 and 48 h of sulforaphane treatment, but not at the
24-h time-point (Fig. 5A).
 | Figure 5.SF modulation of NKG2D and DNAM-1
ligands in HNSCC cells. (A) HNSCC cell line Cal 27 was treated with
vehicle (48 h) or 5 µM SF for 12, 24 or 48 h, followed by
performance of RT-qPCR for RNAs encoding NKG2D ligands (MICA, MICB
and ULBP1-6) and DNAM-1 ligands (CD112 and CD155). Error bars
represent the standard deviation of triplicate assays. The
experiment was performed three times with similar results. (B)
PE/CAPJ 34 or Cal 27 cells were treated with vehicle or 5 µM SF for
the indicated times, followed by immunoblotting for NRF2 or MICA/B.
Densitometric values for NRF2 or MICA/B compared to GAPDH and
normalized to the vehicle group are indicated below each respective
band. (C) Cal 27 cells were treated for 24 h with siNT or siNRF2.
The cells were then treated for an additional 48 h with vehicle or
5 µM SF, followed by performance of RT-qPCR (left panel) or
immunoblotting (right panel). Numbers indicate the ratio of
NRF2/GAPDH or (MICA/B)/GAPDH normalized to siNT/Veh. The experiment
in (B) and (C) was performed three times with similar results.
HNSCC, head and neck squamous cell carcinoma; NRF2, nuclear factor
erythroid 2-related factor 2; RT-qPCR, reverse
transcription-quantitative PCR; SF, sulforaphane; siNT,
non-targeting small interfering RNA; siNRF2, small interfering RNA
targeting NRF2 mRNA; VEH, vehicle. |
Role of NRF2 in sulforaphane induction
of MICA/B
To determine whether sulforaphane induction of
MICA/B was dependent on NRF2 transcription factor, we utilized
siRNA directed against NRF2 mRNA to prevent upregulation of NRF2
protein following sulforaphane treatment (Fig. 5C). As shown, in cells treated with
a non-targeting siRNA (siNT), sulforaphane treatment resulted in
upregulation of NRF2 and MICA/B. Treatment with NRF2 siRNA (siNRF2)
markedly reduced NRF2 RNA levels (Fig.
5C, left panel) and prevented sulforaphane induction of NRF2
protein (right panel). Importantly, siNRF2 treatment also blocked
sulforaphane upregulation of MICA/B, indicating that sulforaphane
effects on MICA/B expression are dependent on NRF2.
Impact of sulforaphane on sensitivity
of HNSCC cells to NK cell-mediated cytotoxicity
The ability of sulforaphane to upregulate MICA/B in
HNSCC suggested that sulforaphane treatment may sensitize HNSCC
cells to NK cell-mediated cytotoxicity. To test this, we first
needed to identify a concentration of sulforaphane that would be
only minimally toxic when used alone. Dose-response analyses were
performed (Fig. 6) with PE/CA-PJ34
(IC25=9.1 µM) and Cal 27 (IC25=7.1 µM), and a
sulforaphane concentration below the IC25's, 5 µM, was
chosen for subsequent experiments. We then pre-treated 6 different
HNSCC cell lines for 48 h with vehicle or 5 µM sulforaphane before
co-culturing for 5 h with the NK cell line NK-92 at different
target to effector ratios. LDH release cytotoxicity assays were
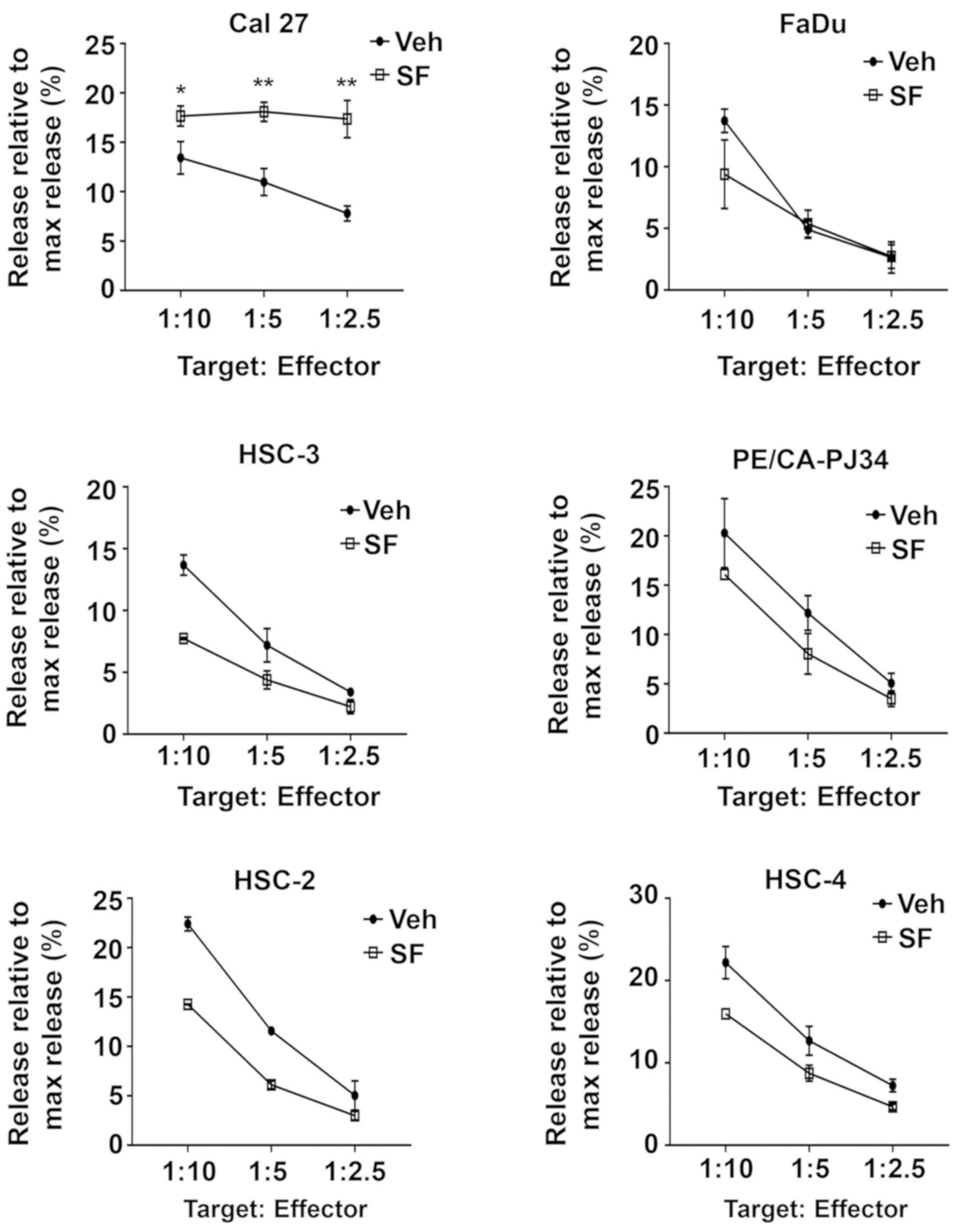
then performed. As Fig. 7
illustrates, sulforaphane treatment resulted in statistically
significant sensitization to NK-92-mediated cytotoxicity in only
one of the six HNSCC cell lines, Cal 27 cells. These findings raise
questions whether sensitization to NK-mediated cytotoxicity
represents a general mechanism contributing to the chemopreventive
activity of sulforaphane against HNSCC.
Discussion
Patients who receive curative-intent therapy for
HNSCC await an uncertain future. SPTs arise at the extraordinarily
high rate of 3–6% per year within this population. There would be
tremendous value in delivering to these patients a chemopreventive
agent that could delay or prevent the development of SPTs. Since
long-term, perhaps chronic, administration would be necessary, such
a chemopreventive agent should be well tolerated and, ideally,
inexpensive. Naturally-occurring compounds derived from vegetables
have promising potential to meet these criteria. Sulforaphane, in
particular, has demonstrated chemopreventive activity against
carcinogen-induced HNSCC in a murine preclinical model (41). Moreover, clinical studies of
broccoli sprout extracts that are rich in glucoraphanin and/or
sulforaphane have shown that they are well tolerated and
demonstrate good bioavailability in healthy human volunteers
(42,43,57).
Consumption of these extracts promoted rapid and sustained
elimination of the common airborne pollutants benzene and acrolein
(44). Hence, there is a strong
basis for evaluating the chemopreventive activity of sulforaphane
in patients who have received curative-intent treatment for HNSCC.
These investigations will require demonstration that the
administered sulforaphane exhibits pharmacodynamic activity in the
target tissue of interest. Although gene targets of sulforaphane
activity have been identified in some normal tissues, as well as
colon, prostate, and breast cancer cell lines, little is known
about the effects of sulforaphane on gene expression in HNSCC or
normal mucosal epithelium. In the current study we identified
biomarkers of sulforaphane activity in HNSCC cells as well as a
normal mucosal epithelial cell line (Het-1A) derived from the upper
aerodigestive tract. Of particular note, we observed robust
sulforaphane induction of HMOX1 and HSPA1A, and suggest their use
as biomarkers of sulforaphane pharmacodynamic activity in future
studies evaluating sulforaphane chemoprevention in HNSCC.
In previous studies we have investigated
sulforaphane pharmacodynamic activity in oral epithelium of healthy
volunteers following consumption of glucoraphanin-rich or
sulforaphane-rich beverages derived from broccoli sprout extracts
(41). Based on RT-qPCR analysis
of buccal cell specimens, >2-fold induction of the target gene
NQO1 was observed in 6 of 9 evaluable participants following
ingestion of glucoraphanin-rich beverage and in 3 of 9 participants
following ingestion of sulforaphane-rich beverage. In most
participants where induction of NQO1 was observed, the level of
induction was only modest, raising concerns about the value of NQO1
as a strong biomarker of sulforaphane activity. Consistent with
this, in our current studies NQO1 was only weakly upregulated by
sulforaphane treatment in the HNSCC cell lines and Het-1A cells we
examined. By contrast, we observed 10- to 20-fold induction of RNAs
for HMOX1 and HSPA1A in both HNSCC cell lines and Het-1A. A lesser,
albeit significant, induction of HMOX1 and HSPA1A, as well as
AKR1C18 was seen in liver tissue in vivo. In future studies
it will be interesting to evaluate sulforaphane target gene
expression in normal oral epithelium from sulforaphane-treated
mice.
Enhanced transcription of genes encoding enzymes
that promote detoxication from carcinogens likely plays a primary
role in the chemopreventive activity of sulforaphane. However,
accumulating evidence suggests that sulforaphane also may impact
immune cells, particularly NK cells, to influence anti-tumor
immunity (51–53). A potential mechanism has been
proposed wherein sulforaphane induces tumor cell expression of NK
cell activating ligands such as MICA/MICB (54). We observed modest
sulforaphane-induced upregulation of MICA/MICB in HNSCC cells, but
did not detect consistent induction of other members of the NKG2D
family. Similarly, we did not detect modulation of the DNAM-1
ligands CD112 and CD155. When we pre-treated a panel of 6 HNSCC
cell lines with sulforaphane, only one of the 6 lines reproducibly
exhibited enhanced sensitivity to cell lysis mediated by NK-92
cells, despite testing a variety of pre-treatment and co-incubation
conditions (data not shown). These findings suggest that direct
effects on NK cells are unlikely to play a broad role in the
chemopreventive activity of sulforaphane against HNSCC.
In summary, our studies identify HMOX1 and HSPA1A as
promising biomarkers of sulforaphane activity in HNSCC and normal
mucosal epithelial cells. Clinical evaluation of sulforaphane
chemopreventive activity against SPT development in HNSCC patients
should consider measurement of these biomarkers to assess
sulforaphane biochemical activity in the relevant target tissues,
namely the epithelial linings of the oral cavity, pharynx, and
larynx. Further clinical studies of sulforaphane in humans seems
warranted given the low cost and tolerability of this agent in
healthy volunteers, and its effectiveness as a chemoprevention
agent in preclinical models of carcinogen-induced cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by National
Institutes of Health (grant no. P50 CA097190).
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH, HL and EDL conducted experiments, analyzed data
and contributed to the writing of the manuscript. JRG and JEB
contributed to the conception of the study design, interpreted the
data, and edited the manuscript. DEJ supervised the study, and
contributed to the study design, analyzed and interpreted the data,
and wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures were performed in strict
accordance with institutional regulations and were approved by the
UCSF Institutional Animal Care and Use Committee.
Patient consent for publication
Not applicable.
Competing interests
DEJ and JRG are co-inventors of cyclic STAT3 decoy
and have financial interests in STAT3 Therapeutics. STAT3
Therapeutics holds an interest in cyclic STAT3 decoy, which is a
not a focus of the studies in this manuscript. The remaining
authors declare that they have no competing interests.
Glossary
Abbreviations
Abbreviations:
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
SPTs
|
second primary tumors
|
|
NRF2
|
nuclear factor erythroid 2-related
factor 2
|
|
NK
|
natural killer
|
|
NKG2D
|
natural killer group 2D
|
|
DNAM-1
|
DNAX accessory molecule-1
|
|
EGFR
|
epidermal growth factor receptor
|
|
NSAIDs
|
nonsteroidal anti-inflammatory
drugs
|
|
NQO1
|
NAD(P)H quinone oxidoreductase 1
|
|
GCLC
|
glutamate-cysteine ligase catalytic
subunit
|
References
|
1
|
Global Burden of Disease Cancer
Collaboration, ; Fitzmaurice C, Allen C, Barber RM, Barregard L,
Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, et al:
Global, regional, and national cancer incidence, mortality, years
of life lost, years lived with disability, and disability-adjusted
life-years for 32 cancer groups, 1990 to 2015: A systematic
analysis for the global burden of disease study. JAMA Oncol.
3:524–458. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hashibe M, Brennan P, Chuang SC, Boccia S,
Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova
E, et al: Interaction between tobacco and alcohol use and the risk
of head and neck cancer: Pooled analysis in the International Head
and Neck Cancer Epidemiology Consortium. Cancer Epidemiol
Biomarkers Prev. 18:541–550. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gillison ML, Koch WM, Capone RB, Spafford
M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, et
al: Evidence for a causal association between human papillomavirus
and a subset of head and neck cancers. J Natl Cancer Inst.
92:709–720. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gillison ML, Chaturvedi AK, Anderson WF
and Fakhry C: Epidemiology of human papillomavirus-positive head
and neck squamous cell carcinoma. J Clin Oncol. 33:3235–3242. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vermorken JB, Mesia R, Rivera F, Remenar
E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol
D, et al: Platinum-based chemotherapy plus cetuximab in head and
neck cancer. N Engl J Med. 359:1116–1127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seiwert TY, Burtness B, Mehra R, Weiss J,
Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, et
al: Safety and clinical activity of pembrolizumab for treatment of
recurrent or metastatic squamous cell carcinoma of the head and
neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial.
Lancet Oncol. 17:956–965. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chow LQ, Haddad R, Gupta S, Mahipal A,
Mehra R, Tahara M, Berger R, Eder JP, Burtness B, Lee SH, et al:
Antitumor activity of pembrolizumab in biomarker-unselected
patients with recurrent and/or metastatic head and neck squamous
cell carcinoma: Results from the phase Ib KEYNOTE-012 expansion
cohort. J Clin Oncol. 34:3838–3845. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferris RL, Blumenschein G Jr, Fayette J,
Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE,
Even C, et al: Nivolumab for recurrent squamous-cell carcinoma of
the head and neck. N Engl J Med. 375:1856–1867. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lippman SM and Hong WK: Second malignant
tumors in head and neck squamous cell carcinoma: The overshadowing
threat for patients with early-stage disease. Int J Radiat Oncol
Biol Phys. 17:691–694. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Day GL, Blot WJ, Shore RE, McLaughlin JK,
Austin DF, Greenberg RS, Liff JM, Preston-Martin S, Sarkar S,
Schoenberg JB, et al: Second cancers following oral and pharyngeal
cancers: Role of tobacco and alcohol. J Natl Cancer Inst.
86:131–137. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
León X, Quer M, Diez S, Orús C,
López-Pousa A and Burgués J: Second neoplasm in patients with head
and neck cancer. Head Neck. 21:204–210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee DH, Roh JL, Baek S, Jung JH, Choi SH,
Nam SY and Kim SY: Second cancer incidence, risk factor, and
specific mortality in head and neck squamous cell carcinoma.
Otolaryngol Head Neck Surg. 149:579–586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sheth SH, Johnson DE, Kensler TW and
Bauman JE: Chemoprevention targets for tobacco-related head and
neck cancer: Past lessons and future directions. Oral Oncol.
51:557–564. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Slaughter DP, Southwick HW and Smejkal W:
Field cancerization in oral stratified squamous epithelium;
clinical implications of multicentric origin. Cancer. 6:963–968.
1953. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hong WK, Endicott J, Itri LM, Doos W,
Batsakis JG, Bell R, Fofonoff S, Byers R, Atkinson EN, Vaughan C,
et al: 13-cis-retinoic acid in the treatment of oral leukoplakia. N
Engl J Med. 315:1501–1505. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hong WK, Lippman SM, Itri LM, Karp DD, Lee
JS, Byers RM, Schantz SP, Kramer AM, Lotan R, Peters LJ, et al:
Prevention of second primary tumors with isotretinoin in
squamous-cell carcinoma of the head and neck. N Engl J Med.
323:795–801. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Khuri FR, Lee JJ, Lippman SM, Kim ES,
Cooper JS, Benner SE, Winn R, Pajak TF, Williams B, Shenouda G, et
al: Randomized phase III trial of low-dose isotretinoin for
prevention of second primary tumors in stage I and II head and neck
cancer patients. J Natl Cancer Inst. 98:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leeman-Neill RJ, Seethala RR, Singh SV,
Freilino ML, Bednash JS, Thomas SM, Panahandeh MC, Gooding WE,
Joyce SC, Lingen MW, et al: Inhibition of EGFR-STAT3 signaling with
erlotinib prevents carcinogenesis in a chemically-induced mouse
model of oral squamous cell carcinoma. Cancer Prev Res (Phila).
4:230–237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gohagan JK, Prorok PC, Hayes RB and Kramer
BS; Prostate Lung, Colorectal and Ovarian Cancer Screening Trial
Project Team, : The prostate, lung, colorectal and ovarian (PLCO)
cancer screening trial of the national cancer institute: History,
organization, and status. Control Clin Trials. 21 (Suppl
6):251S–272S. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mulshine JL, Atkinson JC, Greer RO,
Papadimitrakopoulou VA, Van Waes C, Rudy S, Martin JW, Steinberg
SM, Liewehr DJ, Avis I, et al: Randomized, double-blind,
placebo-controlled phase IIb trial of the cyclooxygenase inhibitor
ketorolac as an oral rinse in oropharyngeal leukoplakia. Clin
Cancer Res. 10:1565–1573. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jayaprakash V, Rigual NR, Moysich KB,
Loree TR, Nasca MA, Menezes RJ and Reid ME: Chemoprevention of head
and neck cancer with aspirin: A case-control study. Arch
Otolaryngol Head Neck Surg. 132:1231–1236. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Papadimitrakopoulou VA, William WN Jr,
Dannenberg AJ, Lippman SM, Lee JJ, Ondrey FG, Peterson DE, Feng L,
Atwell A, El-Naggar AK, et al: Pilot randomized phase II study of
celecoxib in oral premalignant lesions. Clin Cancer Res.
14:2095–2101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahmadi N, Goldman R, Seillier-Moiseiwitsch
F, Noone AM, Kosti O and Davidson BJ: Decreased risk of squamous
cell carcinoma of the head and neck in users of nonsteroidal
anti-inflammatory drugs. Int J Otolaryngol. 2010:4241612010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wilson JC, Murray LJ, Hughes CM, Black A
and Anderson LA: Non-steroidal anti-inflammatory drug and aspirin
use and the risk of head and neck cancer. Br J Cancer.
108:1178–1181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saba NF, Hurwitz SJ, Kono SA, Yang CS,
Zhao Y, Chen Z, Sica G, Muller S, Moreno-Williams R, Lewis M, et
al: Chemoprevention of head and neck cancer with celecoxib and
erlotinib: Results of a phase ib and pharmacokinetic study. Cancer
Prev Res (Phila). 7:283–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hedberg ML, Peyser ND, Bauman JE, Gooding
WE, Li H, Bhola NE, Zhu TR, Zeng Y, Brand TM, Kim MO, et al: Use of
nonsteroidal anti-inflammatory drugs predicts improved patient
survival for PIK3CA-altered head and neck cancer. J Exp Med.
216:419–427. 2019.PubMed/NCBI
|
|
29
|
Day GL, Shore RE, Blot WJ, McLaughlin JK,
Austin DF, Greenberg RS, Liff JM, Preston-Martin S, Sarkar S,
Schoenberg JB, et al: Dietary factors and second primary cancers: A
follow-up of oral and pharyngeal cancer patients. Nutr Cancer.
21:223–232. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chainani-Wu N: Diet and oral, pharyngeal,
and esophageal cancer. Nutr Cancer. 44:104–126. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pavia M, Pileggi C, Nobile CG and
Angelillo IF: Association between fruit and vegetable consumption
and oral cancer: A meta-analysis of observational studies. Am J
Clin Nutr. 83:1126–1134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fowke JH: Head and neck cancer: A case for
inhibition by isothiocyanates and indoles from cruciferous
vegetables. Eur J Cancer Prev. 16:348–356. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bravi F, Bosetti C, Filomeno M, Levi F,
Garavello W, Galimberti S, Negri E and La Vecchia C: Foods,
nutrients and the risk of oral and pharyngeal cancer. Br J Cancer.
109:2904–2910. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kensler TW, Egner PA, Agyeman AS,
Visvanathan K, Groopman JD, Chen JG, Chen TY, Fahey JW and Talalay
P: Keap1-nrf2 signaling: A target for cancer prevention by
sulforaphane. Top Curr Chem. 329:163–177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hong F, Freeman ML and Liebler DC:
Identification of sensor cysteines in human Keap1 modified by the
cancer chemopreventive agent sulforaphane. Chem Res Toxicol.
18:1917–1926. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kensler TW and Wakabayashi N: Nrf2: Friend
or foe for chemoprevention? Carcinogenesis. 31:90–99. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K,
Yamamoto M, Talalay P and Kensler TW: Sensitivity to carcinogenesis
is increased and chemoprotective efficacy of enzyme inducers is
lost in nrf2 transcription factor-deficient mice. Proc Natl Acad
Sci USA. 98:3410–3415. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fahey JW, Haristoy X, Dolan PM, Kensler
TW, Scholtus I, Stephenson KK, Talalay P and Lozniewski A:
Sulforaphane inhibits extracellular, intracellular, and
antibiotic-resistant strains of Helicobacter pylori and prevents
benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci USA.
99:7610–7615. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu C, Huang MT, Shen G, Yuan X, Lin W,
Khor TO, Conney AH and Kong AN: Inhibition of
7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in
C57BL/6 mice by sulforaphane is mediated by nuclear factor
E2-related factor 2. Cancer Res. 66:8293–8296. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cornblatt BS, Ye L, Dinkova-Kostova AT,
Erb M, Fahey JW, Singh NK, Chen MS, Stierer T, Garrett-Mayer E,
Argani P, et al: Preclinical and clinical evaluation of
sulforaphane for chemoprevention in the breast. Carcinogenesis.
28:1485–1490. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bauman JE, Zang Y, Sen M, Li C, Wang L,
Egner PA, Fahey JW, Normolle DP, Grandis JR, Kensler TW and Johnson
DE: Prevention of carcinogen-induced oral cancer by sulforaphane.
Cancer Prev Res (Phila). 9:547–557. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kensler TW, Chen JG, Egner PA, Fahey JW,
Jacobson LP, Stephenson KK, Ye L, Coady JL, Wang JB, Wu Y, et al:
Effects of glucosinolate-rich broccoli sprouts on urinary levels of
aflatoxin-DNA adducts and phenanthrene tetraols in a randomized
clinical trial in He Zuo township, Qidong, People's Republic of
China. Cancer Epidemiol Biomarkers Prev. 14:2605–2613. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kensler TW, Ng D, Carmella SG, Chen M,
Jacobson LP, Munoz A, Egner PA, Chen JG, Qian GS, Chen TY, et al:
Modulation of the metabolism of airborne pollutants by
glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages
in Qidong, China. Carcinogenesis. 33:101–107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Egner PA, Chen JG, Zarth AT, Ng DK, Wang
JB, Kensler KH, Jacobson LP, Munoz A, Johnson JL, Groopman JD, et
al: Rapid and sustainable detoxication of airborne pollutants by
broccoli sprout beverage: Results of a randomized clinical trial in
China. Cancer Prev Res (Phila). 7:813–823. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thimmulappa RK, Mai KH, Srisuma S, Kensler
TW, Yamamoto M and Biswal S: Identification of Nrf2-regulated genes
induced by the chemopreventive agent sulforaphane by
oligonucleotide microarray. Cancer Res. 62:5196–5203.
2002.PubMed/NCBI
|
|
46
|
Hu R, Hebbar V, Kim BR, Chen C, Winnik B,
Buckley B, Soteropoulos P, Tolias P, Hart RP and Kong AN: In vivo
pharmacokinetics and regulation of gene expression profiles by
isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther.
310:263–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Traka M, Gasper AV, Smith JA, Hawkey CJ,
Bao Y and Mithen RF: Transcriptome analysis of human colon Caco-2
cells exposed to sulforaphane. J Nutr. 135:1865–1872. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hu R, Xu C, Shen G, Jain MR, Khor TO,
Gopalkrishnan A, Lin W, Reddy B, Chan JY and Kong AN: Gene
expression profiles induced by cancer chemopreventive
isothiocyanate sulforaphane in the liver of C57BL/6J mice and
C57BL/6J/Nrf2 (−/−) mice. Cancer Lett. 243:170–192. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bhamre S, Sahoo D, Tibshirani R, Dill DL
and Brooks JD: Temporal changes in gene expression induced by
sulforaphane in human prostate cancer cells. Prostate. 69:181–190.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Agyeman AS, Chaerkady R, Shaw PG, Davidson
NE, Visvanathan K, Pandey A and Kensler TW: Transcriptomic and
proteomic profiling of KEAP1 disrupted and sulforaphane-treated
human breast epithelial cells reveals common expression profiles.
Breast Cancer Res Treat. 132:175–187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Thejass P and Kuttan G: Modulation of
cell-mediated immune response in B16F-10 melanoma-induced
metastatic tumor-bearing C57BL/6 mice by sulforaphane.
Immunopharmacol Immunotoxicol. 29:173–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Singh SV, Warin R, Xiao D, Powolny AA,
Stan SD, Arlotti JA, Zeng Y, Hahm ER, Marynowski SW, Bommareddy A,
et al: Sulforaphane inhibits prostate carcinogenesis and pulmonary
metastasis in TRAMP mice in association with increased cytotoxicity
of natural killer cells. Cancer Res. 69:2117–2125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shih YL, Wu LY, Lee CH, Chen YL, Hsueh SC,
Lu HF, Liao NC and Chung JG: Sulforaphane promotes immune responses
in a WEHI-3-induced leukemia mouse model through enhanced
phagocytosis of macrophages and natural killer cell activities
in vivo. Mol Med Rep. 13:4023–4029. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Amin PJ and Shankar BS: Sulforaphane
induces ROS mediated induction of NKG2D ligands in human cancer
cell lines and enhances susceptibility to NK cell mediated lysis.
Life Sci. 126:19–27. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Stoner GD, Kaighn ME, Reddel RR, Resau JH,
Bowman D, Naito Z, Matsukura N, You M, Galati AJ and Harris CC:
Establishment and characterization of SV40 T-antigen immortalized
human esophageal epithelial cells. Cancer Res. 51:365–371.
1991.PubMed/NCBI
|
|
57
|
Egner PA, Chen JG, Wang JB, Wu Y, Sun Y,
Lu JH, Zhu J, Zhang YH, Chen YS, Friesen MD, et al: Bioavailability
of Sulforaphane from two broccoli sprout beverages: Results of a
short-term, cross-over clinical trial in Qidong, China. Cancer Prev
Res (Phila). 4:384–395. 2011. View Article : Google Scholar : PubMed/NCBI
|