Introduction
Mesenchymal stem cells (MSCs) are currently being
increasingly investigated for their therapeutic potential in
regenerative medicine and inflammatory conditions (1-4), due
mainly to their multi-lineage differentiation potential (5) and their limited expression of MHC
class II antigen (6,7), as well as their immunomodulatory
functions (8-11). The former allows MSCs to be applied
to a wide range of clinical conditions, while the latter allows
MSCs to be used not only for autologous but also for allogeneic
transplantations. These application potentials strongly suggest a
need for identifying a stem cell source that is capable of
providing plentiful and ethically acceptable yet easily procured
stem cells. MSCs are traditionally identified and isolated from
bone marrow and bone marrow-derived MSCs (BM-MSCs) remain the most
commonly investigated MSCs (2).
Previous studies have shown some limitations of BM-MSCs to their
wide range of application, including that they have to be isolated
through an invasive procedure, that they exist in bone marrow in a
very low ratio (12), and that
their differentiation capacity decreases as the donor’s age
increases (13). In recent years,
increasing attention has been placed on placenta-derived MSCs
(P-MSCs) (6,14). Besides sharing major properties
with BM-MSCs (15), P-MSCs have
shown certain unique properties that make them a prime candidate
for stem cell-based therapies. Such properties include: i)
Placental tissues originate from the early stage of embryonic
development, which may logically lead to the hypothesis that MSCs
from these tissues may have retained the plasticity of their mother
stem cells (14). This hypothesis
has been supported by recent studies showing that MSCs from
amniotic and chorionic membranes are pluripotent and may
differentiate towards not only a mesodermal lineage but also into
ectodermal (16,17) and endodermal (18) cells. ii) Placenta is the tissue
that maintains fetomaternal immune tolerance during pregnancy,
indicating that cells from this tissue may have immunomodulatory
functions. This is evidenced by the observation that P-MSCs in the
fetomaternal interface show higher immunosuppressive effects than
adult MSCs (19). iii) P-MSCs can
be procured from term placentas in large supply, free of invasive
procedures and ethical complications. A recent scale-up culture
study showed that a range of 7.6-11.6×108 of passage 2
P-MSCs can be obtained from approximately half of a placental
tissue mass, and such cells maintain a 0.6-0.8/day doubling rate
through passage 5 (20),
indicative of placenta as a unique and plentiful source of MSCs for
clinical applications.
Of equal importance to identifying proper stem cell
source for cell-based therapies is the establishment of
clinic-compliant methods for in vitro cell expansion, which
should result in cells free of safety complications, yet maintain
full stemness for expanded cells. Previously, the majority of
expansion protocols for scale-up production of MSCs used medium
supplemented with fetal bovine serum (FBS) (21). It has been observed that patients
transplanted with T cells grown in FBS supplemented medium
developed IgG immunity against FBS proteins (22), and that xenogenic proteins retained
in MSC cytoplasm may have elicited immunological responses in
vivo following cell transplantation (23). The concerns of xenogenic protein
effects have drawn interest in using serum-free conditions.
However, the long-term effects of serum-free medium have yet to be
defined, since cells cultivated in serum-free conditions have
demonstrated contradictory results (21). To this end, the replacement of FBS
with human serum has been explored. While adult human serum has
been found to present several significant limitations (24), autologous human cord blood serum
(hCBS) and autologous adult human serum have been successfully
tested for the in vitro expansion of hematopoietic stem
cells (25), T cells (26), as well as BM-MSCs and cord
blood-derived MSCs (27,28).
Furthermore, the amniotic side and maternal side of
the placenta may isolate fetal and maternal origin P-MSCs;
therefore, it is crucial to verify the origins of P-MSCs. Owing to
short tandem repeats (STRs) having polymorphisms and high mutation
rates, they are broadly used for cell authentication. STRs are
short tandemly repeated DNA sequences that involve a repetitive
unit of 1-6 bp. Strand-slippage replication gives rise to the
mutation of STRs and to different individual DNA samples containing
the length-variant STR alleles, which may be separated by
polyacrylamide gel electrophoresis.
Placental tissues are developmentally different from
other MSC origins, and P-MSCs are developed in vivo in an
environment different from other MSCs. In an attempt to establish a
clinically compliant protocol for P-MSC processing, the
characteristics of P-MSCs cultured in autologous hCBS were
investigated as a replacement for FBS. In this study, we
demonstrate that P-MSCs expanded in autologous hCBS maintain their
self-proliferation capacity and multi-lineage differentiation
potential, express a MSC-specific phenotype, and are genetically
stable.
Materials and methods
Isolation and culture of P-MSCs
Full-term placentas were obtained from healthy
mothers at the time of routine elective caesarean section in the
Affiliated Hospital of Ningxia Medical University. Informed consent
was obtained from each mother prior to delivery. The placental
tissues were immerged under aseptic conditions into protection
solution containing 98% DMEM-low glucose (DMEM-LG; Invitrogen,
Carlsbad, CA, USA), 1% hCBS (prepared in the laboratory) and 1%
penicillin/streptomycin solution (Sigma, St. Louis, MO, USA), and
transferred to the laboratory within 30 min for processing. Pieces
of placental tissue approximately 1-cm-thick were dissected from
the amniotic side of the placentas, and cut into small pieces
approximately 1 mm3 in size, washed in PBS (Invitrogen)
to remove blood cells, and then digested with a combination of 270
U/ml collagenase IV (Roche, Penzberg, Germany) and 240 U/ml dispase
II (Roche) at 37°C for 1 h in a shaker water bath. After digestion,
the cells in suspension were filtered through 150 μm filters (BD,
Franklin Lakes, NJ, USA), collected by centrifugation at 100 × g
for 5 min, washed 3 times, and seeded in 24-cm flasks.
Three different media were used for comparison.
Medium I: DMEM-LG supplemented with 10% FBS (Invitrogen), 2%
L-glutamine and 1% penicillin/streptomycin solution; medium II:
Same as medium I with the exception that the FBS was replaced by
10% autologous hCBS; medium III: Same as medium I with the
exception that the FBS was replaced by 10% non-autologous hCBS.
Cells in all media were cultured in a 37°C, 5% CO2,
humidified incubator. Upon reaching 80% confluence, cells were
harvested with 0.25% trypsin/0.05% EDTA (Invitrogen) and
subcultured by a 1:3 dilution. The cell doubling cycle was
determined by dividing the cell number increase during a given
passage with the length of time (hour) of that passage. After
passage 3, cell samples from each culture were cryopreserved in
solution containing 60% DMEM-LG, 30% hCBS and 10% dimethyl
sulphoxide (DMSO, Invitrogen).
To verify the fetal origin of P-MSCs, maternal
P-MSCs were isolated from tissues 0.5-cm-thick from the maternal
side of the same placentas.
Verification of the fetal origin of
P-MSCs
Blood samples were collected from parents of the
newborn babies whose placentas had been used for isolation of fetal
and maternal P-MSCs. Informed consent was obtained from the parents
prior to sample collection. DNA was prepared from the two types of
P-MSCs as well as the from parental blood samples using an AxyPrep
kit (Axygen). Four known STR polymorphic sites (29,30)
were selected for genetic matching. Polymerase chain reaction (PCR)
primers for these STR sites are shown in Table I. PCR was carried out using an
EasyTaq PCR SuperMix kit (Transgen, Beijing, China) for 30 cycles
under the following conditions: 94°C ×50 sec, annealing temperature
×50 sec, 72°C ×40 sec. PCR results were viewed by polyacrylamide
gel electrophoresis.
 | Table IPCR primers for polymorphic tandem
repeat markers and expected discrimination powers. |
Table I
PCR primers for polymorphic tandem
repeat markers and expected discrimination powers.
| Gene site | Primer
sequence | Tm (°C) | Polymorphic
fragment (bp) | Discrimination
power (DP) | Combined DP |
|---|
| D2S1399 |
5′-CATTGGTCCAGGTAAACTGC-3′ | 55 | 136-176 | 0.958 | 0.9999926 |
|
5′-TTCACAAGGTTCCACAAGGT-3′ | | | |
| D10S2325 |
5′-CTCACGAAAGAAGCCTTCTG-3′ | 58 | 208-233 | 0.948 |
|
5′-GAGCTGAGAGATCACGCACT-3′ | | | |
| GATA198B05 |
5′-GTGGAGCCAATTCACCTAAT-3′ | 55 | 112-152 | 0.9533 |
|
5′-GAGAGACAGAACCTATAGCATGC-3′ | | | |
| D18S535 |
5′-TCATGTGACAAAAGCCACAC-3′ | 55 | 131-163 | 0.927 |
|
5′-AGACAGAAATATAGATGAGAATGCA-3′ | | | |
Phenotypic characterization of P-MSCs by
flow cytometry
All monoclonal antibodies used for flow cytometry
were obtained from BD Pharmingen (Franklin Lakes, NJ, USA), and
prepared in PBS. Single cell suspensions were made from cell
cultures by trysinization and prepared in PBS. Cells
(1×106) were incubated in 150 μl with one of the
following antibodies for 10 min at room temperature: IgG1-PE,
CD29-PE, CD34-PE, CD73-PE, IgG2a-FITC, CD14-FITC, CD44-FITC,
CD45-FITC, CD90-FITC, CD105-FITC or HLA-DR-FITC. After this
incubation, cells were resuspended in 300 μl PBS. Flow cytometry
analysis was performed on a Calibur cytometer (BD Biosciences).
Multi-lineage differentiation of
P-MSCs
Differentiation of P-MSCs towards adipogenic,
osteogenic and neurogenic lineages was performed in vitro.
P-MSCs at passage 3 were used for all differentiation studies.
Adipogenic differentiation was induced by culturing 70% confluent
P-MSCs for 10 days in DMEM-LG medium supplemented with 10% FBS, 1
μM dexamethasone, 1mM 3-isobutyl-1-methyl-xanthine, 10 μg/ml
insulin and 60 μM Indole Miyuki-3-isobutyl-1-methylxanthine
(Sigma). Adipogenic differentiation was assessed by oil Red O
staining. Osteogenic differentiation was induced by culturing 70%
confluent P-MSCs for 14 days in DMEM-LG containing 10% FBS, 100 nM
dexamethasone, 10 mM β-glycerophosphate (Sigma), 0.05 mM ascorbic
acid-2-phosphate (Wako Pure Chemical Industries, Tokyo, Japan) and
2 mM L-glutamine. Osteogenic differentiation was assessed by
alizarin red staining. Neurogenic differentiation was induced by
culturing 50% confluent P-MSCs for 12 days in DMEM-LG containing
10% FBS, 2 mM L-glutamine, 2% DMSO, 200 μM butylated hydroxyanisole
(BHA, Sigma) and 25 ng/ml NGF. Neurogenic differentiation was
assessed by immunostaining for the neural cell-specific proteins
neuron-specific enolase (NSE), glial fibrillary acidic protein
(GFAP), neurofilament (NF-M), growth-associated protein-43 (GAP-43)
and nestin.
Karyotyping and single cell gel
electrophoresis
P-MSCs from passages 2, 14 and 18, and from
cryopreserved passage 6 samples were karyotyped by Giemsa staining
of metaphase chromosomes and analyzed on a Cytovision analyzer
(Applied Imaging, Wetzlar, Germany). DNA stability of P-MSCs was
assessed for passage 2 cells by single cell gel electrophoresis
(SCGE) using an OxiSelect Comet Assay kit (Cell Biolabs, San Diego,
CA, USA) according to the manufacturer’s instructions. For DNA
damage quantification, 50 cells in the SCGE plate of each sample
were randomly selected under a microscope, and diameters of nuclei
(comet head) and tail moment (tail length) were measured. DNA
damage was represented by a parameter of tail moment/head
ratio.
Statistical analysis
Data are expressed as the means ± standard error and
analyzed by the student’s-test. A value of P<0.01 was considered
to indicate a statistically significant difference.
Results
Verification of P-MSCs for their fetal
origin
Placental tissues contain P-MSCs of both fetal and
maternal origin, originating respectively from the amniotic and
maternal side of the placenta. To verify that the P-MSCs isolated
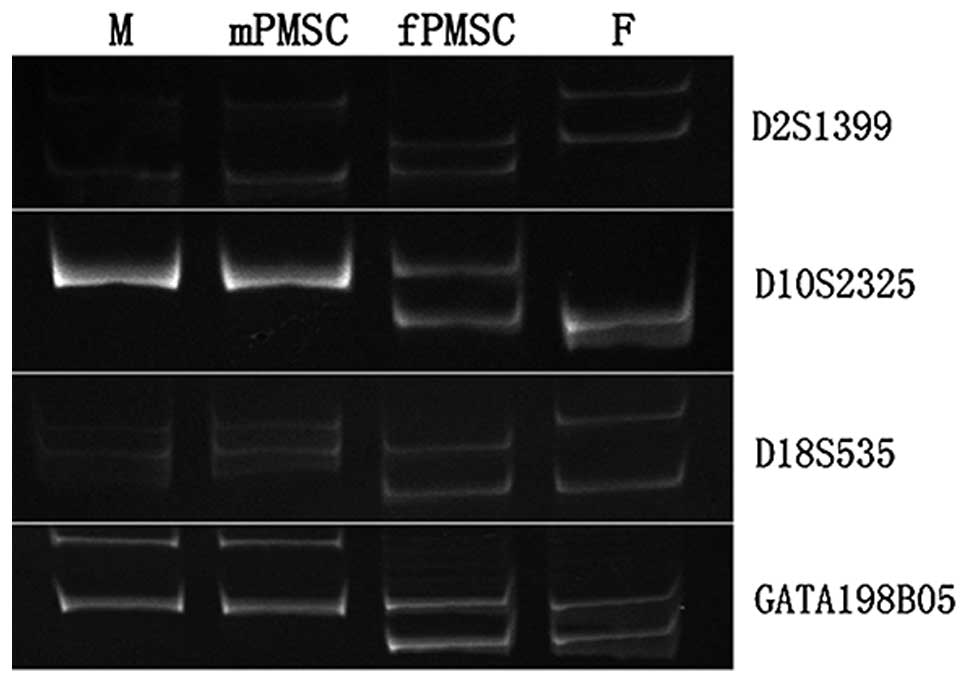
from the amniotic side were of fetal origin, 4 polymorphic STR
markers were used for parent-offspring genetic matching (Table I). These polymorphic markers
individually identify the genetic origin with higher than 90%
discrimination power (DP), and when combined, they give higher than
99.999% DP. As shown in Fig. 1,
for all the markers tested, the P-MSCs from the amniotic side of
the placenta carry alleles of maternal and paternal origin,
indicative of the fetal genome, while the P-MSCs from the maternal
side of the placenta carry alleles of only maternal origin. This
test was performed for multiple samples with consistent results.
Therefore, 1-cm-thick tissue from the amniotic side of placentas
was used for all fetal P-MSC isolations.
P-MSCs growing in hCBS maintained the MSC
phenotype
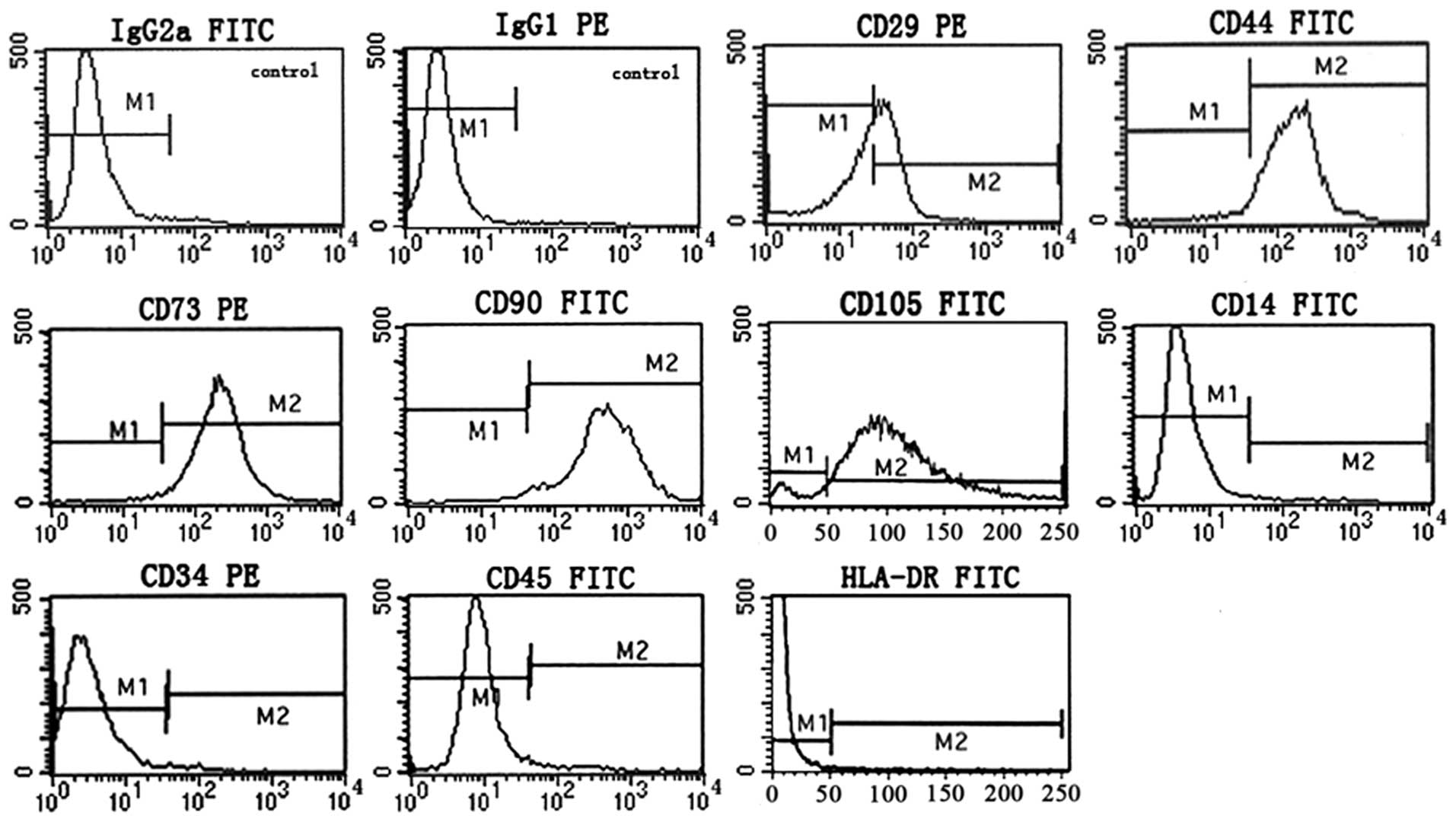
It is generally accepted that MSCs are positive for
surface antigens CD73, CD90 and CD105, and negative for CD34, CD45
and CD14, with restricted expression of HLA-DR (31). Flow cytometric phenotype analysis
of passage 3 P-MSCs grown in hCBS showed that the cells are
positive for CD29, CD44, CD73, CD90 and CD105, and negative for
CD14, CD34, CD45 and HLA-DR, indicative of a typical MSC phenotype
(Fig. 2). These results
demonstrate that hCBS supported the expression of a normal MSC
phenotype.
P-MSCs growing in hCBS maintained
multi-lineage differentiation potential
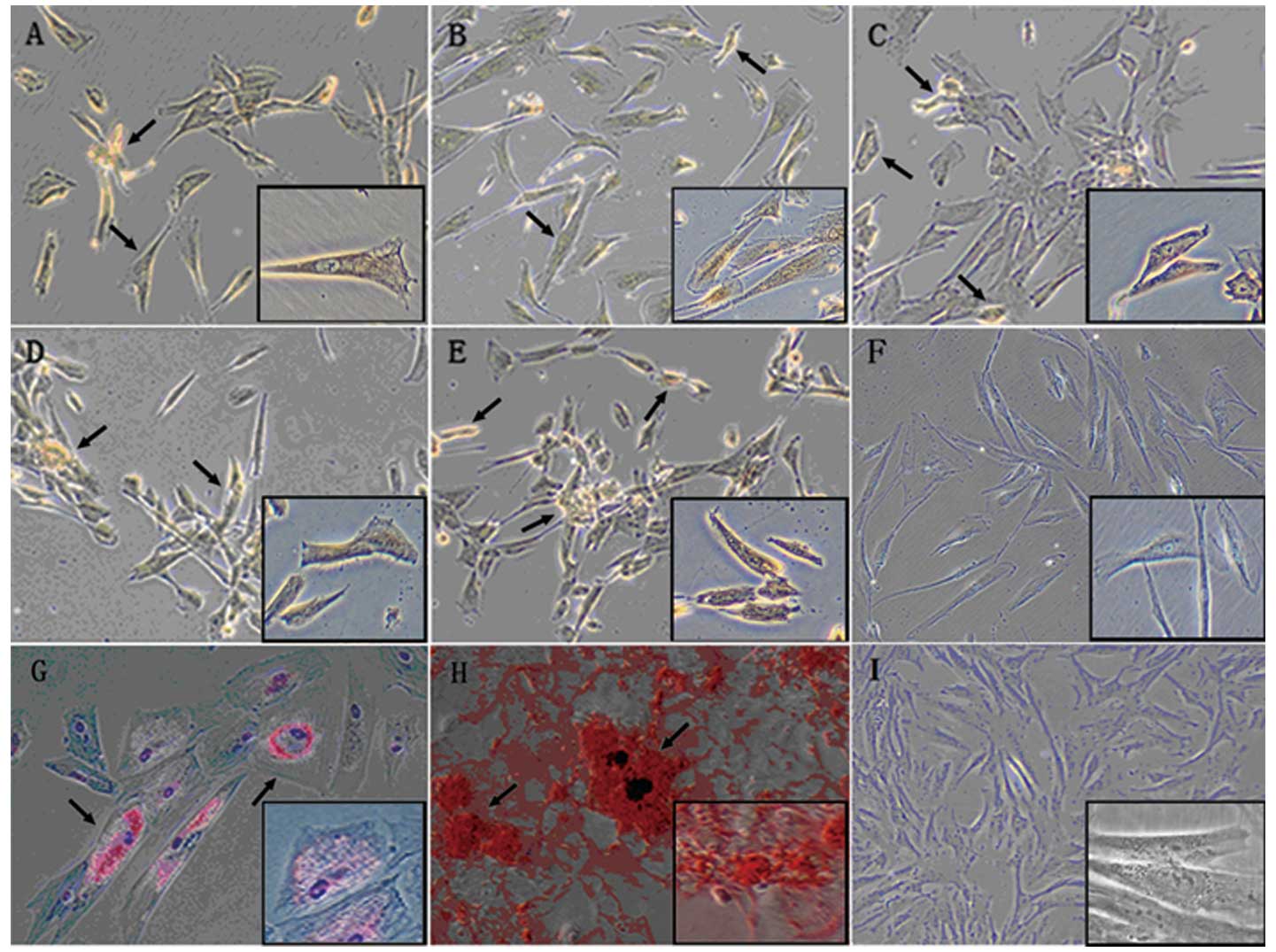
MSCs are characterized by the potential of
differentiation towards mesenchymal lineages (31). Recent studies also showed that
P-MSCs may contain pluripotent cells that are capable of
differentiating towards ectoderm and endoderm lineages (16-18).
To test whether hCBS supports P-MSC differentiation potential, we
performed adipogenic and osteogenic (mesenchymal lineage, Fig. 3G and H, respectively), as well as
neurogenic (ectodermal lineage, Fig.
3A-E), differentiation of P-MSCs. Differentiation protocols for
all the three lineages have been previously used for stem cell
differentiations (see Materials and methods). The results indicate
that P-MSCs grown in hCBS also differentiated into the ectodermal
lineage.
P-MSCs growing in hCBS are genetically
stable
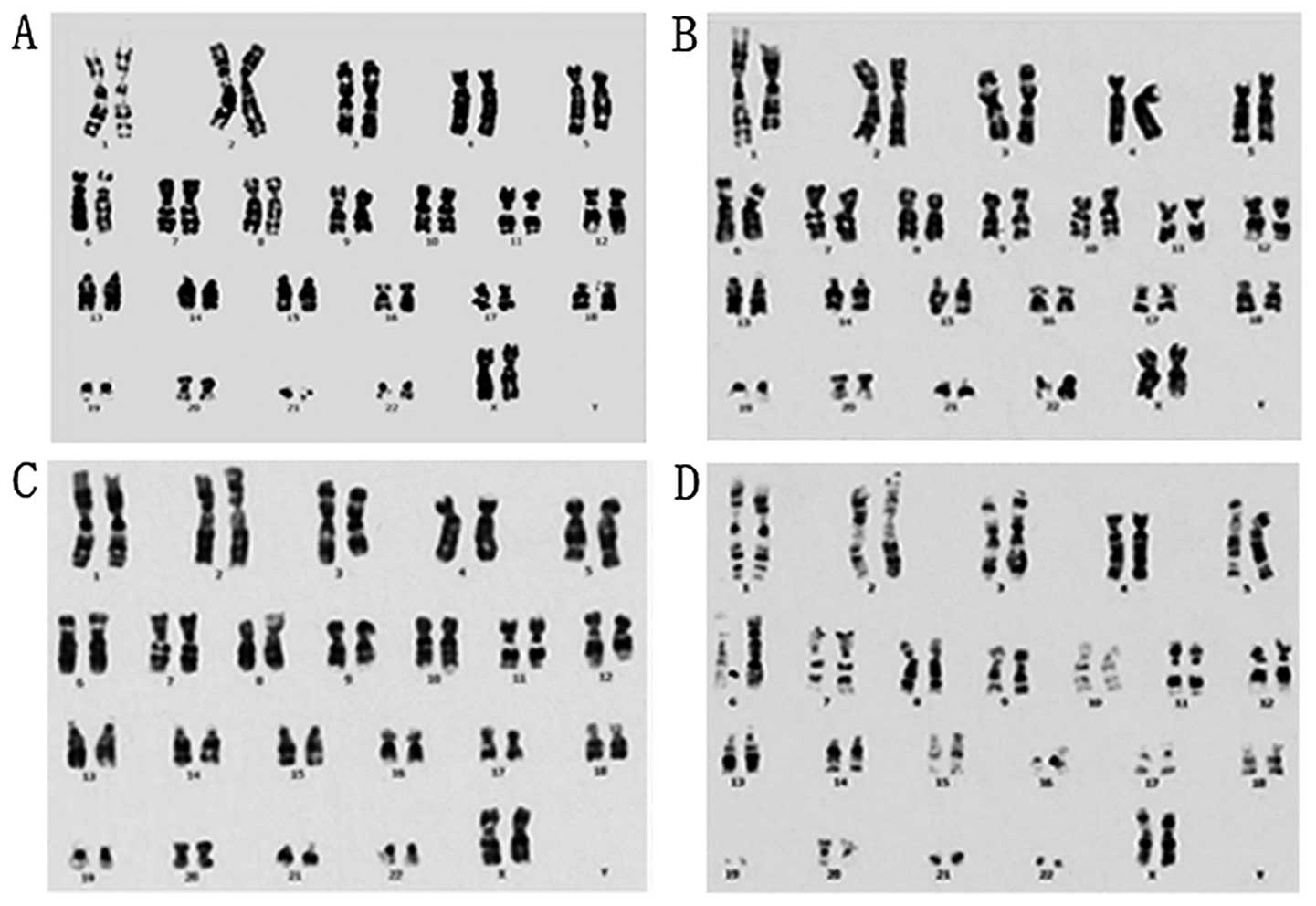
Genetic stability is one of the most important
characteristics of cultured cells when their clinical application
is concerned, and is affected by cell culture conditions. To verify
that hCBS are capable of supporting clinically compliant in
vitro expansion of P-MSCs, we tested the genetic stability of
P-MSCs cultured in hCBS at a cellular level by chromosome
karyotyping and at the DNA level by SCGE. Karyotyping for P-MSCs at
passages 2, 14 and 18 in cultures, and from cryopreserved cells of
passage 6, revealed no detectable genetic variation (Fig. 4). SCGE analysis showed normal
nucleus movement for hCBS cultured P-MSCs, either prior to or
following cryopreservation, and no difference in nucleus movement
between cells cultured in hCBS and in FBS (Fig. 5).
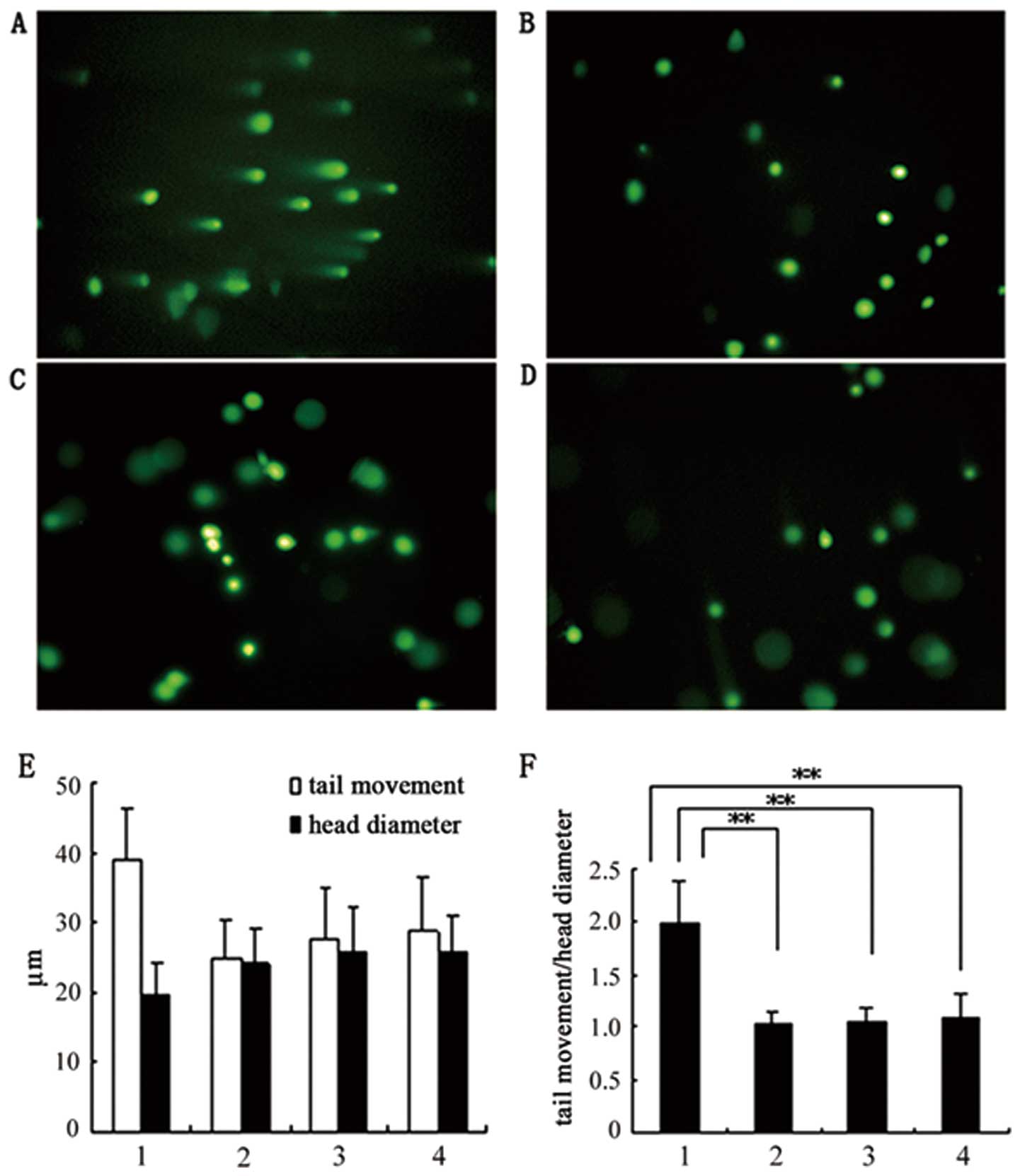 | Figure 5DNA damage analysis by single cell
gel electrophoresis (SCGE) (×200). Passage 2 P-MSCs cultured in
hCBS and FBS were analyzed and quantified. (A) Control. P-MSCs
treated with 5% H2O2 for 15 min prior to
electrophoresis showed DNA damage as detected by comet tail
moments. No DNA damage was detected in the SCGE assay for P-MSCs
either in hCBS (B) prior to and (C) following cryopreservation, or
(D) in FBS. (E and F) DNA damage quantification as measured by
comet tail moment. (E) Measurements of nucleus diameters and tail
moment length. (F) Nucleus moment index as presented by ratios of
nucleus diameters against tail moments (**P<0.01,
group 1 vs. group 2,3,4). (E and F) 1, P-MSCs in FBS treated with
H2O2; 2, P-MSCs in FBS; 3, P-MSCs in hCBS
prior to cryopreservation; 4, P-MSCs in hCBS following
cryopreservation. P-MSCs, human placenta-derived mesenchymal stem
cells; hCBS, human cord blood serum; FBS, fetal bovine serum. |
Discussion
In this study, we demonstrate that homologous hCBS
may be used as a replacement for FBS in the in vitro
expansion of human P-MSCs. Both cell proliferation capacity and
cell differentiation potential, two basic properties of stem cells,
were well maintained in P-MSCs when they were cultured in sustained
passages as well as when recultured following cryopreservation.
P-MSCs growing in hCBS were genetically stable and showed no
difference in DNA damage tests when compared with cells growing in
FBS. These results may serve as a starting point for the
development of a clinically compliant procedure for in vitro
P-MSC expansion, an alternative to FBS-based or serum-free
protocols.
MSCs have drawn increasing interest for their
therapeutic potentials, and hence their cell sources are becoming
an issue of concern. It has been estimated that one MSC may be
found in every 10,000 nucleated cells in the bone marrow of a human
newborn, and this ratio would decrease by 10-fold in the marrow of
the teenage population, and by 100-fold in the marrow of the
elderly population (32). By
contrast, human placentas have proven to be a rich and easy to
procure MSC source. In this study, it was observed that, when
cultured in hCBS, P-MSCs showed a similar phenotype and multiple
differentiation potentials in comparison with BM-MSCs, as
characterized by cell surface antigen typing and as tested for the
generation of mesenchymal and ectodermal cell lineages,
respectively. These observations are in agreement with certain
earlier studies using various culture systems (31). Recently, a study showed that
P-MSCs, when compared with BM-MSCs, appeared to be more immunogenic
and to have less immunomodulatory functions (33). In this study and previous studies
(31), a significant immunogenic
phenotype of P-MSCs was not observed. The different observations in
different studies may be partly due to the difference in cell
populations tested or in the cell culture conditions employed. In
Fazekasova et al’s study (33) a mixture of fetal and maternal
P-MSCs were tested. Although the immunogenic properties of maternal
P-MSCs are less understood, it may be different from that of fetal
P-MSCs, as shown in a previous study where P-MSCs of fetal origin
(from amniotic membranes) revealed higher immunomodulatory
functions than BM-MSCs (19). In
our study, we isolated fetal P-MSCs and verified their genetic
origin. These fetal P-MSCs showed no significant HLA-DR expression
when cultured in hCBS, in agreement with earlier studies with
amniotic membrane cells. The fetal identity of P-MSCs may be
advantageous when clinical application is concerned, since a
homologous genetic background of cells may reduce possible
complications. At the same time, the isolation of fetal P-MSCs
excluded a possibility that the growth of fetal P-MSCs may be
partially supported by maternal cells in the same culture, hence
leading to the conclusion that hCBS may provide sufficient support
to fetal P-MSC growth. The immune properties of maternal P-MSCs
were not analyzed in the present study.
Another property of clinical significance for P-MSCs
is their differentiation potential. In the present study, P-MSCs
cultured in hCBS were differentiated into mesenchymal and
ectodermal lineages, indicative of multiple/pluri-potent
characteristics. A recent study showed that P-MSCS were less prone
to osteogenic differentiation and expressed a lower level of CD146
when compared with MB-MSCs (34),
yet another study showed successful generation of an osteogenic
graft from P-MSCs (35). It
appears that the expression of phenotypes and functions by P-MSCs
may vary when being cultured in different conditions or being
isolated from different tissue origins. Indeed, we have observed
that P-MSCs isolated from different donors may vary significantly
in terms of proliferation capacities even when cultured under the
same conditions (unpublished data). The question of why different
observations have been made for P-MSCs has to be addressed under
tightly controlled and well-defined conditions.
FBS is a well proven tissue culture component for
in vitro cell expansion, but its animal origin limits its
use in clinic-compliant protocols. Our present work demonstrates
that hCBS is an alternative to FBS for in vitro processing
of human P-MSCs. Although the effective components in hCBS are yet
to be defined, it is not difficult to hypothesize that P-MSCs can
be well adapted to this in vitro condition considering that
prior to being isolated the cells in the placenta were nourished
mainly by CBS-borne nutrients and growth factors. This may, at
least in part, explain why cells in homologous hCBS grew better
than in non-homologous hCBS. Taken together, the results in this
study suggest that homologous hCBS may support full stemness for
human P-MSCs, and may hence be used as an alternative to FBS in
clinically compliant procedures for processing human P-MSCs.
Acknowledgements
This study was supported in part by the National
Natural Science Foundation of China (NSFC Program No. 30960176)
(J.W.).
Abbreviations:
|
P-MSCs
|
human placenta-derived mesenchymal
stem cells
|
|
hCBS
|
human cord blood serum
|
|
DP
|
discrimination power
|
|
NSE
|
neuron-specific enolase
|
|
GFAP
|
glial fibrillary acidic protein
|
|
GAP-43
|
growth-associated protein-43
|
|
NF-M
|
neurofilament
|
|
SCGE
|
single cell gel electrophoresis
|
References
|
1
|
Brooke G, Cook M, Blair C, et al:
Therapeutic applications of mesenchymal stromal cells. Semi Cell
Dev Biol. 18:846–858. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Le Blanc K, Frassoni F, Ball L, et al:
Mesenchymal stem cells for treatment of steroid-resistant, severe,
acute graft-versus-host disease: a phase II study. Lancet.
371:1579–1586. 2008.
|
|
3
|
Sun LY, Kentaro A, Zhang HY, et al:
Mesenchymal stem cell transplantation reverses multiorgan
dysfunction in systemic lupus erythematosus mice and humans. Stem
Cells. 27:1421–1432. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Centeno CJ, Busse D, Kisiday J, Keohan C,
Freeman M and Karli D: Increased knee cartilage volume in
degenerative joint disease using percutaneously implanted,
autologous mesenchymal stem cells. Pain Physician. 11:343–353.
2008.
|
|
5
|
Sekiya I, Larson BL, Smith JR, Pochampally
R, Cui JG and Prockop DJ: Expansion of human adult stem cells from
bone marrow stroma: conditions that maximize the yields of early
progenitors and evaluate their quality. Stem Cells. 20:530–541.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parolini O, Alviano F, Bergwerf I, et al:
Toward cell therapy using placenta-derived cells: disease
mechanisms, cell biology, preclinical studies, and regulatory
aspects at the round table. Stem Cells Dev. 19:143–155. 2010.
View Article : Google Scholar
|
|
7
|
Chang CJ, Yen ML, Chen YC, et al:
Placenta-derived multipotent cells exhibit immunosuppressive
properties that are enhanced in the presence of interferon-gamma.
Stem Cells. 24:2466–2477. 2006. View Article : Google Scholar
|
|
8
|
Magatti M, De Munari S, Vertua E, Gibelli
L, Wengler GS and Parolini O: Human amnion mesenchyme harbors cells
with allogeneic T-cell suppression and stimulation capabilities.
Stem Cells. 26:182–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bailo M, Soncini M, Vertua E, et al:
Engraftment potential of human amnion and chorion cells derived
from term placenta. Transplantation. 78:1439–1448. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Niederkorn JY, Neelam S, et al:
Immunosuppressive factors secreted by human amniotic epithelial
cells. Invest Ophthalmol Vis Sci. 46:900–907. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Magatti M, De Munari S, Vertua E, et al:
Amniotic mesenchymal tissue cells inhibit dendritic cell
differentiation of peripheral blood and amnion resident monocytes.
Cell Transplant. 18:899–914. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pittenger MF, Mackay AM, Beck SC, et al:
Multilineage potential of mesenchymal cells. Science. 284:143–147.
1999. View Article : Google Scholar
|
|
13
|
Mareschi K, Ferrero I, Rustichelli D, et
al: Expansion of mesenchymal stem cells isolated from pediatric and
adult donor bone marrow. J Cell Biochem. 97:744–754. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Evangelista M, Soncini M and Parolini O:
Placenta-derived stem cells: new hope for cell therapy.
Cytotechnology. 58:33–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barlow S, Brooke G, Chatterjee K, et al:
Comparison of human placenta- and bone marrow-derived multipotent
mesenchymal stem cells. Stem Cells Dev. 17:1095–1107. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Portmann-Lanz CB, Schoeberlein A, Huber A,
et al: Placental mesenchymal stem cells as potential autologous
graft for preand perinatal neuroregeneration. Am J Obstet Gynecol.
194:664–673. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sakuragawa N, Kakinuma K, Kikuchi A, et
al: Human amnion mesenchyme cells express phenotypes of neuroglial
progenitor cells. J Neurosci Res. 78:208–214. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei JP, Zhang TS, Kawa S, et al: Human
amnion-isolated cells normalize blood glucose in
streptozotocin-induced diabetic mice. Cell Transplant. 12:545–552.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roelen DL, van der Mast BJ, in’t Anker PS,
et al: Differential immunomodulatory effects of fetal versus
maternal multipotent stromal cells. Hum Immunol. 70:16–23. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brooke G, Rossetti T, Pelekanos R, et al:
Manufacturing of human placenta-derived mesenchymal stem cells for
clinical trials. Br J Haematol. 144:571–579. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mannello F and Tonti GA: No breakthroughs
for human mesenchymal and embryonic stem cell culture: conditioned
medium, feeder layer, or feeder-free; medium with fetal calf serum,
human serum, or enriched plasma; serum-free, serum replacement
nonconditioned medium, or ad hoc formula? all that glitters is not
gold! Stem Cells. 25:1603–1609. 2007.
|
|
22
|
Tuschong L, Soenen SL, Blaese RM, Candotti
F and Muul LM: Immune response to fetal calf serum by two adenosine
deaminase-deficient patients after T cell gene therapy. Hum Gene
Ther. 13:1605–1610. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Spees JL, Gregory CA, Singh H, et al:
Internalized antigens must be removed to prepare hypoimmunogenic
mesenchymal stem cells for cell and gene therapy. Mol Ther.
9:747–756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Berger MG, Veyrat-Masson R, Rapatel C,
Descamps S, Chassagne J and Boiret-Dupre N: Cell culture medium
composition and translational adult bone marrow-derived stem cell
research. Stem Cells. 24:2888–2990. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lam AC, Li K, Zang ZB, et al: Preclinical
ex vivo expansion of cord blood hematopoietic stem and progenitor
cells: duration of culture; the media, serum supplements, and
growth factors used; and engraftment in NOD/SCID mice. Transfusion.
41:1567–1576. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim YM, Jung MH, Song HY, et al: Ex vivo
expansion of Human umbilical cord blood derived T lymphocytes with
homologous cord blood plasma. Tohoku J Exp Med. 205:115–122. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shetty P, Bharucha K and Tanavde V: Human
umbilical cord blood serum can replace fetal bovine serum in the
culture of mesenchymal stem cells. Cell Biol Int. 31:293–298. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mizuno N, Shiba H, Ozeki Y, et al: Human
autologous serum obtained using a completely closed bag system as a
substitute for foetal calf serum in human mesenchymal stem cell
cultures. Cell Biol Int. 30:521–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xia SX, Gao YZ, Bian SZ, et al: Genetic
polymorphisms of two STR loci D2S1399 and D5S2500 in eastern
Chinese Han population. Fa Yi Xue Za Zhi. 20:200–201.
2004.PubMed/NCBI
|
|
30
|
Nievas P, Martinez Jarreta B, Abecia E and
Lareu MV: Fluorescence based amplification of the STR loci D18S535,
D1S1656 and D12S391 in a population sample from Aragon (North
Spain). Int J Legal Med. 113:58–59. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parolini O, Alviano F, Bagnara GP, et al:
Concise review: isolation and characterization of cells from human
term placenta: outcome of the first international Workshop on
Placenta Derived Stem Cells. Stem Cells. 26:300–311. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Caplan AL: Why are MSCs therapeutic? New
data: new insight. J Pathol. 217:318–324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fazekasova H, Lechler R, Langford K and
Lombardi G: Placenta-derived MSCs are partially immunogenic and
less immunomodulatory than bone marrow-derived MSCs. J Tissue Eng
Regen Med. 5:684–694. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pilz GA, Ulrich C, Ruh M, et al: Human
term placenta-derived mesenchymal stromal cells are less prone to
osteogenic differentiation than bone marrow-derived mesenchymal
stromal cells. Stem Cells Dev. 20:635–646. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mohr S, Portmann-Lanz CB, Schoeberlein A,
Sager R and Surbek DV: Generation of an osteogenic graft from human
placenta and placenta-derived mesenchymal stem cells. Reprod Sci.
17:1006–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|